Evolution Shapes the Gene Expression Response to Oxidative Stress


{kind=link}
{kind=link}
Abstract
:1. Background
1.1. ROS Regulation of Gene Expression in Vertebrate Systems
1.2. Circadian Clocks and Timing of the Response to ROS
1.3. Stress Response and Cytoplasmic Granules
2. Fish as Models for Studying ROS Responses
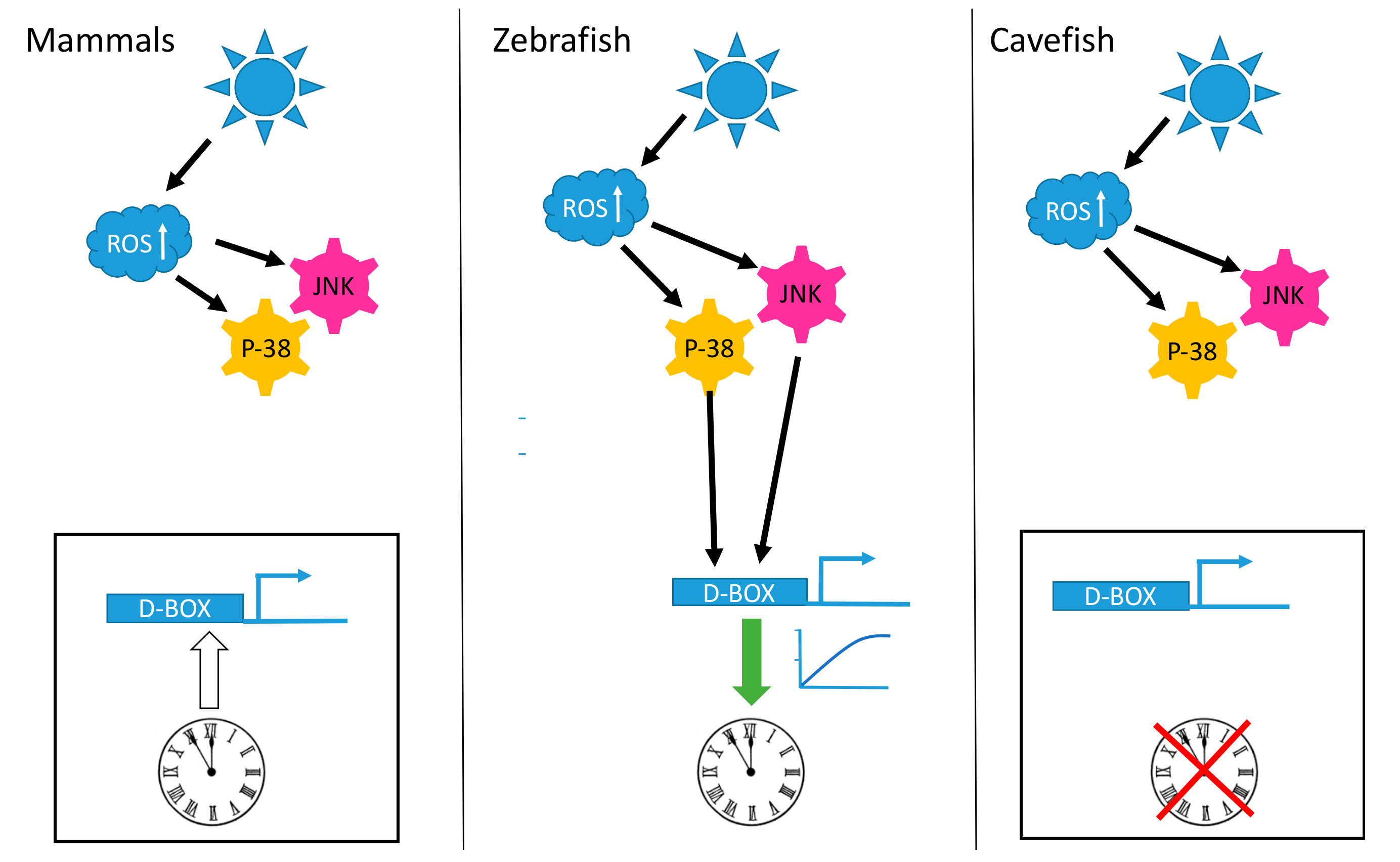
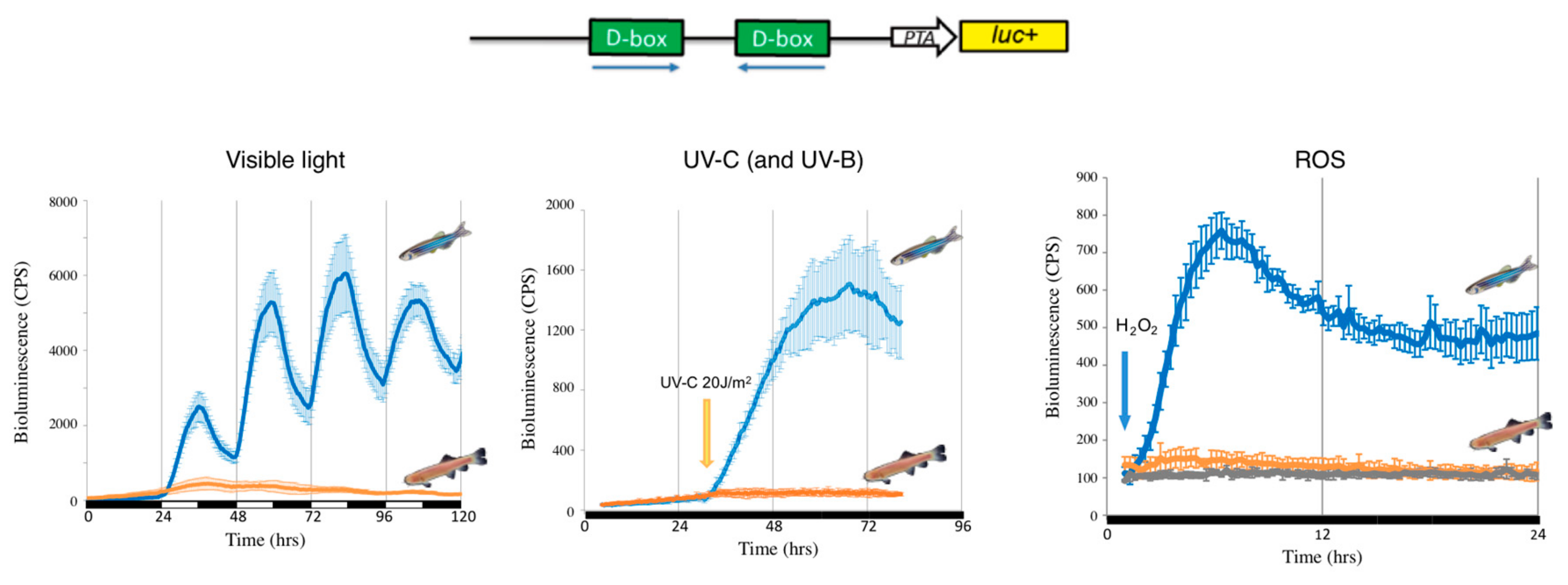
2.1. Sunlight, ROS and Regulation of the Circadian Clock in Fish
2.2. The D-Box and the Transcriptional Response to ROS
2.3. Adaptation of Mechanisms Responding to Oxidative Stress during Evolution
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hayyan, M.; Hashim, M.A.; AlNashef, I.M. Superoxide Ion: Generation and Chemical Implications. Chem. Rev. 2016, 116, 3029–3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, C.; Ding, A. SnapShot: Reactive Oxygen Intermediates (ROI). Cell 2010, 140, 951. [Google Scholar] [CrossRef] [PubMed]
- Ralser, M.; Wamelink, M.M.; Kowald, A.; Gerisch, B.; Heeren, G.; Struys, E.A.; Klipp, E.; Jakobs, C.; Breitenbach, M.; Lehrach, H.; et al. Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J. Biol. 2007, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Janero, D.R.; Hreniuk, D.; Sharif, H.M. Hydroperoxide-induced oxidative stress impairs heart muscle cell carbohydrate metabolism. Am. J. Physiol. Cell Physiol. 1994, 266, C179–C188. [Google Scholar] [CrossRef] [PubMed]
- Le Goffe, C.; Vallette, G.; Charrier, L.; Candelon, T.; Bou-Hanna, C.; Bouhours, J.F.; Laboisse, C.L. Metabolic control of resistance of human epithelial cells to H2O2 and NO stresses. Biochem. J. 2002, 364, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Elchuri, S.; Oberley, T.D.; Qi, W.; Eisenstein, R.S.; Jackson Roberts, L.; Van Remmen, H.; Epstein, C.J.; Huang, T.T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 2005, 24, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Muid, K.A.; Karakaya, H.C.; Koc, A. Absence of superoxide dismutase activity causes nuclear DNA fragmentation during the aging process. Biochem. Biophys. Res. Commun. 2014, 444, 260–263. [Google Scholar] [CrossRef] [Green Version]
- Chelikani, P.; Fita, I.; Loewen, P.C. Diversity of structures and properties among catalases. Cell. Mol. Life Sci. 2004, 61, 192–208. [Google Scholar] [CrossRef]
- Sinitsyna, O.; Krysanova, Z.; Ishchenko, A.; Dikalova, A.E.; Stolyarov, S.; Kolosova, N.; Vasunina, E.; Nevinsky, G. Age-associated changes in oxidative damage and the activity of antioxidant enzymes in rats with inherited overgeneration of free radicals. J. Cell. Mol. Med. 2006, 10, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef]
- Margis, R.; Dunand, C.; Teixeira, F.K.; Margis-Pinheiro, M. Glutathione peroxidase family—An evolutionary overview. FEBS J. 2008, 275, 3959–3970. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, K.; Iwai, S.; Tainer, J.A. The intricate structural chemistry of base excision repair machinery: Implications for DNA damage recognition, removal, and repair. DNA Repair 2007, 6, 410–428. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA repair in mammalian cells: Base excision repair: The long and short of it. Cell. Mol. Life Sci. 2009, 66, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Karisch, R.; Fernandez, M.; Taylor, P.; Virtanen, C.; St-Germain, J.R.; Jin, L.L.; Harris, I.S.; Mori, J.; Mak, T.W.; Senis, Y.A.; et al. Global proteomic assessment of the classical protein-tyrosine phosphatome and “Redoxome”. Cell 2011, 146, 826–840. [Google Scholar] [CrossRef] [PubMed]
- Mehdy, M.C. Active Oxygen Species in Plant Defense against Pathogens. Plant Physiol. 1994, 105, 467–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imlay, J.A. Transcription Factors That Defend Bacteria against Reactive Oxygen Species. Annu. Rev. Microbiol. 2015, 69, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Storz, G.; Tartaglia, L.A.; Ames, B.N. Transcriptional regulator of oxidative stress-inducible genes: Direct activation by oxidation. Science 1990, 248, 189–194. [Google Scholar] [CrossRef]
