Epigenetic Regulation of Plant Gametophyte Development


{kind=link}
Abstract
:1. Introduction
2. Basics of Epigenetic Regulation in Plants
2.1. DNA Methylation
2.2. Chromatin Modification
2.3. Small Non-Coding RNAs
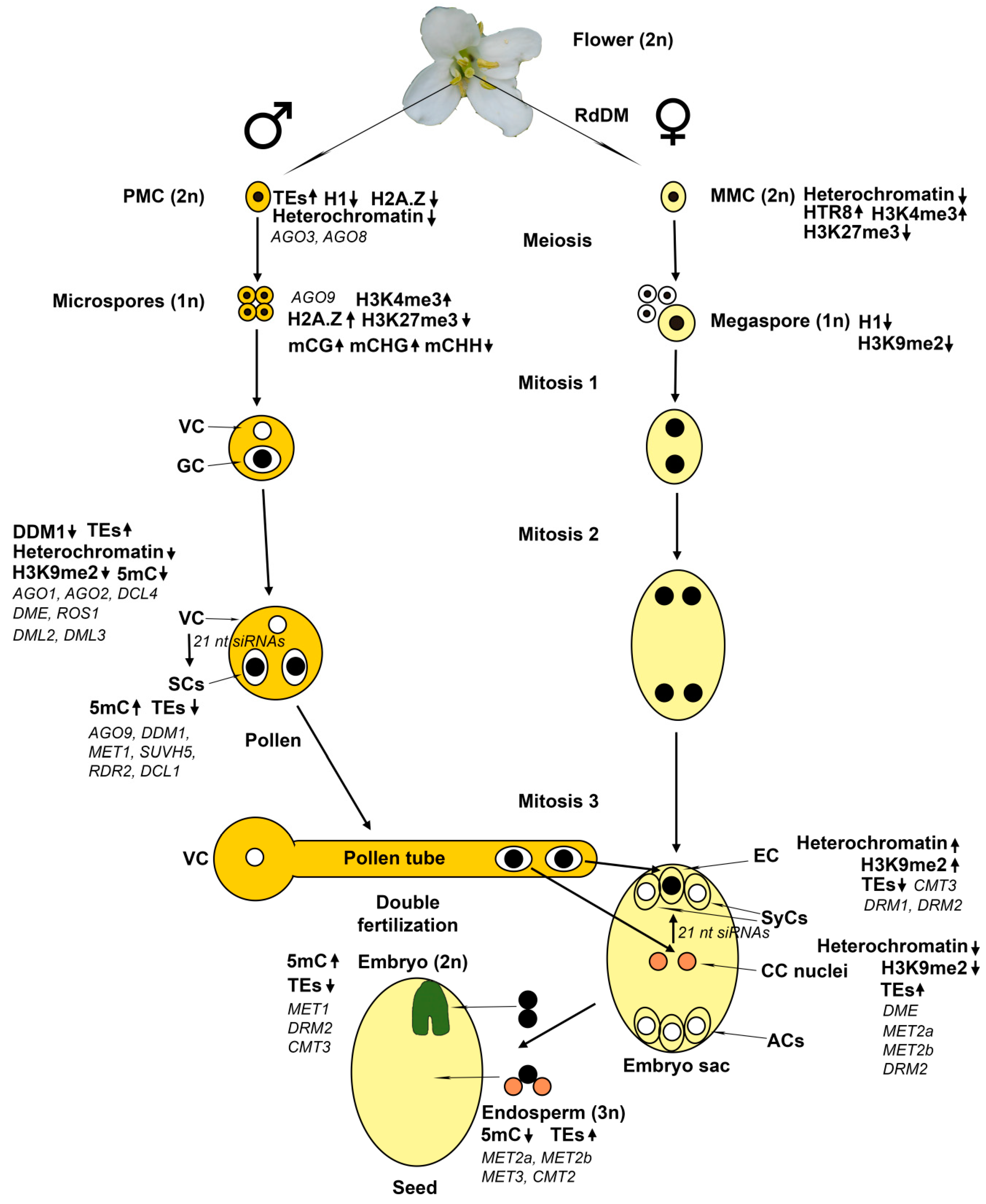
3. Epigenetic Regulation at the Meiotic Stage of Gametophyte Development
4. Epigenetic Regulation at the Post-Meiotic Stage of Gametophyte Development
4.1. The Male Gametophyte
4.2. The Female Gametophyte
4.3. Common Epigenetic Mechanisms in Male and Female Gametophytes
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Ma, H.; Sundaresan, V. Development of flowering plant gametophytes. Curr. Top. Dev. Biol. 2010, 91, 379–412. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.C.; Shi, D.Q.; Chen, Y.H. Female gametophyte development in flowering plants. Annu. Rev. Plant Biol. 2010, 61, 89–108. [Google Scholar] [CrossRef] [PubMed]
- Friedman, W.E.; Madrid, E.N.; Williams, J.H. Origin of the fittest and survival of the fittest: Relating female gametophyte development to endosperm genetics. Int. J. Plant Sci. 2008, 169, 79–92. [Google Scholar] [CrossRef]
- Diboll, A.; Larson, D. An electron microscopic study of the mature megagametophyte in Zea mays. Am. J. Bot. 1996, 53, 391–402. [Google Scholar] [CrossRef]
- Pagnussat, G.C.; Yu, H.J.; Sundaresan, V. Cell-fate switch of synergid to egg cell in Arabidopsis eostre mutant embryo sacs arises from misexpression of the BEL1-like homeodomain gene BLH1. Plant Cell 2007, 19, 3578–3592. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Huang, B.Q.; Han, Y.; Zee, S.Y. Fertilization in maize indeterminate gametophyte1 mutant. Protoplasma 2004, 223, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Pagnussat, G.C.; Alandete-Saez, M.; Bowman, J.L.; Sundaresan, V. Auxin-dependent patterning and gamete specification in the Arabidopsis female gametophyte. Science 2009, 324, 1684–1689. [Google Scholar] [CrossRef] [PubMed]
- Gross-Hardt, R.; Kagi, C.; Baumann, N.; Moore, J.M.; Baskar, R.; Gagliano, W.B.; Jurgens, G.; Grossniklaus, U. LACHESIS restricts gametic cell fate in the female gametophyte of Arabidopsis. PLoS Biol. 2007, 5, e47. [Google Scholar] [CrossRef] [PubMed]
- Moll, C.; von Lyncker, L.; Zimmermann, S.; Kagi, C.; Baumann, N.; Twell, D.; Grossniklaus, U.; Gross-Hardt, R. CLO/GFA1 and ATO are novel regulators of gametic cell fate in plants. Plant J. 2008, 56, 913–921. [Google Scholar] [CrossRef]
- Bleckmann, A.; Alter, S.; Dresselhaus, T. The beginning of a seed: Regulatory mechanisms of double fertilization. Front. Plant Sci. 2014, 5, 452. [Google Scholar] [CrossRef]
