NOS2 inhibitor 1400W Induces Autophagic Flux and Influences Extracellular Vesicle Profile in Human Glioblastoma U87MG Cell Line

 ,
, 

 ,
,  and
and 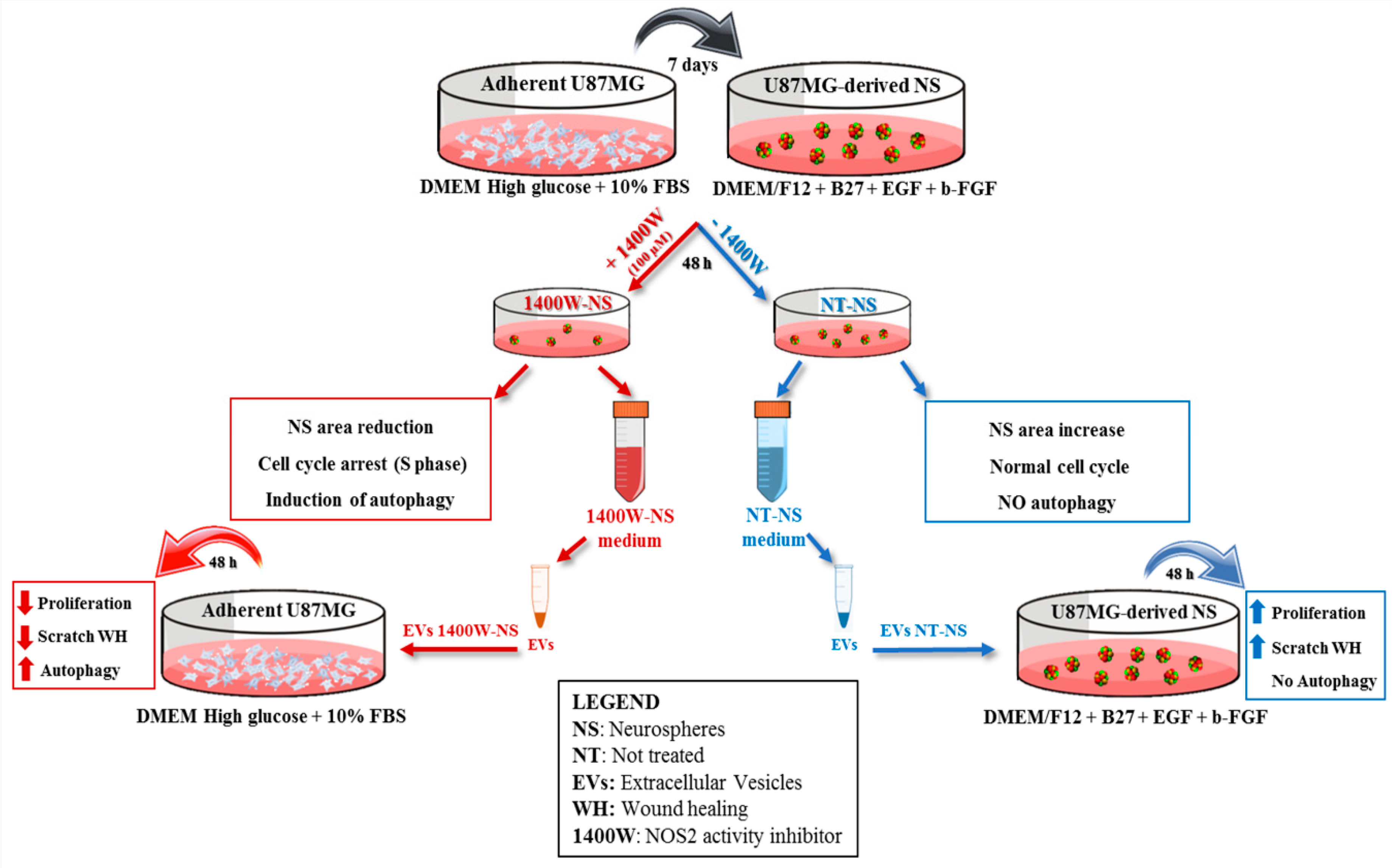{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. 1400W Reduces Growth of Glioma Stem Cells by Inducing S-phase Cell Cycle Arrest
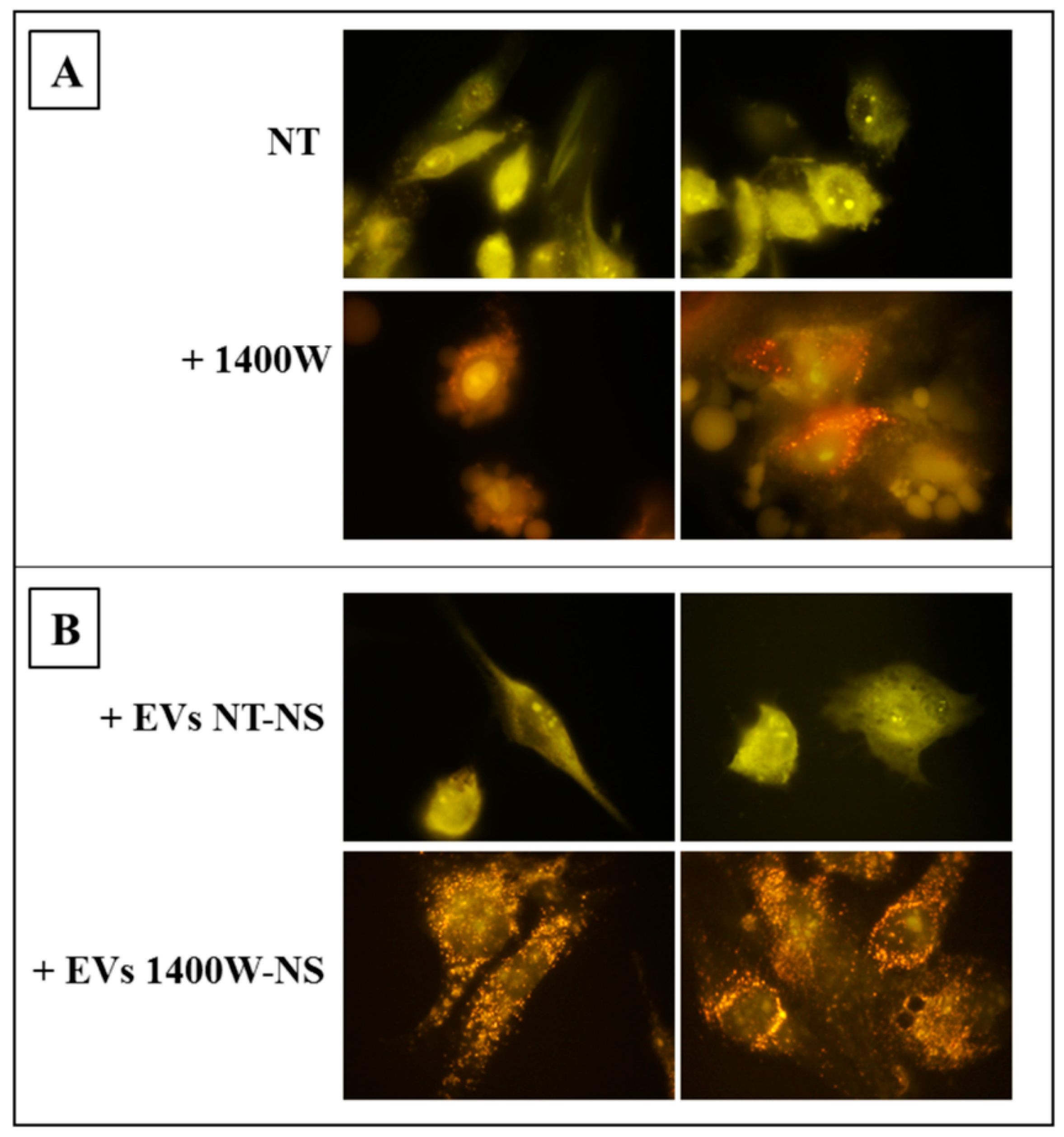
2.2. 1400W Induces Autophagy of Glioma Stem Cells
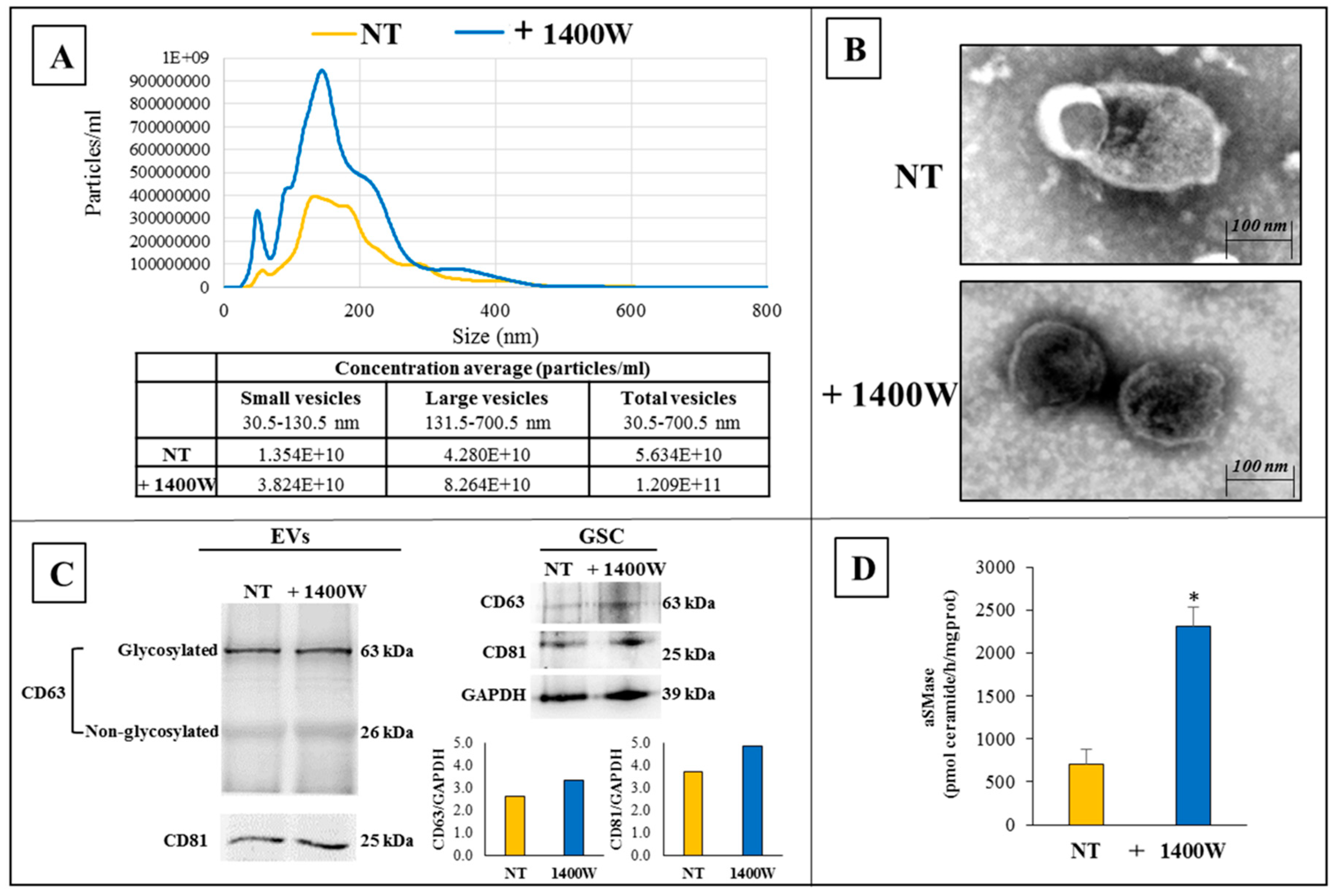
2.3. 1400.W Influences the Release of Extracellular Vesicles by Glioma Stem Cells
2.4. EVs Released by 1400W-Treated Glioma Stem Cells Negatively Influence Proliferation and Migration of Adherent U87MG Cells
2.5. EVs Released by 1400W-Treated Glioma Stem Cells Promote Autophagy of Recipient Adherent U87MG Cells
3. Discussion
4. Materials and Methods
4.1. U87MG Cell line, Glioma Stem Cells, and Treatments
4.2. Scanning Electron Microscopy
4.3. Cell Cycle Profile and Apoptosis Analysis by Flow Cytometer
4.4. Detection and Quantification of Acidic Vesicular Organelles (AVO) by Fluorescent Staining with Acridine Orange
4.5. Isolation and Characterization of NS-Derived Extracellular Vesicles
4.6. Nanoparticle Tracking Analysis (NTA)
4.7. Transmission Electron Microscopy
4.8. Western Blot Analysis
4.9. Acid Sphingomyelinase Activity
4.10. Cell Proliferation Assay
4.11. Scratch Wound Healing Assays
4.12. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NOS2 | Nitric Oxide Synthase 2 |
| GBM | Glioblastoma Multiforme |
| 1400W | N-(3-(Aminomethyl)benzyl)acetamidine |
| GSCs | Glioma Stem Cells |
| EVs | Extracellular vesicles |
| NS | Neurospheres |
| CDK4 | Cyclin dependent kinase 4 |
| AVO | Acidic vesicular organelles |
| AO | Acridine orange |
| GSC-M | Glioma stem cell-specific medium |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [Green Version]
