Proteasome and Autophagy-Mediated Impairment of Late Long-Term Potentiation (l-LTP) after Traumatic Brain Injury in the Somatosensory Cortex of Mice

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
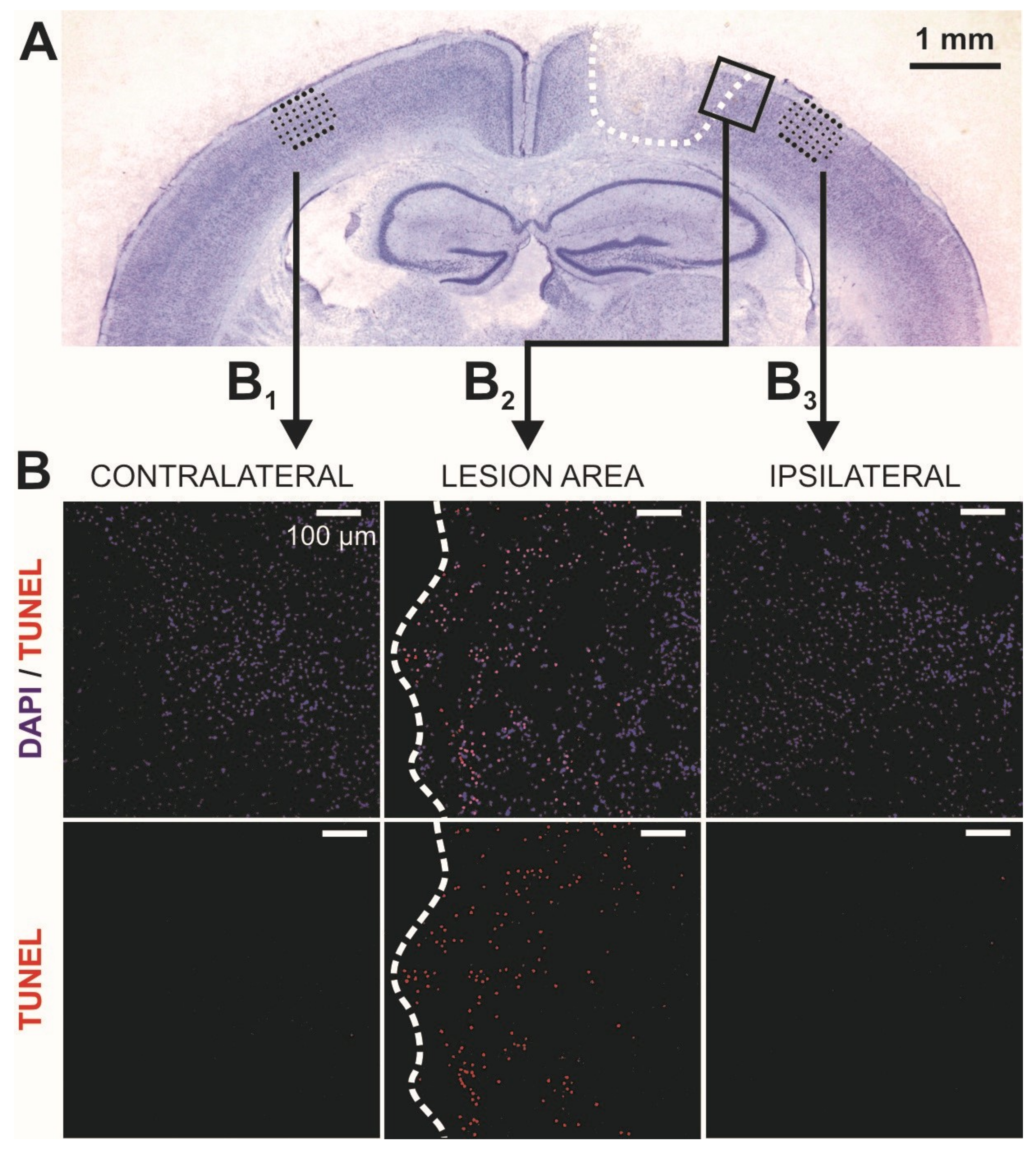
2.1. Lesion Expansion in the Acute Phase after Controlled Cortical Impact (CCI)
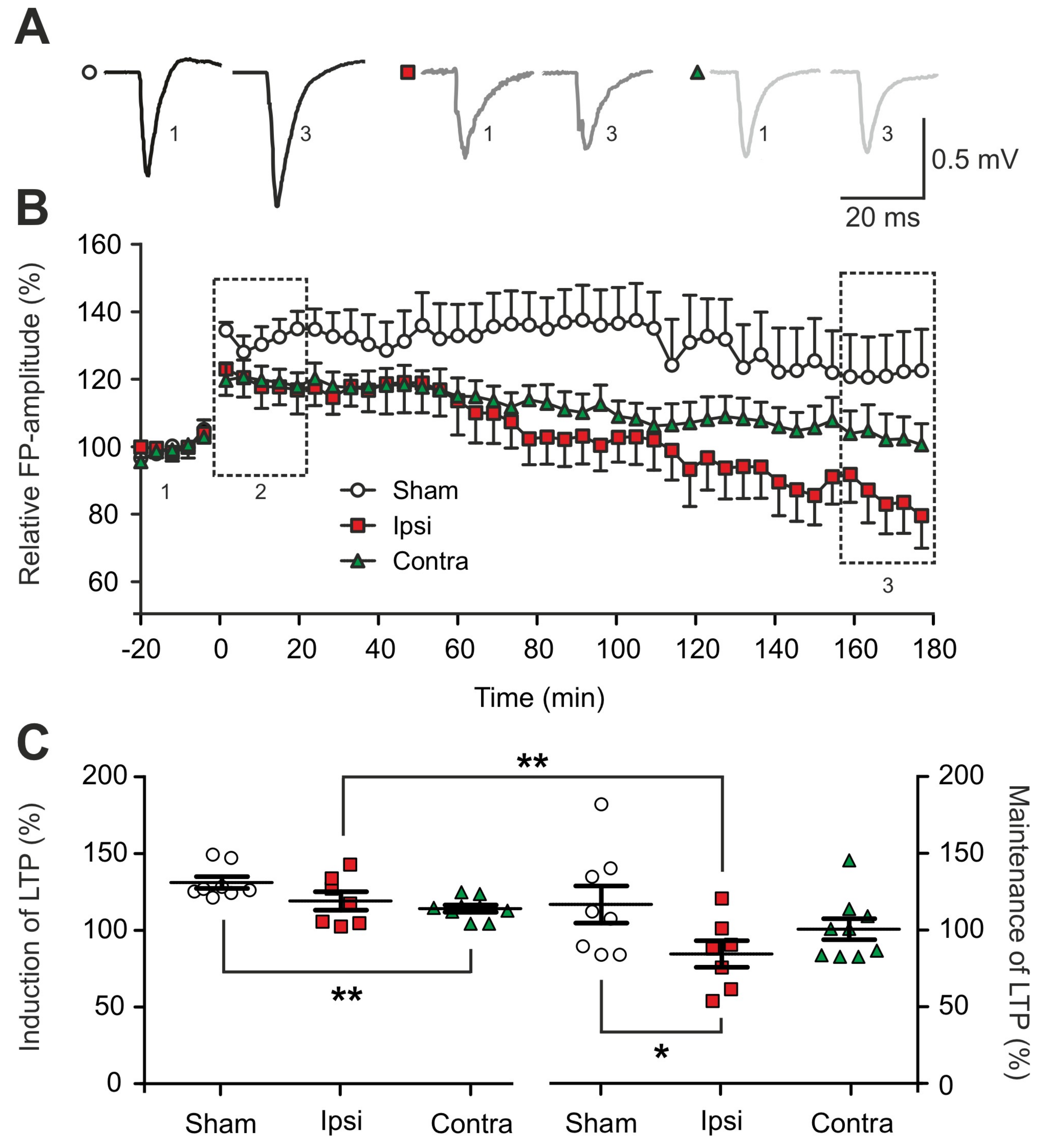
2.2. Impairment of Long-term Synaptic Plasticity 1 to 2 Days Post-Lesion
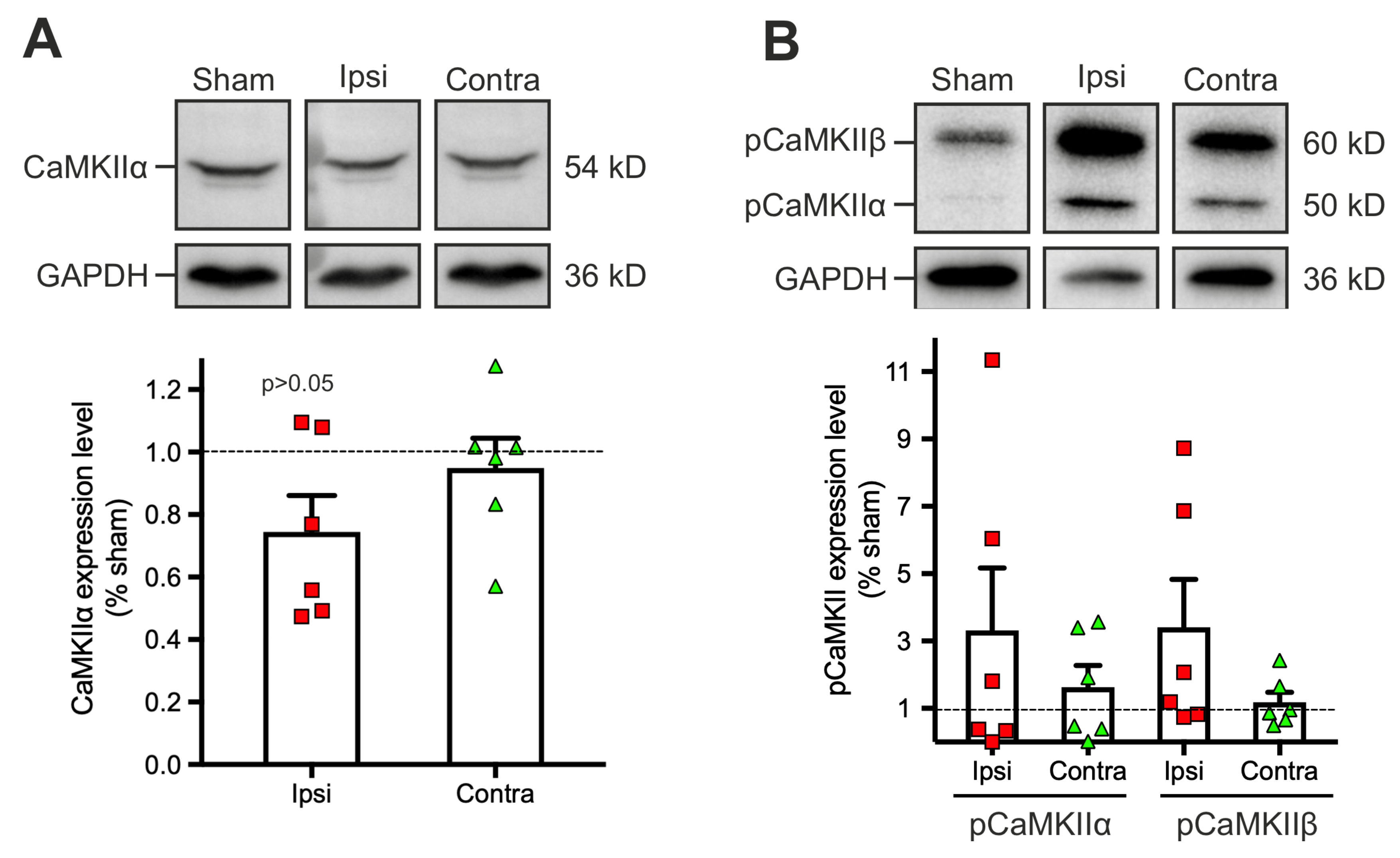
2.3. Dysregulation of the Phosphorylation Level of Calcium-Calmoduline Dependent Kinase II α (CaMKIIα) 1 to 2 Days Post-Traumatic Brain Injury (TBI)
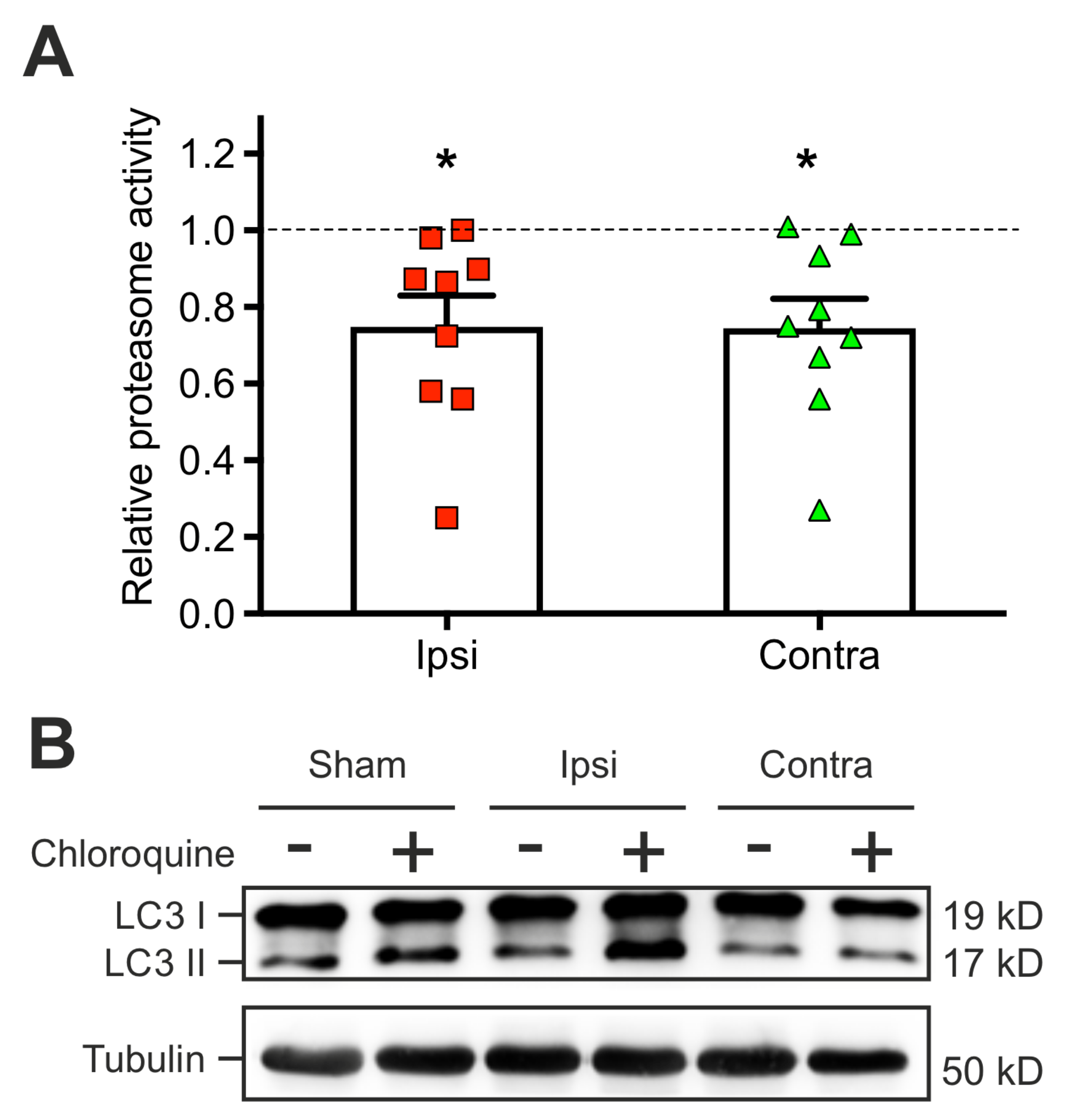
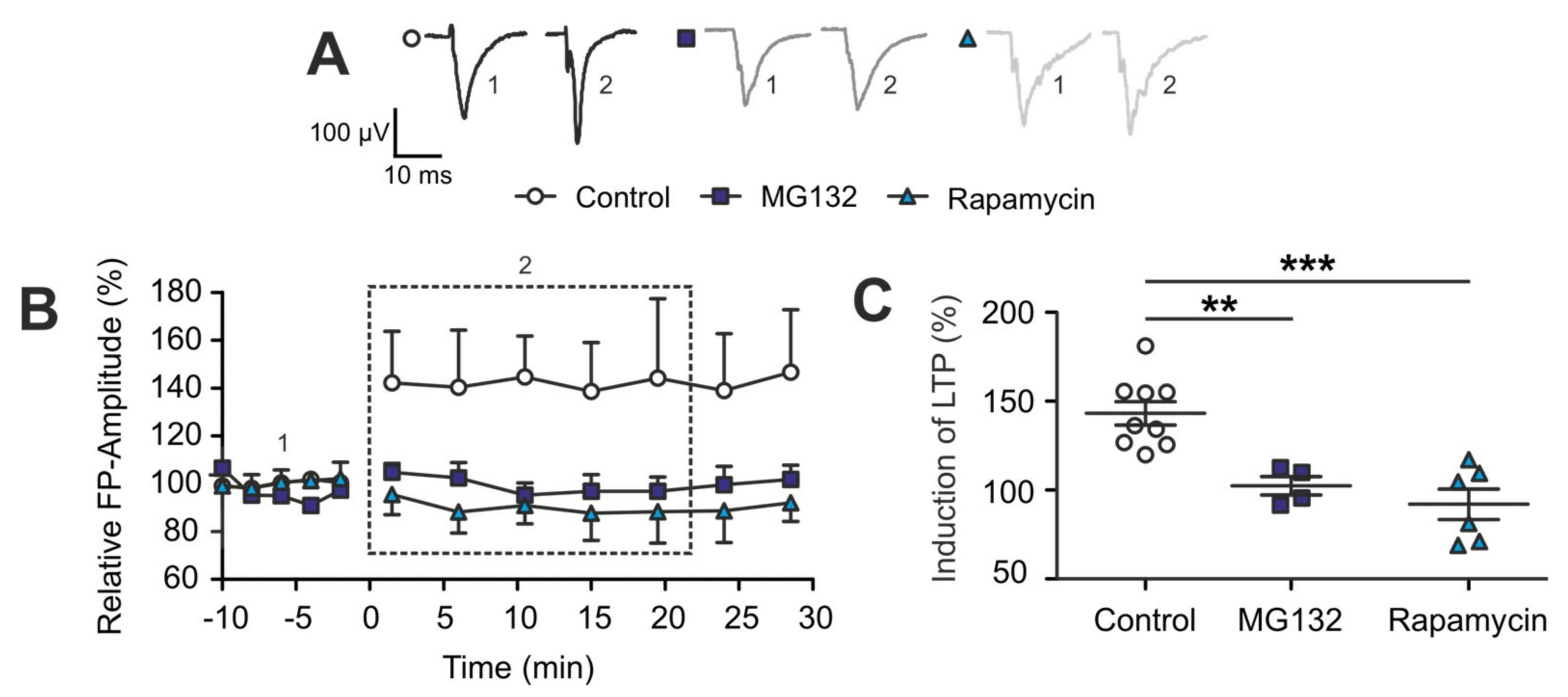
2.4. The TBI-Induced Impairment of Late Long-Term Potentiation (l-LTP) is Mediated by Altered Protein Degradation Systems
3. Discussion
3.1. Traumatic Brain Injury Induced an Early Impairment of Long-Term Synaptic Plasticity
3.2. Expression of Plasticity Related Proteins Is Not Significantly Altered Early after TBI
3.3. TBI-Induced Early Bidirectional Changes in the Activity of Two Protein Degradation Systems
3.4. Applicability and Future Directions
4. Materials and Methods
4.1. Animals and Ethical Statement
4.2. Induction of Traumatic Brain Injury
4.3. Electrophysiology In Vitro
4.3.1. Tissue Preparation
4.3.2. Multi-Electrode Array Recordings
4.4. Histology
4.5. Western Blots
4.6. Enzymatic Assays
4.6.1. Cell Death (TUNEL) Assay
4.6.2. Proteasome Activity Assay
4.6.3. Autophagic Degradation Activity Assay
4.7. Statistical Analysis
4.8. Limitations
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| aCSF | artificial cerebrospinal fluid |
| AMC | 7-amino-4-methylcoumarin |
| ATP | Adenosine triphosphate |
| BAG | Beclin2-associated anthanogene |
| BDNF | brain-derived neurotrophic factor |
| CaCl2 | calcium chloride |
| CaMKII | calcium/calmodulin-dependent protein kinase II |
| CCI | controlled cortical impact |
| CO2 | carbon dioxide |
| DNA | deoxyribonucleic acid |
| DTT | dithiothreitol |
| EFP | extracellular field potentials |
| GABA | gamma-aminobutyric acid |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| Hsp | heat shock protein |
| KCl | potassium chloride |
| LC3 | light chain 3 protein |
| LFP | local field potential |
| L-LTP | late long-term plasticity |
| LTP | long-term plasticity |
| MEA | multi electrode array |
| MgCl2 | magnesium chloride |
| mTOR | mammalian target of rapamycin |
| n | number |
| N2 | nitrogen |