- Feng, S.; Jacobsen, S.E. Epigenetic modifications in plants: An evolutionary perspective. Curr. Opin. Plant Biol. 2011, 14, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Vanyushin, B.F.; Ashapkin, V.V. DNA methylation in higher plants: Past, present and future. Biochim. Biophys. Acta 2011, 1809, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulfite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef]
- Woo, H.R.; Dittmer, T.A.; Richards, E.J. Three SRA-domain methylcytosine-binding proteins cooperate to maintain global CpG methylation and epigenetic silencing in Arabidopsis. PLoS Genet. 2008, 4, e1000156. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhong, X.; Bernatavichute, Y.V.; Stroud, H.; Feng, S.; Caro, E.; Vashisht, A.A.; Terragni, J.; Chin, H.G.; Tu, A.; et al. Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell 2012, 151, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.M.; Bostick, M.; Zhang, X.; Kraft, E.; Henderson, I.; Callis, J.; Jacobsen, S.E. The SRA methylcytosine-binding domain links DNA and histone methylation. Curr. Biol. 2007, 17, 379–384. [Google Scholar] [CrossRef]
- Cao, X.; Jacobsen, S.E. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 2002, 12, 1138–1144. [Google Scholar] [CrossRef]
- Jullien, P.E.; Susaki, D.; Yelagandula, R.; Higashiyama, T.; Berger, F. DNA methylation dynamics during sexual reproduction in Arabidopsis thaliana. Curr. Biol. 2012, 22, 1825–1830. [Google Scholar] [CrossRef]
- Zemach, A.; Kim, M.Y.; Hsieh, P.H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Matzke, M.A.; Kanno, T.; Matzke, A.J. RNA-directed DNA methylation: The evolution of a complex epigenetic pathway in flowering plants. Annu. Rev. Plant Biol. 2015, 66, 243–267. [Google Scholar] [CrossRef] [PubMed]
- Wendte, J.M.; Pikaard, C.S. The RNAs of RNA-directed DNA methylation. Biochim. Biophys. Acta 2017, 1860, 140–148. [Google Scholar] [CrossRef]
- Johnson, L.M.; Du, J.; Hale, C.J.; Bischof, S.; Feng, S.; Chodavarapu, R.K.; Zhong, X.; Marson, G.; Pellegrini, M.; Segal, D.J.; et al. SRA- and SET-domain-containing proteins link RNA polymerase V occupancy to DNA methylation. Nature 2014, 507, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Carroll, B.J. Evolution and diversification of small RNA pathways in flowering plants. Plant Cell Physiol. 2018, 59, 2169–2187. [Google Scholar] [CrossRef] [PubMed]
- Nuthikattu, S.; McCue, A.D.; Panda, K.; Fultz, D.; Defraia, C.; Thomas, E.N.; Slotkin, R.K. The initiation of epigenetic silencing of active transposable elements is triggered by RDR6 and 21–22 nucleotide small interfering RNAs. Plant Physiol. 2013, 162, 116–131. [Google Scholar] [CrossRef]
- McCue, A.D.; Panda, K.; Nuthikattu, S.; Choudury, S.G.; Thomas, E.N.; Slotkin, R.K. ARGONAUTE 6 bridges transposable element mRNA-derived siRNAs to the establishment of DNA methylation. EMBO J. 2015, 34, 20–35. [Google Scholar] [CrossRef]