- Stoyanov, G.S.; Dzhenkov, D.; Ghenev, P.; Iliev, B.; Enchev, Y.; Tonchev, A.B. Cell biology of glioblastoma multiforme: From basic science to diagnosis and treatment. Med. Oncol. 2018, 35. [Google Scholar] [CrossRef]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Garnier, D.; Renoult, O.; Alves-Guerra, M.C.; Paris, F.; Pecqueur, C. Glioblastoma Stem-Like Cells, Metabolic Strategy to Kill a Challenging Target. Front. Oncol. 2019, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Solis, C.; Serrano-Garcia, N.; Escamilla-Ramirez, A.; Castillo-Rodriguez, R.A.; Jimenez-Farfan, D.; Palencia, G.; Calvillo, M.; Alvarez-Lemus, M.A.; Flores-Najera, A.; Cruz-Salgado, A.; et al. Autophagic and Apoptotic Pathways as Targets for Chemotherapy in Glioblastoma. Int. J. Mol. Sci. 2018, 19, 3773. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Noda, N.N.; Inagaki, F. Mechanisms of Autophagy. Annu. Rev. Biophys. 2015, 44, 101–122. [Google Scholar] [CrossRef]
- Singh, S.S.; Vats, S.; Chia, A.Y.Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Xu, J.; Camfield, R.; Gorski, S.M. The interplay between exosomes and autophagy—Partners in crime. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Eyler, C.E.; Wu, Q.; Yan, K.; MacSwords, J.M.; Chandler-Militello, D.; Misuraca, K.L.; Lathia, J.D.; Forrester, M.T.; Lee, J.; Stamler, J.S.; et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell 2011, 146, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Jahani-Asl, A.; Bonni, A. iNOS: A potential therapeutic target for malignant glioma. Curr. Mol. Med. 2013, 13, 1241–1249. [Google Scholar] [CrossRef]
- Shen, S.C.; Wu, M.S.; Lin, H.Y.; Yang, L.Y.; Chen, Y.H.; Chen, Y.C. Reactive Oxygen Species-Dependent Nitric Oxide Production in Reciprocal Interactions of Glioma and Microglial Cells. J. Cell. Physiol. 2014, 229, 2015–2026. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Wink, D.A. NOS2 as an Emergent Player in Progression of Cancer. Antioxid Red. Sign 2017, 26, 963–965. [Google Scholar] [CrossRef]