| NaCl | sodium chloride |
| NaH2PO4 | monosodium phosphate |
| NaHCO3 | sodium hydrogene carbonate |
| NMDA | N-methyl-d-aspartic acid |
| O2 | oxygen |
| pMEA | perforated multi-electrode array |
| TBI | traumatic brain injury |
| TBS | theta-burst stimulation |
References
- Tagliaferri, F.; Compagnone, C.; Korsic, M.; Servadei, F.; Kraus, J. A systematic review of brain injury epidemiology in Europe. Acta Neurochir. 2006, 148, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Rozenbeek, B.; Maas, A.I.R.; Menon, D.K. Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 2013, 9, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Mckee, A.C.; Daneshvar, D.H. The neuropathology of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 45–66. [Google Scholar] [PubMed] [Green Version]
- Nortje, J.; Menon, D.K. Traumatic brain injury: Physiology, mechanisms, and outcome. Curr. Opin. Neurol. 2004, 17, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Robbins and Cotran. Pathologic Basis of Disease, 8th ed.; Saunders by Elsevier: Philadelphia, PA, USA, 2010; pp. 1287–1290. [Google Scholar]
- Gadani, S.P.; Walsh, J.T.; Lukens, J.R.; Kipnis, J. Dealing with Danger in the CNS: The Response of the Immune System to Injury. Neuron 2015, 87, 47–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chen, Y.; Jenkins, L.W.; Kochanek, P.M.; Clark, R.S.B. Bench-to-bedside review: Apoptosis/programmed cell death triggered by traumatic brain injury. Crit. Care 2015, 9, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.R.; Tesco, G. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front. Aging Neurosci. 2013, 5, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerriero, R.M.; Giza, C.C.; Rotenberg, A. Glutamate and GABA imbalance following traumatic brain injury. Curr. Neurol. Neurosci. Rep. 2015, 15, 27. [Google Scholar] [CrossRef]
- Imbrosci, B.; Wang, Y.; Arckens, L.; Mittmann, T. Neuronal mechanisms underlying transhemispheric diaschasis following focal cortical injuries. Brain Struct. Funct. 2015, 220, 1649–1664. [Google Scholar] [CrossRef]
- Le Prieult, F.; Thal, S.C.; Engelhard, K.; Imbrosci, B.; Mittmann, T. Acute Cortical Transhemispheric Diaschisis after Unilateral Traumatic Brain Injury. J. Neurotrauma 2017, 34, 1097–1110. [Google Scholar] [CrossRef]
- Buetefisch, C.M.; Kleiser, R.; Seitz, R.J. Post-lesional cerebral reorganization: Evidence from functional neuroimaging and transcranial magnetic stimulation. J. Physiol. Paris 2006, 99, 437–454. [Google Scholar] [CrossRef] [PubMed]