- Christman, M.F.; Morgan, R.W.; Jacobson, F.S.; Ames, B.N. Positive control of a regulon for defenses against oxidative stress and some heat-shock proteins in Salmonella typhimurium. Cell 1985, 41, 753–762. [Google Scholar] [CrossRef]
- Demple, B.; Amabile-Cuevas, C.F. Redox redux: The control of oxidative stress responses. Cell 1991, 67, 837–839. [Google Scholar] [CrossRef]
- Elsen, S.; Swem, L.R.; Swem, D.L.; Bauer, C.E. RegB/RegA, a highly conserved redox-responding global two-component regulatory system. Microbiol. Mol. Biol. Rev. 2004, 68, 263–279. [Google Scholar] [CrossRef]
- Wu, J.; Bauer, C.E. RegB/RegA, a global redox-responding two-component system. Adv. Exp. Med. Biol. 2008, 631, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.Y.; Chae, H.Z.; Rhee, S.G.; Jeang, K.T. Regulatory role for a novel human thioredoxin peroxidase in NF-kappaB activation. J. Biol. Chem. 1997, 272, 30952–30961. [Google Scholar] [CrossRef] [PubMed]
- Biteau, B.; Labarre, J.; Toledano, M.B. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 2003, 425, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.; Tenhaken, R.; Dixon, R.; Lamb, C. H2O2 from the oxidative burst orchestrates the plant hypersensitive disease resistance response. Cell 1994, 79, 583–593. [Google Scholar] [CrossRef]
- Alvarez, M.E.; Pennell, R.I.; Meijer, P.J.; Ishikawa, A.; Dixon, R.A.; Lamb, C. Reactive oxygen intermediates mediate a systemic signal network in the establishment of plant immunity. Cell 1998, 92, 773–784. [Google Scholar] [CrossRef]
- Foyer, C.H.; Shigeoka, S. Understanding oxidative stress and antioxidant functions to enhance photosynthesis. Plant Physiol. 2011, 155, 93–100. [Google Scholar] [CrossRef]
- Shapiro, B.M. The control of oxidant stress at fertilization. Science 1991, 252, 533–536. [Google Scholar] [CrossRef]
- Wu, S.C.; Liao, C.W.; Pan, R.L.; Juang, J.L. Infection-induced intestinal oxidative stress triggers organ-to-organ immunological communication in Drosophila. Cell Host Microbe 2012, 11, 410–417. [Google Scholar] [CrossRef]
- Ferrandon, D.; Imler, J.L.; Hetru, C.; Hoffmann, J.A. The Drosophila systemic immune response: Sensing and signalling during bacterial and fungal infections. Nat. Rev. Immunol. 2007, 7, 862–874. [Google Scholar] [CrossRef]
- Buchon, N.; Broderick, N.A.; Chakrabarti, S.; Lemaitre, B. Invasive and indigenous microbiota impact intestinal stem cell activity through multiple pathways in Drosophila. Genes Dev. 2009, 23, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.W.S.; Lee, Q.Y.; Wong, B.S.E.; Cai, Y.; Baeg, G.H. Redox Homeostasis Plays Important Roles in the Maintenance of the Drosophila Testis Germline Stem Cells. Stem Cell Rep. 2017, 9, 342–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohania, D.; Chandel, S.; Kumar, P.; Verma, V.; Digvijay, K.; Tripathi, D.; Choudhury, K.; Mitten, S.K.; Shah, D. Ultraviolet Radiations: Skin Defense-Damage Mechanism. Adv. Exp. Med. Biol. 2017, 996, 71–87. [Google Scholar] [CrossRef]
- Klotz, L.O.; Steinbrenner, H. Cellular adaptation to xenobiotics: Interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox Biol. 2017, 13, 646–654. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Aggeli, I.K.; Gaitanaki, C.; Beis, I. Involvement of JNKs and p38-MAPK/MSK1 pathways in H2O2-induced upregulation of heme oxygenase-1 mRNA in H9c2 cells. Cell. Signal. 2006, 18, 1801–1812. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Lahair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox Signal. 2006, 8, 1775–1789. [Google Scholar] [CrossRef]