- Palumbo, P.; Miconi, G.; Cinque, B.; Lombardi, F.; La Torre, C.; Dehcordi, S.R.; Galzio, R.; Cimini, A.; Giordano, A.; Cifone, M.G. NOS2 expression in glioma cell lines and glioma primary cell cultures: Correlation with neurosphere generation and SOX-2 expression. Oncotarget 2017, 8, 25582–25598. [Google Scholar] [CrossRef]
- Miconi, G.; Palumbo, P.; Dehcordi, S.R.; La Torre, C.; Lombardi, F.; Evtoski, Z.; Cimini, A.M.; Galzio, R.; Cifone, M.G.; Cinque, B. Immunophenotypic characterization of human glioblastoma stem cells: Correlation with clinical outcome. J. Cell. Biochem. 2015, 116, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, P.; Lombardi, F.; Siragusa, G.; Dehcordi, S.R.; Luzzi, S.; Cimini, A.; Cifone, M.G.; Cinque, B. Involvement of NOS2 Activity on Human Glioma Cell Growth, Clonogenic Potential, and Neurosphere Generation. Int. J. Mol. Sci. 2018, 19, 2801. [Google Scholar] [CrossRef]
- Fedorov, R.; Hartmann, E.; Ghosh, D.K.; Schlichting, I. Structural basis for the specificity of the nitric-oxide synthase inhibitors W1400 and Nomega-propyl-L-Arg for the inducible and neuronal isoforms. J. Obiol. Chem. 2003, 278, 45818–45825. [Google Scholar] [CrossRef] [PubMed]
- Maccallini, C.; Di Matteo, M.; Gallorini, M.; Montagnani, M.; Graziani, V.; Ammazzalorso, A.; Amoia, P.; De Filippis, B.; Di Silvestre, S.; Fantacuzzi, M.; et al. Discovery of N-{3-[(ethanimidoylamino)methyl]benzyl}-l-prolinamide dihydrochloride: A new potent and selective inhibitor of the inducible nitric oxide synthase as a promising agent for the therapy of malignant glioma. Eur. J. Med. Chem. 2018, 152, 53–64. [Google Scholar] [CrossRef]
- Re, N.; Fantacuzzi, M.; Maccallini, C.; Paciotti, R.; Amoroso, R. Recent developments of amidine-like compounds as selective NOS inhibitors. Curr. Enzyme Inhib. 2016, 12, 30–39. [Google Scholar] [CrossRef]
- Zheng, K.; He, Z.; Kitazato, K.; Wang, Y. Selective Autophagy Regulates Cell Cycle in Cancer Therapy. Theranostics 2019, 9, 104–125. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Overholtzer, M.; Thompson, C.B. Autophagy in cellular metabolism and cancer. J. Clin. Investig. 2015, 125, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Andreu, Z.; Yanez-Mo, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef] [PubMed]