- Wakade, C.; SukumariRamesh, S.; Laird, M.D.; Dhandapani, K.M.; Vender, J.R. Delayed reduction in hippocampal postsynaptic density protein-95 expression temporally correlates with cognitive dysfunction following controlled cortical impact in mice. J. Neurosurg. 2011, 113, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.H. Neurotransmitters. In Principles of Neural Science, 4th ed.; Elsevier Science Publishing Co., Inc.: New York, NY, USA, 2000; pp. 280–297. [Google Scholar]
- Castillo, P.E. Presynaptic LTP and LTD of Excitatory and Inhibitory Synapses. Cold Spring Harb. Perspect. Biol. 2012, 4, a005728. [Google Scholar] [CrossRef] [PubMed]
- Nowak, L.; Bregestovski, P.; Ascher, P.; Herbet, A.; Prochiantz, A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature 2011, 307, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.P.; Cooke, S.F. Long-term potentiation and long-term depression: A clinical perspective. Clin. (Sao Paulo) 2011, 66, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Redondo, R.L.; Morris, R.G. Making memories last: The synaptic tagging and capture hypothesis. Nat. Rev. Neurosci. 2011, 12, 17–30. [Google Scholar] [CrossRef]
- Malinow, R.; Malenka, R.C. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 2002, 25, 103–126. [Google Scholar] [CrossRef]
- Lisman, J.; Schulman, H.; Cline, H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 2002, 3, 175–190. [Google Scholar] [CrossRef]
- Byth, L.A. Ca2+- and CaMKIImediated processes in early LTP. Ann. Neurosci. 2014, 21, 151–153. [Google Scholar] [CrossRef]
- Fonseca, R.; Vabulas, R.M.; Hartl, F.U.; Bonhoeffer, T.; Nägerl, U.V. A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron 2006, 52, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.; Frey, J.U.; Sanna, P.P.; Behnisch, T. Protein degradation by the proteasome is required for synaptic tagging and the heterosynaptic stabilization of hippocampal latephase longterm potentiation. Neuroscience 2010, 169, 1520–1526. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Bach, S.V.; Haynes, K.A.; Hegde, A.N. Proteasome modulates positive and negative translational regulators in long-term synaptic plasticity. J. Neurosci. 2014, 34, 3171–3182. [Google Scholar] [CrossRef] [PubMed]