- Yong, H.Y.; Koh, M.S.; Moon, A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin. Investig. Drugs 2009, 18, 1893–1905. [Google Scholar] [CrossRef]
- Davies, C.; Tournier, C. Exploring the function of the JNK (c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem. Soc. Trans. 2012, 40, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, K.; Matsuzawa, A.; Ichijo, H. Regulation of apoptosis signal-regulating kinase 1 in redox signaling. Methods Enzymol. 2010, 474, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Takeda, K.; Ichijo, H. ASK family proteins in stress response and disease. Mol. Biotechnol. 2007, 37, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, A.; Ichijo, H. Redox control of cell fate by MAP kinase: Physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim. Biophys. Acta 2008, 1780, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Hai, T.; Curran, T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc. Natl. Acad. Sci. USA 1991, 88, 3720–3724. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.; Ronai, Z.A.; Lau, E. ATF2, a paradigm of the multifaceted regulation of transcription factors in biology and disease. Pharmacol. Res. 2017, 119, 347–357. [Google Scholar] [CrossRef]
- Bohmann, D.; Bos, T.J.; Admon, A.; Nishimura, T.; Vogt, P.K.; Tjian, R. Human proto-oncogene c-jun encodes a DNA binding protein with structural and functional properties of transcription factor AP-1. Science 1987, 238, 1386–1392. [Google Scholar] [CrossRef]
- Hayakawa, J.; Mittal, S.; Wang, Y.; Korkmaz, K.S.; Adamson, E.; English, C.; Ohmichi, M.; McClelland, M.; Mercola, D. Identification of promoters bound by c-Jun/ATF2 during rapid large-scale gene activation following genotoxic stress. Mol. Cell 2004, 16, 521–535. [Google Scholar] [CrossRef]
- Maekawa, T.; Sakura, H.; Kanei-Ishii, C.; Sudo, T.; Yoshimura, T.; Fujisawa, J.; Yoshida, M.; Ishii, S. Leucine zipper structure of the protein CRE-BP1 binding to the cyclic AMP response element in brain. EMBO J. 1989, 8, 2023–2028. [Google Scholar] [CrossRef]
- Li, X.Y.; Green, M.R. Intramolecular inhibition of activating transcription factor-2 function by its DNA-binding domain. Genes Dev. 1996, 10, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Vogt, P.K. Fortuitous convergences: The beginnings of JUN. Nat. Rev. Cancer 2002, 2, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Angel, P.; Allegretto, E.A.; Okino, S.T.; Hattori, K.; Boyle, W.J.; Hunter, T.; Karin, M. Oncogene jun encodes a sequence-specific trans-activator similar to AP-1. Nature 1988, 332, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Binetruy, B.; Smeal, T.; Karin, M. Ha-Ras augments c-Jun activity and stimulates phosphorylation of its activation domain. Nature 1991, 351, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Smeal, T.; Binetruy, B.; Mercola, D.A.; Birrer, M.; Karin, M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature 1991, 354, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Schreiber, M.; Piu, F.; Beeche, M.; Wagner, E.F.; Karin, M. The mammalian UV response: C-Jun induction is required for exit from p53-imposed growth arrest. Cell 2000, 103, 897–907. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef]
- Pineda-Molina, E.; Klatt, P.; Vazquez, J.; Marina, A.; Garcia de Lacoba, M.; Perez-Sala, D.; Lamas, S. Glutathionylation of the p50 subunit of NF-kappaB: A mechanism for redox-induced inhibition of DNA binding. Biochemistry 2001, 40, 14134–14142. [Google Scholar] [CrossRef]
- Droge, W. Redox regulation in anabolic and catabolic processes. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 190–195. [Google Scholar] [CrossRef]