- Gehring, M.; Reik, W.; Henikoff, S. DNA demethylation by DNA repair. Trends Genet. 2009, 25, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Gehring, M.; Johnson, L.; Hannon, M.; Harada, J.J.; Goldberg, R.B.; Jacobsen, S.E.; Fischer, R.L. DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 2002, 110, 33–42. [Google Scholar] [CrossRef]
- Tang, K.; Lang, Z.; Zhang, H.; Zhu, J.-K. The DNA demethylase ROS1 targets genomic regions with distinct chromatin modifications. Nat. Plants 2016, 2, 16169. [Google Scholar] [CrossRef]
- Lang, Z.; Lei, M.; Wang, X.; Tang, K.; Miki, D.; Zhang, H.; Mangrauthia, S.K.; Liu, W.; Nie, W.; Ma, G.; et al. The methyl-CpG-binding protein MBD7 facilitates active DNA demethylation to limit DNA hyper-methylation and transcriptional gene silencing. Mol. Cell 2015, 57, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Zhang, H.; Julian, R.; Tang, K.; Xie, S.; Zhu, J.K. Regulatory link between DNA methylation and active demethylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, 3553–3557. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.P.; Pignatta, D.; Henikoff, S.; Gehring, M. Methylation-sensitive expression of a DNA demethylase gene serves as an epigenetic rheostat. PLoS Genet. 2015, 11, e1005142. [Google Scholar] [CrossRef] [PubMed]
- Penterman, J.; Zilberman, D.; Huh, J.H.; Ballinger, T.; Henikoff, S.; Fischer, R.L. DNA demethylation in the Arabidopsis genome. Proc. Natl. Acad. Sci. USA 2007, 104, 6752–6757. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Galisteo, A.P.; Morales-Ruiz, T.; Ariza, R.R.; Roldán-Arjona, T. Arabidopsis DEMETER-LIKE proteins DML2 and DML3 are required for appropriate distribution of DNA methylation marks. Plant Mol. Biol. 2008, 67, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Berr, A.; Shafiq, S.; Shen, W.-H. Histone modifications in transcriptional activation during plant development. Biochim. Biophys. Acta 2011, 1809, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bernatavichute, Y.V.; Cokus, S.; Pellegrini, M.; Jacobsen, S.E. Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol. 2009, 10, R62. [Google Scholar] [CrossRef]
- Zhang, X.; Clarenz, O.; Cokus, S.; Bernatavichute, Y.V.; Pellegrini, M.; Goodrich, J.; Jacobsen, S.E. Whole-genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis. PLoS Biol. 2007, 5, e129. [Google Scholar] [CrossRef]
- Zhang, X.; Germann, S.; Blus, B.J.; Khorasanizadeh, S.; Gaudin, V.; Jacobsen, S.E. The Arabidopsis LHP1 protein colocalizes with histone H3 Lys27 trimethylation. Nat. Struct. Mol. Biol. 2007, 14, 869–871. [Google Scholar] [CrossRef]
- Bernatavichute, Y.V.; Zhang, X.; Cokus, S.; Pellegrini, M.; Jacobsen, S.E. Genome-wide association of histone H3 lysine nine methylation with CHG DNA methylation in Arabidopsis thaliana. PLoS ONE 2008, 3, e3156. [Google Scholar] [CrossRef]