- Ghighlieri, V.; Pendolino, V.; Bagetta, V.; Sgobio, C.; Calabresi, P.; Picconi, B. mTOR inhibitor rapamycin suppresses striatal postischemic LTP. Exp. Neurol. 2010, 226, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Miao, Y.; Chen, L.; Jin, P.; Zha, Y.; Chai, Y.; Zheng, F.; Zhang, Y.; Zhuo, W.; Zhand, J.; et al. The role of elevated autophagy on the synaptic plasticity impairment caused by CdSe/ZnS quantum dots. Biomaterials 2013, 34, 10172–10181. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.; Torres, C.A.; Setlik, W.; Cebrián, C.; Mosharov, E.V.; Tang, G.; Cheng, H.C.; Yarygina, O.; Burke, R.E.; Gershon, M.; et al. Regulation of Presynaptic Neurotransmission by Macroautophagy. Neuron 2012, 47, 277–284. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Carra, S.; Behl, C. Emerging roles of molecular chaperones and co-chaperones in selective autophagy: Focus on BAG proteins. J. Mol. Med. 2011, 89, 1175–1182. [Google Scholar] [CrossRef]
- Behl, C. Breaking BAG: The Co-Chaperone BAG3 in Health and Disease. Trends Pharm. Sci. 2016, 37, 672–688. [Google Scholar] [CrossRef]
- Morawe, T.; Hiebel, C.; Kern, A.; Behl, C. Protein Homeostasis, Aging and Alzheimer’s Disease. Mol. Neurobiol. 2012, 46, 41–54. [Google Scholar] [CrossRef]
- Behl, C. BAG3 and friends Co-chaperones in selective autophagy during aging and disease. Autophagy 2011, 7, 795–798. [Google Scholar] [CrossRef]
- Yao, X.; Liu, J.; McCabe, J.T. Alterations of cerebral cortex and hippocampal proteasome subunit expression and function in a traumatic brain injury rat model. J. Neurochem. 2007, 104, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Fukuda, T.; Iwadate, K. Immunohistochemical analysis of the ubiquitin proteasome system and autophagy lysosome system induced after traumatic intracranial injury: Association with time between the injury and death. Am. J. Forensic Med. Pathol. 2014, 35, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.S.; Bayir, H.; Chu, C.T.; Alber, S.M.; Watkins, S.C. Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy 2008, 4, 88–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, C.; Zhao, Z.; Aungst, S.; Sabirzhanov, B.; Faden, A.I.; Lipinski, M.M. Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy 2014, 10, 2208–2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Li, E.; Sun, G.; Yan, H.; Foley, L.; Andrzejczuk, L.; Attarwala, I.; Hitchens, T.; Kiselyov, K.; Dixon, C.; et al. Effects of DHA on Hippocampal Autophagy and Lysosome Function After Traumatic Brain Injury. Mol. Neurobiol. 2017, 55, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Cajigas, I.J.; Will, T.; Schuman, E.M. Protein homeostasis and synaptic plasticity. EMBO J. 2010, 29, 2746–2752. [Google Scholar] [CrossRef] [PubMed]