- Nioi, P.; McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: Reassessment of the ARE consensus sequence. Biochem. J. 2003, 374, 337–348. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Mimura, J.; Inose-Maruyama, A.; Taniuchi, S.; Kosaka, K.; Yoshida, H.; Yamazaki, H.; Kasai, S.; Harada, N.; Kaufman, R.J.; Oyadomari, S.; et al. Concomitant Nrf2- and ATF4-activation by Carnosic Acid Cooperatively Induces Expression of Cytoprotective Genes. Int. J. Mol. Sci. 2019, 20, 1706. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- De Nicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Sobotta, M.C.; Liou, W.; Stocker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015, 11, 64–70. [Google Scholar] [CrossRef]
- Rampon, C.; Volovitch, M.; Joliot, A.; Vriz, S. Hydrogen Peroxide and Redox Regulation of Developments. Antioxidants 2018, 7, 159. [Google Scholar] [CrossRef]
- Johnson, T.M.; Yu, Z.X.; Ferrans, V.J.; Lowenstein, R.A.; Finkel, T. Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93, 11848–11852. [Google Scholar] [CrossRef]
- Gambino, V.; De Michele, G.; Venezia, O.; Migliaccio, P.; Dall’Olio, V.; Bernard, L.; Minardi, S.P.; Della Fazia, M.A.; Bartoli, D.; Servillo, G.; et al. Oxidative stress activates a specific p53 transcriptional response that regulates cellular senescence and aging. Aging Cell 2013, 12, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef]
- Trinei, M.; Giorgio, M.; Cicalese, A.; Barozzi, S.; Ventura, A.; Migliaccio, E.; Milia, E.; Padura, I.M.; Raker, V.A.; Maccarana, M.; et al. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene 2002, 21, 3872–3878. [Google Scholar] [CrossRef] [Green Version]
- Bhat, S.S.; Anand, D.; Khanday, F.A. p66Shc as a switch in bringing about contrasting responses in cell growth: Implications on cell proliferation and apoptosis. Mol. Cancer 2015, 14, 76. [Google Scholar] [CrossRef]
- Loudon, A.S. Circadian biology: A 2.5 billion year old clock. Curr. Biol. 2012, 22, R570–R571. [Google Scholar] [CrossRef]
- Reppert, S.M.; Weaver, D.R. Molecular analysis of mammalian circadian rhythms. Annu. Rev. Physiol. 2001, 63, 647–676. [Google Scholar] [CrossRef]
- Dushay, M.S.; Rosbash, M.; Hall, J.C. Mapping the clock rhythm mutation to the period locus of Drosophila melanogaster by germline transformation. J. Neurogenet. 1992, 8, 173–179. [Google Scholar] [CrossRef]
- Reddy, P.; Jacquier, A.C.; Abovich, N.; Petersen, G.; Rosbash, M. The period clock locus of D. melanogaster codes for a proteoglycan. Cell 1986, 46, 53–61. [Google Scholar] [CrossRef]
- Zehring, W.A.; Wheeler, D.A.; Reddy, P.; Konopka, R.J.; Kyriacou, C.P.; Rosbash, M.; Hall, J.C. P-element transformation with period locus DNA restores rhythmicity to mutant, arrhythmic Drosophila melanogaster. Cell 1984, 39, 369–376. [Google Scholar] [CrossRef]
- McClung, C.R.; Fox, B.A.; Dunlap, J.C. The Neurospora clock gene frequency shares a sequence element with the Drosophila clock gene period. Nature 1989, 339, 558–562. [Google Scholar] [CrossRef]
- Bargiello, T.A.; Jackson, F.R.; Young, M.W. Restoration of circadian behavioural rhythms by gene transfer in Drosophila. Nature 1984, 312, 752–754. [Google Scholar] [CrossRef]
- Dunlap, J.C. Molecular bases for circadian clocks. Cell 1999, 96, 271–290. [Google Scholar] [CrossRef]
- Ko, C.H.; Takahashi, J.S. Molecular components of the mammalian circadian clock. Hum. Mol. Genet 2006, 15 (Suppl. 2), R271–R277. [Google Scholar] [CrossRef]
- Bellet, M.M.; Sassone-Corsi, P. Mammalian circadian clock and metabolism—The epigenetic link. J. Cell Sci. 2010, 123, 3837–3848. [Google Scholar] [CrossRef]
- Buhr, E.D.; Takahashi, J.S. Molecular components of the Mammalian circadian clock. Handb. Exp. Pharmacol. 2013, 3–27. [Google Scholar] [CrossRef]
- Peirson, S.N.; Halford, S.; Foster, R.G. The evolution of irradiance detection: Melanopsin and the non-visual opsins. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2849–2865. [Google Scholar] [CrossRef]
- Roenneberg, T.; Foster, R.G. Twilight times: Light and the circadian system. Photochem. Photobiol. 1997, 66, 549–561. [Google Scholar] [CrossRef]