- Ma, Z.; Zhang, X. Actions of plant Argonautes: Predictable or unpredictable? Curr. Opin. Plant Biol. 2018, 45, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Qi, Y. RNAi in plants: An Argonaute-centered view. Plant Cell 2016, 28, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Tucker, M.R.; Okada, T.; Hu, Y.; Scholefield, A.; Taylor, J.M.; Koltunow, A.M. Somatic small RNA pathways promote the mitotic events of megagametogenesis during female reproductive development in Arabidopsis. Development 2012, 139, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, X.; Guo, X.; Wang, X.J.; Zhang, X. Arabidopsis AGO3 predominantly recruits 24-nt small RNAs to regulate epigenetic silencing. Nat. Plants 2016, 2, 16049. [Google Scholar] [CrossRef] [PubMed]
- Havecker, E.R.; Wallbridge, L.M.; Hardcastle, T.J.; Bush, M.S.; Kelly, K.A.; Dunn, R.M.; Schwach, F.; Doonan, J.H.; Baulcombe, D.C. The Arabidopsis RNA-directed DNA methylation argonautes functionally diverge based on their expression and interaction with target loci. Plant Cell 2010, 22, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.G.; Zhang, H.; Tang, K.; Zhu, X.; Qian, W.; Hou, Y.J.; Wang, B.; Lang, Z.; Zhao, Y.; Wang, X.; et al. Specific but interdependent functions for Arabidopsis AGO4 and AGO6 in RNA-directed DNA methylation. EMBO J. 2015, 34, 581–592. [Google Scholar] [CrossRef]
- Chen, C.; Farmer, A.D.; Langley, R.J.; Mudge, J.; Crow, J.A.; May, G.D.; Huntley, J.; Smith, A.G.; Retzel, E.F. Meiosis-specific gene discovery in plants: RNA-Seq applied to isolated Arabidopsis male meiocytes. BMC Plant Biol. 2010, 10, 280. [Google Scholar] [CrossRef]
- Yang, H.; Lu, P.; Wang, Y.; Ma, H. The transcriptome landscape of Arabidopsis male meiocytes from high-throughput sequencing: The complexity and evolution of the meiotic process. Plant J. 2011, 65, 503–516. [Google Scholar] [CrossRef]
- Schmidt, A.; Wuest, S.E.; Vijverberg, K.; Baroux, C.; Kleen, D.; Grossniklaus, U. Transcriptome analysis of the Arabidopsis megaspore mother cell uncovers the importance of RNA helicases for plant germline development. PLoS Biol. 2011, 9, e1001155. [Google Scholar] [CrossRef]
- Olmedo-Monfil, V.; Duran-Figueroa, N.; Arteaga-Vazquez, M.; Demesa-Arévalo, E.; Autran, D.; Grimanelli, D.; Slotkin, R.K.; Martienssen, R.A.; Vielle-Calzada, J.P. Control of female gamete formation by a small RNA pathway in Arabidopsis. Nature 2010, 464, 628–632. [Google Scholar] [CrossRef]
- Hernández-Lagana, E.; Rodríguez-Leal, D.; Lúa, J.; Vielle-Calzada, J.P. A Multigenic Network of ARGONAUTE4 Clade Members Controls Early Megaspore Formation in Arabidopsis. Genetics 2016, 204, 1045–1056. [Google Scholar] [CrossRef]