- Graves, A.B.; White, E.; Koepsell, T.D.; Reifler, B.V.; van Belle, G.; Larson, E.B.; Raskind, M. The association between head trauma and Alzheimer’s disease. Am. J. Epidemiol. 1990, 131, 491–501. [Google Scholar] [CrossRef]
- Mayeux, R.; Ottman, R.; Maestre, G.; Ngai, C.; Tang, M.X.; Ginsberg, H.; Tycko, B.; Shelanski, M. Synergistic effects of traumatic head injury and apolipoprotein-epsilon 4 in pateints with Alzheimer’s disease. Neurology 1995, 45, 555–557. [Google Scholar] [CrossRef]
- Graham, D.I.; Gentleman, S.M.; Lynch, A.; Roberts, G.W. Distribution of beta-amyloid protein in the brain following severe head injury. Neuropathol. Appl. Neurobiol. 1995, 21, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Cupples, L.A.; Kurz, A.; Auerbach, S.H.; Volicer, L.; Chui, H.; Green, R.C.; Sadovnick, A.D.; Duara, R.; DeCarli, C.; et al. Head injury and risk of AD in the MIRAGE study. Neurology 2000, 54, 1316–1323. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Widespread τ and amyloid-β pathology many years after traumatic brain injury in humans. Brain Pathol. 2012, 22, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Ojo, J.O.; Mouzon, B.; Greenberg, M.B.; Bachmeier, C.; Mullan, M.; Crawford, F. Repetitive mild traumatic brain injury augments tau pathology and glial activation in aged hTau mice. J. Neuropathol. Exp. Neurol. 2013, 72, 137–151. [Google Scholar] [CrossRef]
- Mittmann, T.; Eysel, U.T. Increased synaptic plasticity in the surround of visual cortex lesions in rats. Neuroreport 2001, 12, 3341–3347. [Google Scholar] [CrossRef]
- Huemmeke, M.; Eysel, U.T.; Mittmann, T. Metabotropic glutamate receptors mediate expression of LTP in slices of rat visual cortex. Eur. J. Neurosci. 2002, 15, 1641–1645. [Google Scholar] [CrossRef]
- Dohle, C.I.; Eysel, U.T.; Mittmann, T. Spatial distribution of long-term potentiation in the surround of visual cortex lesions in vitro. Exp. Brain Res. 2009, 199, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Imbrosci, B.; Mittmann, T. Functional consequences of the disturbances in the GABA-mediated inhibition induced by injuries in the cerebral cortex. Neural Plast. 2011, 2011, 614329. [Google Scholar] [CrossRef]
- Cantu, D.; Walter, K.; Andresen, L.; TaylorWeiner, A.; Hampton, D.; Tesco, G.; Dulla, C.G. Traumatic brain injury increases cortical glutamate network activity by compromising GABAergic control. Cereb. Cortex 2015, 25, 2306–2320. [Google Scholar] [CrossRef] [PubMed]