- Whitmore, D.; Foulkes, N.S.; Strahle, U.; Sassone-Corsi, P. Zebrafish Clock rhythmic expression reveals independent peripheral circadian oscillators. Nat. Neurosci. 1998, 1, 701–707. [Google Scholar] [CrossRef]
- Turek, F.W.; Joshu, C.; Kohsaka, A.; Lin, E.; Ivanova, G.; McDearmon, E.; Laposky, A.; Losee-Olson, S.; Easton, A.; Jensen, D.R.; et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science 2005, 308, 1043–1045. [Google Scholar] [CrossRef]
- Asher, G.; Sassone-Corsi, P. Time for food: The intimate interplay between nutrition, metabolism, and the circadian clock. Cell 2015, 161, 84–92. [Google Scholar] [CrossRef]
- Zarrinpar, A.; Chaix, A.; Panda, S. Daily Eating Patterns and Their Impact on Health and Disease. Trends Endocrinol. Metab. 2016, 27, 69–83. [Google Scholar] [CrossRef]
- DiAngelo, J.R.; Erion, R.; Crocker, A.; Sehgal, A. The central clock neurons regulate lipid storage in Drosophila. PLoS ONE 2011, 6, e19921. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Moulik, M.; Fang, Z.; Saha, P.; Zou, F.; Xu, Y.; Nelson, D.L.; Ma, K.; Moore, D.D.; Yechoor, V.K. Bmal1 and beta-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced beta-cell failure in mice. Mol. Cell. Biol. 2013, 33, 2327–2338. [Google Scholar] [CrossRef]
- Kondratov, R.V.; Kondratova, A.A.; Gorbacheva, V.Y.; Vykhovanets, O.V.; Antoch, M.P. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006, 20, 1868–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamaru, T.; Hattori, M.; Ninomiya, Y.; Kawamura, G.; Vares, G.; Honda, K.; Mishra, D.P.; Wang, B.; Benjamin, I.; Sassone-Corsi, P.; et al. ROS stress resets circadian clocks to coordinate pro-survival signals. PLoS ONE 2013, 8, e82006. [Google Scholar] [CrossRef] [PubMed]
- Gyongyosi, N.; Kaldi, K. Interconnections of reactive oxygen species homeostasis and circadian rhythm in Neurospora crassa. Antioxid. Redox Signal. 2014, 20, 3007–3023. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.A. Circadian Metabolism: From Mechanisms to Metabolomics and Medicine. Trends Endocrinol. Metab. 2016, 27, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.A.; Gaspar, L. Circadian Metabolomics: Insights for Biology and Medicine. In A Time for Metabolism and Hormones; Sassone-Corsi, P., Christen, Y., Eds.; Springer: Cham, Switzerland, 2016; pp. 79–85. [Google Scholar] [CrossRef] [Green Version]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef]
- Peek, C.B.; Affinati, A.H.; Ramsey, K.M.; Kuo, H.Y.; Yu, W.; Sena, L.A.; Ilkayeva, O.; Marcheva, B.; Kobayashi, Y.; Omura, C.; et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 2013, 342, 1243417. [Google Scholar] [CrossRef]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef]
- Wilking, M.; Ndiaye, M.; Mukhtar, H.; Ahmad, N. Circadian rhythm connections to oxidative stress: Implications for human health. Antioxid. Redox Signal. 2013, 19, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Putker, M.; O’Neill, J.S. Reciprocal Control of the Circadian Clock and Cellular Redox State—A Critical Appraisal. Mol. Cells 2016, 39, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Davis, A.J.; Giebultowicz, J.M. Circadian regulation of response to oxidative stress in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2008, 374, 299–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomas-Zapico, C.; Coto-Montes, A.; Martinez-Fraga, J.; Rodriguez-Colunga, M.J.; Tolivia, D. Effects of continuous light exposure on antioxidant enzymes, porphyric enzymes and cellular damage in the Harderian gland of the Syrian hamster. J. Pineal Res. 2003, 34, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Fanjul-Moles, M.L.; Lopez-Riquelme, G.O. Relationship between Oxidative Stress, Circadian Rhythms, and AMD. Oxid. Med. Cell. Longev. 2016, 2016, 7420637. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Sainz, R.M.; Lopez-Burillo, S.; Mayo, J.C.; Manchester, L.C.; Tan, D.X. Melatonin ameliorates neurologic damage and neurophysiologic deficits in experimental models of stroke. Ann. N.Y. Acad. Sci. 2003, 993, 35–47, discussion 48–53. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. RNA granules: Post-transcriptional and epigenetic modulators of gene expression. Nat. Rev. Mol. Cell Biol. 2009, 10, 430–436. [Google Scholar] [CrossRef]