- She, W.; Grimanelli, D.; Rutowicz, K.; Whitehead, M.W.; Puzio, M.; Kotlinski, M.; Jerzmanowski, A.; Baroux, C. Chromatin reprogramming during the somatic-to-reproductive cell fate transition in plants. Development 2013, 140, 4008–4019. [Google Scholar] [CrossRef] [Green Version]
- Pillot, M.; Baroux, C.; Vazquez, M.A.; Autran, D.; Leblanc, O.; Vielle-Calzada, J.P.; Grossniklaus, U.; Grimanelli, D. Embryo and endosperm inherit distinct chromatin and transcriptional states from the female gametes in Arabidopsis. Plant Cell 2010, 22, 307–320. [Google Scholar] [CrossRef]
- Ravi, M.; Shibata, F.; Ramahi, J.S.; Nagaki, K.; Chen, C.; Murata, M.; Chan, S.W. Meiosis-specific loading of the centromere-specific histone CENH3 in Arabidopsis thaliana. PLoS Genet. 2011, 7, e1002121. [Google Scholar] [CrossRef]
- She, W.; Baroux, C. Chromatin dynamics in pollen mother cells underpin a common scenario at the somatic-to-reproductive fate transition of both the male and female lineages in Arabidopsis. Front. Plant Sci. 2015, 6, 294. [Google Scholar] [CrossRef]
- Walker, J.; Gao, H.; Zhang, J.; Aldridge, B.; Vickers, M.; Higgins, J.D.; Feng, X. Sexual-lineage-specific DNA methylation regulates meiosis in Arabidopsis. Nat. Genet. 2018, 50, 130–137. [Google Scholar] [CrossRef]
- Pina, C.; Pinto, F.; Feijó, J.A.; Becker, J.D. Gene family analysis of the Arabidopsis pollen transcriptome reveals biological implications for cell growth, division control, and gene expression regulation. Plant Physiol. 2005, 138, 744–756. [Google Scholar] [CrossRef]
- Borges, F.; Gomes, G.; Gardner, R.; Moreno, N.; McCormick, S.; Feijó, J.A.; Becker, J.D. Comparative transcriptomics of Arabidopsis sperm cells. Plant Physiol. 2008, 148, 1168–1181. [Google Scholar] [CrossRef]
- Grant-Downton, R.; Hafidh, S.; Twell, D.; Dickinson, H.G. Small RNA pathways are present and functional in the angiosperm male gametophyte. Mol. Plant. 2009, 2, 500–512. [Google Scholar] [CrossRef]
- Grant-Downton, R.; Le Trionnaire, G.; Schmid, R.; Rodriguez-Enriquez, J.; Hafidh, S.; Mehdi, S.; Twell, D.; Dickinson, H. MicroRNA and tasiRNA diversity in mature pollen of Arabidopsis thaliana. BMC Genom. 2009, 10, 643. [Google Scholar] [CrossRef]
- Slotkin, R.K.; Vaughn, M.; Borges, F.; Tanurdzić, M.; Becker, J.D.; Feijó, J.A.; Martienssen, R.A. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 2009, 136, 461–472. [Google Scholar] [CrossRef]
- Martínez, G.; Panda, K.; Köhler, C.; Slotkin, R.K. Silencing in sperm cells is directed by RNA movement from the surrounding nurse cell. Nat. Plants 2016, 2, 16030. [Google Scholar] [CrossRef]
- Dunoyer, P.; Himber, C.; Voinnet, O. DICER-LIKE 4 is required for RNA interference and produces the 21-nucleotide small interfering RNA component of the plant cell-to-cell silencing signal. Nat. Genet. 2005, 37, 1356–1360. [Google Scholar] [CrossRef]