- Record, M.; Silvente-Poirot, S.; Poirot, M.; Wakelam, M.J.O. Extracellular vesicles: Lipids as key components of their biogenesis and functions. J. Lipid Res. 2018, 59, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Giusti, I.; Di Francesco, M.; Dolo, V. Extracellular Vesicles in Glioblastoma: Role in Biological Processes and in Therapeutic Applications. Curr. Cancer Drug Targ. 2017, 17, 221–235. [Google Scholar] [CrossRef]
- D’Alessio, A.; Proietti, G.; Sica, G.; Scicchitano, B.M. Pathological and Molecular Features of Glioblastoma and Its Peritumoral Tissue. Cancers 2019, 11, 469. [Google Scholar] [CrossRef]
- Sarkar, S.; Korolchuk, V.I.; Renna, M.; Imarisio, S.; Fleming, A.; Williams, A.; Garcia-Arencibia, M.; Rose, C.; Luo, S.; Underwood, B.R.; et al. Complex inhibitory effects of nitric oxide on autophagy. Mol. Cell 2011, 43, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Cell death-based treatment of glioblastoma. Cell Death Dis. 2018, 9, 121. [Google Scholar] [CrossRef]
- Jawhari, S.; Ratinaud, M.H.; Verdier, M. Glioblastoma, hypoxia and autophagy: A survival-prone ‘menage-a-trois’. Cell Death Dis. 2016, 7, e2434. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boya, P.; Gonzalez-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Metivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef]
- Huang, X.; Bai, H.M.; Chen, L.; Li, B.; Lu, Y.C. Reduced expression of LC3B-II and Beclin 1 in glioblastoma multiforme indicates a down-regulated autophagic capacity that relates to the progression of astrocytic tumors. J. Clin. Neurosci. 2010, 17, 1515–1519. [Google Scholar] [CrossRef] [PubMed]