- Reeves, T.M.; Lyeth, B.G.; Povlishock, J.T. Longterm potentiation deficits and excitability changes following traumatic brain injury. Exp. Brain Res. 1995, 106, 248–256. [Google Scholar] [CrossRef]
- Sanders, M.J.; Sick, T.J.; PerezPinzon, M.A.; Dietrich, W.D.; Green, E.J. Chronic failure in the maintenance of longterm potentiation following fluid percussion injury in the rat. Brain Res. 2000, 861, 69–76. [Google Scholar] [CrossRef]
- Schwarzbach, E.; Bonislawski, D.P.; Xiong, G.; Cohen, A.S. Mechanisms underlying the inability to induce area CA1 LTP in the mouse after traumatic brain injury. Hippocampus 2006, 16, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Fineman, I.; Hovda, D.A.; Smith, M.M.; Yoshino, A.; Becker, D.P. Concussive brain injury is associated with a prolonged accumulation of calcium: A 45Ca2+ autoradiographic study. Brain Res. 1993, 624, 94–102. [Google Scholar] [CrossRef]
- Atkins, C.M.; Chen, S.; Alonso, O.F.; Dietrich, W.D.; Hu, B.R. Activation of calcium/calmodulin-dependent protein kinases after traumatic brain injury. J. Cereb. Blood Flow Metab. 2006, 26, 1507–1518. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.Y.; Baussi, O.; Levine, A.; Chen, Y.; Schacher, S. Persistent longterm synaptic plasticity requires activation of a new signaling pathway by additional stimuli. J. Neurosci. 2011, 31, 8841–8850. [Google Scholar] [CrossRef]
- Hu, J.Y.; Adler, K.; Farah, C.A.; Hastings, M.H.; Sossin, W.S.; Schacher, S. CellSpecific PKM Isoforms Contribute to the Maintenance of Different Forms of Persistent Long-Term Synaptic Plasticity. J. Neurosci. 2017, 37, 2476–2763. [Google Scholar] [CrossRef]
- Shao, C.Y.; Sondhi, R.; van de Nes, P.S.; Sacktor, T.C. PKMζ is necessary and sufficient for synaptic clustering of PSD95. Hippocampus 2012, 22, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Volk, L.J.; Bachmann, J.L.; Johnson, R.; Yu, Y.; Huganir, R.L. PKMζ is not required for hippocampal synaptic plasticity, learning and memory. Nature 2013, 493, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Salon, M.; Alonso, M.; Vianna, M.R.; Viola, H.; Mello e Souza, T.; Izguierdo, I.; Pasquini, J.M.; Medina, J.H. The ubiquitin-proteasome cascade is required for mammalian long-term memory formation. Eur. J. Neurosci. 2001, 14, 1820–1826. [Google Scholar] [CrossRef]
- Tai, H.C.; Schuman, E.M. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 2008, 9, 826–838. [Google Scholar] [CrossRef]