- Le Trionnaire, G.; Grant-Downton, R.T.; Kourmpetli, S.; Dickinson, H.G.; Twell, D. Small RNA activity and function in angiosperm gametophytes. J. Exp. Bot. 2011, 62, 1601–1610. [Google Scholar] [CrossRef]
- Borges, F.; Pereira, P.A.; Slotkin, R.K.; Martienssen, R.A.; Becker, J.D. MicroRNA activity in the Arabidopsis male germline. J. Exp. Bot. 2011, 62, 1611–1620. [Google Scholar] [CrossRef]
- Brownfield, L.; Hafidh, S.; Borg, M.; Sidorova, A.; Mori, T.; Twell, D. A plant germline-specific integrator of sperm specification and cell cycle progression. PLoS Genet. 2009, 5, e1000430. [Google Scholar] [CrossRef]
- Schoft, V.K.; Chumak, N.; Mosiolek, M.; Slusarz, L.; Komnenovic, V.; Brownfield, L.; Twell, D.; Kakutani, T.; Tamaru, H. Induction of RNA-directed DNA methylation upon decondensation of constitutive heterochromatin. EMBO Rep. 2009, 10, 1015–1021. [Google Scholar] [CrossRef]
- Ingouff, M.; Rademacher, S.; Holec, S.; Soljić, L.; Xin, N.; Readshaw, A.; Foo, S.H.; Lahouze, B.; Sprunck, S.; Berger, F. Zygotic resetting of the HISTONE 3 variant repertoire participates in epigenetic reprogramming in Arabidopsis. Curr. Biol. 2010, 20, 2137–2143. [Google Scholar] [CrossRef]
- Calarco, J.P.; Borges, F.; Donoghue, M.T.; Van Ex, F.; Jullien, P.E.; Lopes, T.; Gardner, R.; Berger, F.; Feijó, J.A.; Becker, J.D.; et al. Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 2012, 151, 194–205. [Google Scholar] [CrossRef]
- Schoft, V.K.; Chumak, N.; Choi, Y.; Hannon, M.; Garcia-Aguilar, M.; Machlicova, A.; Slusarz, L.; Mosiolek, M.; Park, J.S.; Park, G.T.; et al. Function of the DEMETER DNA glycosylase in the Arabidopsis thaliana male gametophyte. Proc. Natl. Acad. Sci. USA 2011, 108, 8042–8047. [Google Scholar] [CrossRef]
- Hsieh, T.-F.; Ibarra, C.A.; Silva, P.; Zemach, A.; Eshed-Williams, L.; Fischer, R.L.; Zilberman, D. Genome-wide demethylation of Arabidopsis endosperm. Science 2009, 324, 1451–1454. [Google Scholar] [CrossRef]
- Mosher, R.A.; Melnyk, C.W.; Kelly, K.A.; Dunn, R.M.; Studholme, D.J.; Baulcombe, D.C. Uniparental expression of PolIV-dependent siRNAs in developing endosperm of Arabidopsis. Nature 2009, 460, 283–286. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, C.; Baulcombe, D.C.; Chen, Z.J. Maternal siRNAs as regulators of parental genome imbalance and gene expression in endosperm of Arabidopsis seeds. Proc. Natl. Acad. Sci. USA 2012, 109, 5529–5534. [Google Scholar] [CrossRef]
- Henderson, I.R.; Jacobsen, S.E. Tandem repeats upstream of the Arabidopsis endogene SDC recruit non-CG DNA methylation and initiate siRNA spreading. Genes Dev. 2008, 22, 1597–1606. [Google Scholar] [CrossRef]
- Makarevich, G.; Villar, C.B.R.; Erilova, A.; Koehler, C. Mechanism of PHERES1 imprinting in Arabidopsis. J. Cell Sci. 2008, 121, 906–912. [Google Scholar] [CrossRef]