- Heberle, A.M.; Razquin Navas, P.; Langelaar-Makkinje, M.; Kasack, K.; Sadik, A.; Faessler, E.; Hahn, U.; Marx-Stoelting, P.; Opitz, C.A.; Sers, C.; et al. The PI3K and MAPK/p38 pathways control stress granule assembly in a hierarchical manner. Life Sci. Alliance 2019, 2, e201800257. [Google Scholar] [CrossRef]
- Khong, A.; Matheny, T.; Jain, S.; Mitchell, S.F.; Wheeler, J.R.; Parker, R. The Stress Granule Transcriptome Reveals Principles of mRNA Accumulation in Stress Granules. Mol. Cell 2017, 68, 808–820. [Google Scholar] [CrossRef]
- Takahashi, M.; Higuchi, M.; Matsuki, H.; Yoshita, M.; Ohsawa, T.; Oie, M.; Fujii, M. Stress granules inhibit apoptosis by reducing reactive oxygen species production. Mol. Cell. Biol. 2013, 33, 815–829. [Google Scholar] [CrossRef]
- Guarino, A.M.; Troiano, A.; Pizzo, E.; Bosso, A.; Vivo, M.; Pinto, G.; Amoresano, A.; Pollice, A.; La Mantia, G.; Calabro, V. Oxidative Stress Causes Enhanced Secretion of YB-1 Protein that Restrains Proliferation of Receiving Cells. Genes 2018, 9, 513. [Google Scholar] [CrossRef] [PubMed]
- Pagano, C.; di Martino, O.; Ruggiero, G.; Maria Guarino, A.; Mueller, N.; Siauciunaite, R.; Reischl, M.; Simon Foulkes, N.; Vallone, D.; Calabro, V. The tumor-associated YB-1 protein: New player in the circadian control of cell proliferation. Oncotarget 2017, 8, 6193–6205. [Google Scholar] [CrossRef] [PubMed]
- Pagano, C.; Siauciunaite, R.; Idda, M.L.; Ruggiero, G.; Ceinos, R.M.; Pagano, M.; Frigato, E.; Bertolucci, C.; Foulkes, N.S.; Vallone, D. Evolution shapes the responsiveness of the D-box enhancer element to light and reactive oxygen species in vertebrates. Sci. Rep. 2018, 8, 13180. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, J.; Cho, S.; Sassone-Corsi, P. Circadian control by the reduction/oxidation pathway: Catalase represses light-dependent clock gene expression in the zebrafish. Proc. Natl. Acad. Sci. USA 2007, 104, 15747–15752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallone, D.; Gondi, S.B.; Whitmore, D.; Foulkes, N.S. E-box function in a period gene repressed by light. Proc. Natl. Acad. Sci. USA 2004, 101, 4106–4111. [Google Scholar] [CrossRef] [PubMed]
- Vallone, D.; Santoriello, C.; Gondi, S.B.; Foulkes, N.S. Basic protocols for zebrafish cell lines: Maintenance and transfection. Methods Mol. Biol. 2007, 362, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Vallone, D.; Lahiri, K.; Dickmeis, T.; Foulkes, N.S. Zebrafish cell clocks feel the heat and see the light! Zebrafish 2005, 2, 171–187. [Google Scholar] [CrossRef]
- Menaker, M.; Takahashi, J.S.; Eskin, A. The physiology of circadian pacemakers. Annu. Rev. Physiol. 1978, 40, 501–526. [Google Scholar] [CrossRef]
- Davies, W.I.; Tamai, T.K.; Zheng, L.; Fu, J.K.; Rihel, J.; Foster, R.G.; Whitmore, D.; Hankins, M.W. An extended family of novel vertebrate photopigments is widely expressed and displays a diversity of function. Genome Res. 2015, 25, 1666–1679. [Google Scholar] [CrossRef] [Green Version]
- Hockberger, P.E.; Skimina, T.A.; Centonze, V.E.; Lavin, C.; Chu, S.; Dadras, S.; Reddy, J.K.; White, J.G. Activation of flavin-containing oxidases underlies light-induced production of H2O2 in mammalian cells. Proc. Natl. Acad. Sci. USA 1999, 96, 6255–6260. [Google Scholar] [CrossRef]