- Jo, G.H.; Bogler, O.; Chwae, Y.J.; Yoo, H.; Lee, S.H.; Park, J.B.; Kim, Y.J.; Kim, J.H.; Gwak, H.S. Radiation-induced autophagy contributes to cell death and induces apoptosis partly in malignant glioma cells. Cancer Res. Treat. 2015, 47, 221–241. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Luo, Y.; Fu, S.; Ji, X.; Ling, F. Autophagy: A strategy for malignant gliomas’ resistance to therapy. Med. Hypotheses 2009, 73, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Daido, S.; Kanzawa, T.; Kondo, S.; Kondo, Y. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int. J. Oncol. 2005, 26, 1401–1410. [Google Scholar] [CrossRef]
- Lomonaco, S.L.; Finniss, S.; Xiang, C.; Decarvalho, A.; Umansky, F.; Kalkanis, S.N.; Mikkelsen, T.; Brodie, C. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int. J. Cancer 2009, 125, 717–722. [Google Scholar] [CrossRef]
- Padmakrishnan, C.J.; Easwer, H.V.; Vijayakurup, V.; Menon, G.R.; Nair, S.; Gopala, S. High LC3/Beclin Expression Correlates with Poor Survival in Glioma: A Definitive Role for Autophagy as Evidenced by In Vitro Autophagic Flux. Pathol. Oncol. Res. 2019, 25, 137–148. [Google Scholar] [CrossRef]
- Colella, B.; Faienza, F.; Di Bartolomeo, S. EMT Regulation by Autophagy: A New Perspective in Glioblastoma Biology. Cancers 2019, 11, 312. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Galluzzi, L.; Kroemer, G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat. Immunol. 2012, 13, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Verderio, C.; Gabrielli, M.; Giussani, P. Role of sphingolipids in the biogenesis and biological activity of extracellular vesicles. J. Lipid Res. 2018, 59, 1325–1340. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef]
- Li, X.Y.; Zhang, L.Q.; Zhang, X.G.; Li, X.; Ren, Y.B.; Ma, X.Y.; Li, X.G.; Wang, L.X. Association between AKT/mTOR signalling pathway and malignancy grade of human gliomas. J. Neuro-Oncol. 2011, 103, 453–458. [Google Scholar] [CrossRef]