- Hsieh, P.H.; He, S.; Buttress, T.; Gao, H.; Couchman, M.; Fischer, R.L.; Zilberman, D.; Feng, X. Arabidopsis male sexual lineage exhibits more robust maintenance of CG methylation than somatic tissues. Proc. Natl. Acad. Sci. USA 2016, 113, 15132–15137. [Google Scholar] [CrossRef]
- Ibarra, C.A.; Feng, X.; Schoft, V.K.; Hsieh, T.F.; Uzawa, R.; Rodrigues, J.A.; Zemach, A.; Chumak, N.; Machlicova, A.; Nishimura, T.; et al. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 2012, 337, 1360–1364. [Google Scholar] [CrossRef]
- Houben, A.; Kumke, K.; Nagaki, K.; Hause, G. CENH3 distribution and differential chromatin modifications during pollen development in rye (Secale cereale L.). Chromosome Res. 2011, 19, 471–480. [Google Scholar] [CrossRef]
- Schmid, M.W.; Schmidt, A.; Klostermeier, U.C.; Barann, M.; Rosenstiel, P.; Grossniklaus, U. A powerful method for transcriptional profiling of specific cell types in eukaryotes: Laser-assisted microdissection and RNA sequencing. PLoS ONE 2012, 7, e29685. [Google Scholar] [CrossRef]
- Ashapkin, V.V.; Kutueva, L.I.; Vanyushin, B.F. Plant DNA methyltransferase genes: Multiplicity, expression, methylation patterns. Biochemistry (Mosc) 2016, 81, 141–151. [Google Scholar] [CrossRef]
- Gehring, M.; Bubb, K.L.; Henikoff, S. Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science 2009, 324, 1447–1451. [Google Scholar] [CrossRef]
- Ingouff, M.; Selles, B.; Michaud, C.; Vu, T.M.; Berger, F.; Schorn, A.J.; Autran, D.; Van Durme, M.; Nowack, M.K.; Martienssen, R.A.; et al. Live-cell analysis of DNA methylation during sexual reproduction in Arabidopsis reveals context and sex-specific dynamics controlled by noncanonical RdDM. Genes Dev. 2017, 31, 72–83. [Google Scholar] [CrossRef]
- Park, K.; Kim, M.Y.; Vickers, M.; Park, J.S.; Hyun, Y.; Okamoto, T.; Zilberman, D.; Fischer, R.L.; Feng, X.; Choi, Y.; et al. DNA demethylation is initiated in the central cells of Arabidopsis and rice. Proc. Natl. Acad. Sci. USA 2016, 113, 15138–15143. [Google Scholar] [CrossRef]
- Gent, J.I.; Ellis, N.A.; Guo, L.; Harkess, A.E.; Yao, Y.; Zhang, X.; Dawe, R.K. CHH islands: De novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 2013, 23, 628–637. [Google Scholar] [CrossRef]
- Erdmann, R.M.; Hoffmann, A.; Walter, H.K.; Wagenknecht, H.A.; Groß-Hardt, R.; Gehring, M. Molecular movement in the Arabidopsis thaliana female gametophyte. Plant Reprod. 2017, 30, 141–146. [Google Scholar] [CrossRef]
- Wuest, S.E.; Vijverberg, K.; Schmidt, A.; Weiss, M.; Gheyselinck, J.; Lohr, M.; Wellmer, F.; Rahnenführer, J.; von Mering, C.; Grossniklaus, U. Arabidopsis female gametophyte gene expression map reveals similarities between plant and animal gametes. Curr. Biol. 2010, 20, 506–512. [Google Scholar] [CrossRef]