- Mabb, A.M.; Ehlers, M.D. Ubiquitination in postsynaptic function and plasticity. Annu. Rev. Cell. Dev. Biol. 2010, 26, 179–210. [Google Scholar] [CrossRef]
- Jarome, T.J.; Helmstetter, F.J. The ubiquitin-proteasome system as a critical regulator of synaptic plasticity and long-term memory formation. Neurobiol. Learn. Mem. 2013, 105, 107–116. [Google Scholar] [CrossRef]
- Tsai, N.P. Ubiquitin proteasome system-mediated degradation of synaptic proteins: An update from the postsynaptic side. Biochim. Biophys. Acta 2014, 1843, 2838–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, 10–17. [Google Scholar] [CrossRef]
- Sunkaria, A.; Yadav, A.; Bhardwaj, S.; Sandhir, R. Postnatal Proteasome Inhibtion Promotes Amyloid-β Aggregation in Hippocampus and Impairs Spatial Learning in Adult Mice. Neuroscience 2017, 367, 47–59. [Google Scholar] [CrossRef]
- Dong, C.; Upadhya, S.C.; Ding, L.; Smith, T.K.; Hegde, A.N. Proteasome inhibition enhances the induction and impairs the maintenance of late-phase long-term potentiation. Learn. Mem. 2008, 15, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, E.; Lessmann, V.; Brigadski, T. Pre- and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology 2014, 76, 610–627. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.R.; Mele, M.; Vaz, S.H.; Kellermayer, B.; Grimaldi, M.; Colino-Oliveira, M.; Rombo, D.M.; Comprido, D.; Sebastiao, A.M.; Duarte, C.B. Differential Role of the Proteasome in the Early and Late Phases of BDNF-Induced Facilitation of LTP. J. Neurosci. 2015, 35, 3319–3329. [Google Scholar] [CrossRef] [Green Version]
- Stoica, L.; Zhu, P.J.; Huang, W.; Zhou, H.; Kozma, S.C.; CostaMattioli, M. Selective pharmacogenetic inhibition of mammalian target of Rapamycin complex I (mTORC1) blocks longterm synaptic plasticity and memory storage. Proc. Natl. Acad. Sci. USA 2011, 108, 3791–3796. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Li, W.; Wang, H.; Zhang, J.; Yu, C.; Tan, S.; Wang, H.; Xu, X.; Dong, J.; Yao, B.; et al. Autophagy mediates the degradation of synaptic vesicles: A potential mechanism of synaptic plasticity injury induced by microwave exposure in rats. Physiol. Behav. 2018, 188, 119–127. [Google Scholar] [CrossRef]
- Blockaert, J.; Marin, P. mTOR in Brain Physiology and Pathologies. Physiol. Rev. 2015, 95, 1157–1187. [Google Scholar] [CrossRef] [PubMed]