- Giusti, I.; Di Francesco, M.; Cantone, L.; D'Ascenzo, S.; Bollati, V.; Carta, G.; Dolo, V. Time-dependent release of extracellular vesicle subpopulations in tumor CABA I cells. Oncol. Rep. 2015, 34, 2752–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loidl, A.; Claus, R.; Deigner, H.P.; Hermetter, A. High-precision fluorescence assay for sphingomyelinase activity of isolated enzymes and cell lysates. J. Lipid Res. 2002, 43, 815–823. [Google Scholar] [PubMed]
- La Torre, C.; Cinque, B.; Lombardi, F.; Miconi, G.; Palumbo, P.; Evtoski, Z.; Placidi, G.; Fanini, D.; Cimini, A.M.; Benedetti, E.; et al. Nitric Oxide Chemical Donor Affects the Early Phases of In Vitro Wound Healing Process. J. Cell. Physiol. 2016, 231, 2185–2195. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palumbo, P.; Lombardi, F.; Augello, F.R.; Giusti, I.; Luzzi, S.; Dolo, V.; Cifone, M.G.; Cinque, B. NOS2 inhibitor 1400W Induces Autophagic Flux and Influences Extracellular Vesicle Profile in Human Glioblastoma U87MG Cell Line. Int. J. Mol. Sci. 2019, 20, 3010. https://doi.org/10.3390/ijms20123010
Palumbo P, Lombardi F, Augello FR, Giusti I, Luzzi S, Dolo V, Cifone MG, Cinque B. NOS2 inhibitor 1400W Induces Autophagic Flux and Influences Extracellular Vesicle Profile in Human Glioblastoma U87MG Cell Line. International Journal of Molecular Sciences. 2019; 20(12):3010. https://doi.org/10.3390/ijms20123010
Chicago/Turabian StylePalumbo, Paola, Francesca Lombardi, Francesca Rosaria Augello, Ilaria Giusti, Sabino Luzzi, Vincenza Dolo, Maria Grazia Cifone, and Benedetta Cinque. 2019. "NOS2 inhibitor 1400W Induces Autophagic Flux and Influences Extracellular Vesicle Profile in Human Glioblastoma U87MG Cell Line" International Journal of Molecular Sciences 20, no. 12: 3010. https://doi.org/10.3390/ijms20123010
APA StylePalumbo, P., Lombardi, F., Augello, F. R., Giusti, I., Luzzi, S., Dolo, V., Cifone, M. G., & Cinque, B. (2019). NOS2 inhibitor 1400W Induces Autophagic Flux and Influences Extracellular Vesicle Profile in Human Glioblastoma U87MG Cell Line. International Journal of Molecular Sciences, 20(12), 3010. https://doi.org/10.3390/ijms20123010