- Jullien, P.E.; Mosquna, A.; Ingouff, M.; Sakata, T.; Ohad, N.; Berger, F. Retinoblastoma and its binding partner MSI1 control imprinting in Arabidopsis. PLoS Biol. 2008, 6, e194:1–e194:13. [Google Scholar] [CrossRef]
- Anderson, S.N.; Johnson, C.S.; Jones, D.S.; Conrad, L.J.; Gou, X.; Russell, S.D.; Sundaresan, V. Transcriptomes of isolated Oryza sativa gametes characterized by deep sequencing: Evidence for distinct sex-dependent chromatin and epigenetic states before fertilization. Plant J. 2013, 76, 729–741. [Google Scholar] [CrossRef]
- Ono, A.; Yamaguchi, K.; Fukada-Tanaka, S.; Terada, R.; Mitsui, T.; Iida, S. A null mutation of ROS1a for DNA demethylation in rice is not transmittable to progeny. Plant J. 2012, 71, 564–574. [Google Scholar] [CrossRef]
- Hsieh, T.-F.; Shin, J.; Uzawa, R.; Silva, P.; Cohen, S.; Bauer, M.J.; Hashimoto, M.; Kirkbride, R.C.; Harada, J.J.; Zilberman, D.; et al. Regulation of imprinted gene expression in Arabidopsis endosperm. Proc. Natl. Acad. Sci. USA 2011, 108, 1755–1762. [Google Scholar] [CrossRef]
- Zheng, B.; He, H.; Zheng, Y.; Wu, W.; McCormick, S. An ARID domain-containing protein within nuclear bodies is required for sperm cell formation in Arabidopsis thaliana. PLoS Genet. 2014, 10, e1004421. [Google Scholar] [CrossRef]
- Li, L.; Wu, W.; Zhao, Y.; Zheng, B. A reciprocal inhibition between ARID1 and MET1 in male and female gametes in Arabidopsis. J. Integr. Plant Biol. 2017, 59, 657–668. [Google Scholar] [CrossRef]
- Gutierrez-Marcos, J.F.; Dickinson, H.G. Epigenetic reprogramming in plant reproductive lineages. Plant Cell Physiol. 2012, 53, 817–823. [Google Scholar] [CrossRef]
- She, W.; Baroux, C. Chromatin dynamics during plant sexual reproduction. Front. Plant Sci. 2014, 5, 354. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashapkin, V.V.; Kutueva, L.I.; Aleksandrushkina, N.I.; Vanyushin, B.F. Epigenetic Regulation of Plant Gametophyte Development. Int. J. Mol. Sci. 2019, 20, 3051. https://doi.org/10.3390/ijms20123051
Ashapkin VV, Kutueva LI, Aleksandrushkina NI, Vanyushin BF. Epigenetic Regulation of Plant Gametophyte Development. International Journal of Molecular Sciences. 2019; 20(12):3051. https://doi.org/10.3390/ijms20123051
Chicago/Turabian StyleAshapkin, Vasily V., Lyudmila I. Kutueva, Nadezhda I. Aleksandrushkina, and Boris F. Vanyushin. 2019. "Epigenetic Regulation of Plant Gametophyte Development" International Journal of Molecular Sciences 20, no. 12: 3051. https://doi.org/10.3390/ijms20123051
APA StyleAshapkin, V. V., Kutueva, L. I., Aleksandrushkina, N. I., & Vanyushin, B. F. (2019). Epigenetic Regulation of Plant Gametophyte Development. International Journal of Molecular Sciences, 20(12), 3051. https://doi.org/10.3390/ijms20123051