- Glatigny, M.; Moriceau, S.; Rivagorda, M.; Ramos-Brossier, M.; Nascimbeni, A.C.; Lante, F.; Shanley, M.R.; Boudarene, N.; Rousseaud, A.; Friedman, A.K.; et al. Autophagy Is Required for Memory Formation and Reverses Age-Related Memory Decline. Curr. Biol. 2019, 29, 435–448. [Google Scholar] [CrossRef]
- Karpova, A.; Mikhaylova, M.; Thomas, U.; Knöpfel, T.; Behnisch, T. Involvement of Protein Synthesis and Degradation in Long-Term Potentiation of Schaffer Collateral CA1 Synapses. J. Neurosci. 2006, 26, 4949–4955. [Google Scholar] [CrossRef] [PubMed]
- Thal, S.C.; Wyschkon, S.; Pieter, D.; Engelhard, K.; Werner, C. Selection of endogenous control genes for normalization of gene expression analysis after experimental brain trauma in mice. J. Neurotrauma 2008, 25, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Renziehausen, J.; Hiebel, C.; Nagel, H.; Kundu, A.; Kins, S.; Kögel, D.; Behl, C.; Hajieva, P. The cleavage product of amyloid-β protein precursor sAβPPα modulates BAG3-dependent aggresome formation and enhances cellular proteasomal activity. J. Alzheimers Dis. 2015, 44, 879–896. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–139. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feldmann, L.K.; Le Prieult, F.; Felzen, V.; Thal, S.C.; Engelhard, K.; Behl, C.; Mittmann, T. Proteasome and Autophagy-Mediated Impairment of Late Long-Term Potentiation (l-LTP) after Traumatic Brain Injury in the Somatosensory Cortex of Mice. Int. J. Mol. Sci. 2019, 20, 3048. https://doi.org/10.3390/ijms20123048
Feldmann LK, Le Prieult F, Felzen V, Thal SC, Engelhard K, Behl C, Mittmann T. Proteasome and Autophagy-Mediated Impairment of Late Long-Term Potentiation (l-LTP) after Traumatic Brain Injury in the Somatosensory Cortex of Mice. International Journal of Molecular Sciences. 2019; 20(12):3048. https://doi.org/10.3390/ijms20123048
Chicago/Turabian StyleFeldmann, Lucia K., Florie Le Prieult, Vanessa Felzen, Serge C. Thal, Kristin Engelhard, Christian Behl, and Thomas Mittmann. 2019. "Proteasome and Autophagy-Mediated Impairment of Late Long-Term Potentiation (l-LTP) after Traumatic Brain Injury in the Somatosensory Cortex of Mice" International Journal of Molecular Sciences 20, no. 12: 3048. https://doi.org/10.3390/ijms20123048
APA StyleFeldmann, L. K., Le Prieult, F., Felzen, V., Thal, S. C., Engelhard, K., Behl, C., & Mittmann, T. (2019). Proteasome and Autophagy-Mediated Impairment of Late Long-Term Potentiation (l-LTP) after Traumatic Brain Injury in the Somatosensory Cortex of Mice. International Journal of Molecular Sciences, 20(12), 3048. https://doi.org/10.3390/ijms20123048