- Zhao, H.; Di Mauro, G.; Lungu-Mitea, S.; Negrini, P.; Guarino, A.M.; Frigato, E.; Braunbeck, T.; Ma, H.; Lamparter, T.; Vallone, D.; et al. Modulation of DNA Repair Systems in Blind Cavefish during Evolution in Constant Darkness. Curr. Biol. 2018, 28, 3229–3243. [Google Scholar] [CrossRef] [PubMed]
- Lichtsteiner, S.; Wuarin, J.; Schibler, U. The interplay of DNA-binding proteins on the promoter of the mouse albumin gene. Cell 1987, 51, 963–973. [Google Scholar] [CrossRef]
- Mueller, C.R.; Maire, P.; Schibler, U. DBP, a liver-enriched transcriptional activator, is expressed late in ontogeny and its tissue specificity is determined posttranscriptionally. Cell 1990, 61, 279–291. [Google Scholar] [CrossRef]
- Drolet, D.W.; Scully, K.M.; Simmons, D.M.; Wegner, M.; Chu, K.T.; Swanson, L.W.; Rosenfeld, M.G. TEF, a transcription factor expressed specifically in the anterior pituitary during embryogenesis, defines a new class of leucine zipper proteins. Genes Dev. 1991, 5, 1739–1753. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Ohyashiki, K.; Toyama, K.; Cleary, M.L. Hlf, a novel hepatic bZIP protein, shows altered DNA-binding properties following fusion to E2A in t(17;19) acute lymphoblastic leukemia. Genes Dev. 1992, 6, 1608–1620. [Google Scholar] [CrossRef]
- Lin, S.C.; Lin, M.H.; Horvath, P.; Reddy, K.L.; Storti, R.V. PDP1, a novel Drosophila PAR domain bZIP transcription factor expressed in developing mesoderm, endoderm and ectoderm, is a transcriptional regulator of somatic muscle genes. Development 1997, 124, 4685–4696. [Google Scholar]
- Xu, X.; Liu, L.; Wong, K.C.; Ge, R. Cloning and characterization of two isoforms of the zebrafish thyrotroph embryonic factor (tef alpha and tefbeta). Biochim. Biophys. Acta 1998, 1395, 13–20. [Google Scholar] [CrossRef]
- Cowell, I.G. E4BP4/NFIL3, a PAR-related bZIP factor with many roles. Bioessays 2002, 24, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Falvey, E.; Marcacci, L.; Schibler, U. DNA-binding specificity of PAR and C/EBP leucine zipper proteins: A single amino acid substitution in the C/EBP DNA-binding domain confers PAR-like specificity to C/EBP. Biol. Chem. 1996, 377, 797–809. [Google Scholar]
- Li, S.; Hunger, S.P. The DBP transcriptional activation domain is highly homologous to that of HLF and TEF and is not responsible for the tissue type-specific transcriptional activity of DBP. Gene 2001, 263, 239–245. [Google Scholar] [CrossRef]
- Gachon, F. Physiological function of PARbZip circadian clock-controlled transcription factors. Ann. Med. 2007, 39, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Wuarin, J.; Schibler, U. Expression of the liver-enriched transcriptional activator protein DBP follows a stringent circadian rhythm. Cell 1990, 63, 1257–1266. [Google Scholar] [CrossRef]
- Falvey, E.; Fleury-Olela, F.; Schibler, U. The rat hepatic leukemia factor (HLF) gene encodes two transcriptional activators with distinct circadian rhythms, tissue distributions and target preferences. EMBO J. 1995, 14, 4307–4317. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.S.; Lee, M.H.; Lee, S.H.; Bae, K. Cu/Zn superoxide dismutase is differentially regulated in period gene-mutant mice. Biochem. Biophys. Res. Commun. 2011, 409, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R.; Coto-Montes, A.; Poeggeler, B. Circadian rhythms, oxidative stress, and antioxidative defense mechanisms. Chronobiol. Int. 2003, 20, 921–962. [Google Scholar] [CrossRef] [PubMed]
- Beaver, L.M.; Klichko, V.I.; Chow, E.S.; Kotwica-Rolinska, J.; Williamson, M.; Orr, W.C.; Radyuk, S.N.; Giebultowicz, J.M. Circadian regulation of glutathione levels and biosynthesis in Drosophila melanogaster. PLoS ONE 2012, 7, e50454. [Google Scholar] [CrossRef]
- Blanco, R.A.; Ziegler, T.R.; Carlson, B.A.; Cheng, P.Y.; Park, Y.; Cotsonis, G.A.; Accardi, C.J.; Jones, D.P. Diurnal variation in glutathione and cysteine redox states in human plasma. Am. J. Clin. Nutr. 2007, 86, 1016–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gachon, F.; Olela, F.F.; Schaad, O.; Descombes, P.; Schibler, U. The circadian PAR-domain basic leucine zipper transcription factors DBP, TEF, and HLF modulate basal and inducible xenobiotic detoxification. Cell Metab. 2006, 4, 25–36. [Google Scholar] [CrossRef]
- Ben-Moshe, Z.; Vatine, G.; Alon, S.; Tovin, A.; Mracek, P.; Foulkes, N.S.; Gothilf, Y. Multiple PAR and E4BP4 bZIP transcription factors in zebrafish: Diverse spatial and temporal expression patterns. Chronobiol. Int. 2010, 27, 1509–1531. [Google Scholar] [CrossRef]
- Cavallari, N.; Frigato, E.; Vallone, D.; Frohlich, N.; Lopez-Olmeda, J.F.; Foa, A.; Berti, R.; Sanchez-Vazquez, F.J.; Bertolucci, C.; Foulkes, N.S. A blind circadian clock in cavefish reveals that opsins mediate peripheral clock photoreception. PLoS Biol. 2011, 9, e1001142. [Google Scholar] [CrossRef]
- Ceinos, R.M.; Frigato, E.; Pagano, C.; Frohlich, N.; Negrini, P.; Cavallari, N.; Vallone, D.; Fuselli, S.; Bertolucci, C.; Foulkes, N.S. Mutations in blind cavefish target the light-regulated circadian clock gene, period 2. Sci. Rep. 2018, 8, 8754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerkema, M.P.; Davies, W.I.; Foster, R.G.; Menaker, M.; Hut, R.A. The nocturnal bottleneck and the evolution of activity patterns in mammals. Proc Biol Sci. 2013, 280, 20130508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maor, R.; Dayan, T.; Ferguson-Gow, H.; Jones, K.E. Temporal niche expansion in mammals from a nocturnal ancestor after dinosaur extinction. Nat. Ecol. Evol. 2017, 1, 1889–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heesy, C.P.; Hall, M.I. The nocturnal bottleneck and the evolution of mammalian vision. Brain Behav. Evol. 2010, 75, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Domankevich, V.; Lijuan, H.; Xiaodong, F.; Korol, A.; Avivi, A.; Shams, I. Genome maintenance and bioenergetics of the long-lived hypoxia-tolerant and cancer-resistant blind mole rat, Spalax: A cross-species analysis of brain transcriptome. Sci. Rep. 2016, 6, 38624. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.J.; Glazer, P.M. The tumor microenvironment and DNA repair. Semin. Radiat. Oncol. 2010, 20, 282–287. [Google Scholar] [CrossRef]
- Gorbunova, V.; Hine, C.; Tian, X.; Ablaeva, J.; Gudkov, A.V.; Nevo, E.; Seluanov, A. Cancer resistance in the blind mole rat is mediated by concerted necrotic cell death mechanism. Proc. Natl. Acad. Sci. USA 2012, 109, 19392–19396. [Google Scholar] [CrossRef] [Green Version]
- Ashur-Fabian, O.; Avivi, A.; Trakhtenbrot, L.; Adamsky, K.; Cohen, M.; Kajakaro, G.; Joel, A.; Amariglio, N.; Nevo, E.; Rechavi, G. Evolution of p53 in hypoxia-stressed Spalax mimics human tumor mutation. Proc. Natl. Acad. Sci. USA 2004, 101, 12236–12241. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siauciunaite, R.; Foulkes, N.S.; Calabrò, V.; Vallone, D. Evolution Shapes the Gene Expression Response to Oxidative Stress. Int. J. Mol. Sci. 2019, 20, 3040. https://doi.org/10.3390/ijms20123040
Siauciunaite R, Foulkes NS, Calabrò V, Vallone D. Evolution Shapes the Gene Expression Response to Oxidative Stress. International Journal of Molecular Sciences. 2019; 20(12):3040. https://doi.org/10.3390/ijms20123040
Chicago/Turabian StyleSiauciunaite, Rima, Nicholas S. Foulkes, Viola Calabrò, and Daniela Vallone. 2019. "Evolution Shapes the Gene Expression Response to Oxidative Stress" International Journal of Molecular Sciences 20, no. 12: 3040. https://doi.org/10.3390/ijms20123040
APA StyleSiauciunaite, R., Foulkes, N. S., Calabrò, V., & Vallone, D. (2019). Evolution Shapes the Gene Expression Response to Oxidative Stress. International Journal of Molecular Sciences, 20(12), 3040. https://doi.org/10.3390/ijms20123040