Mitochondria as a Source and a Target for Uremic Toxins


 ,
, 

Abstract
:1. Introduction
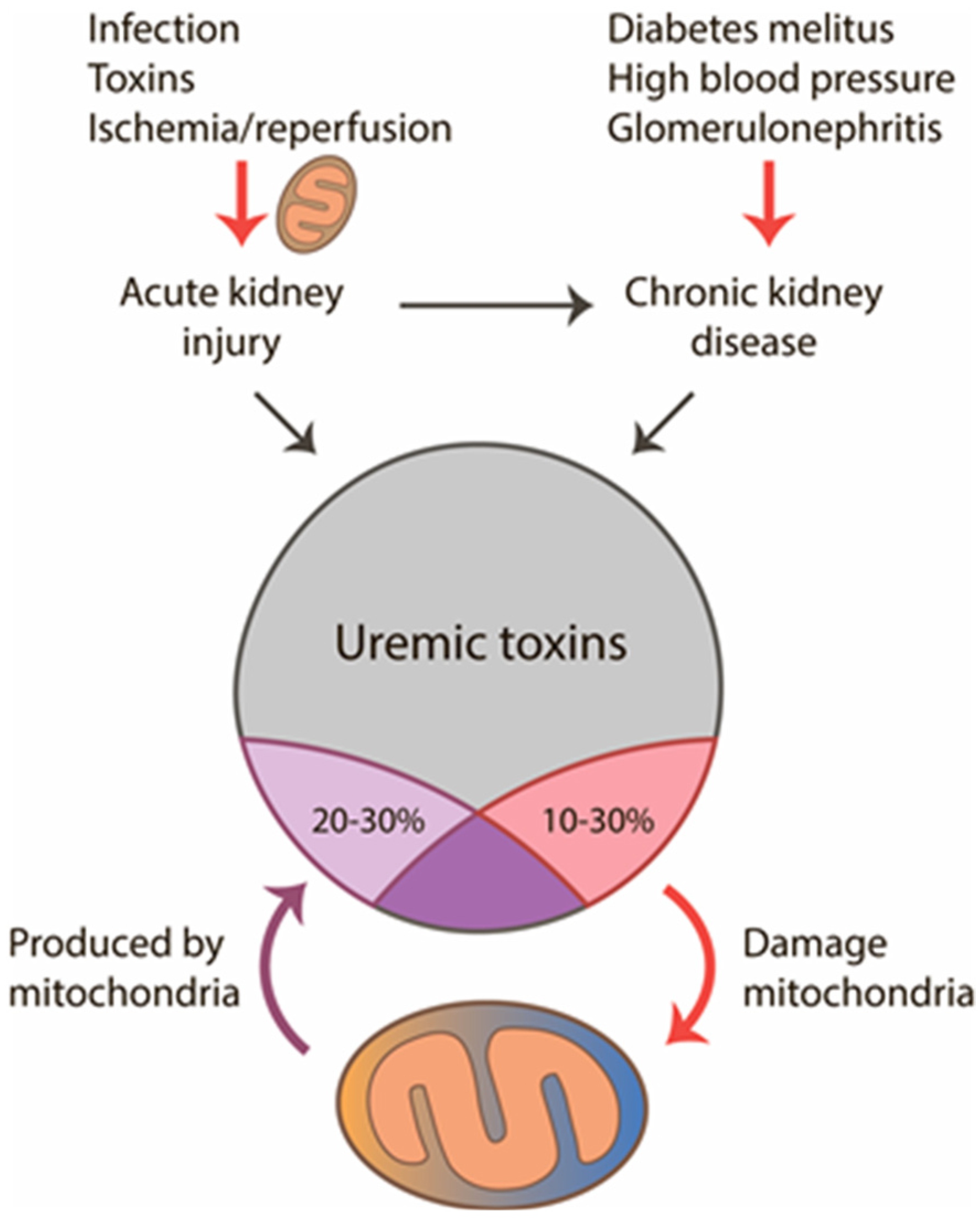
2. Mitochondria in Acute and Chronic Kidney Pathologies
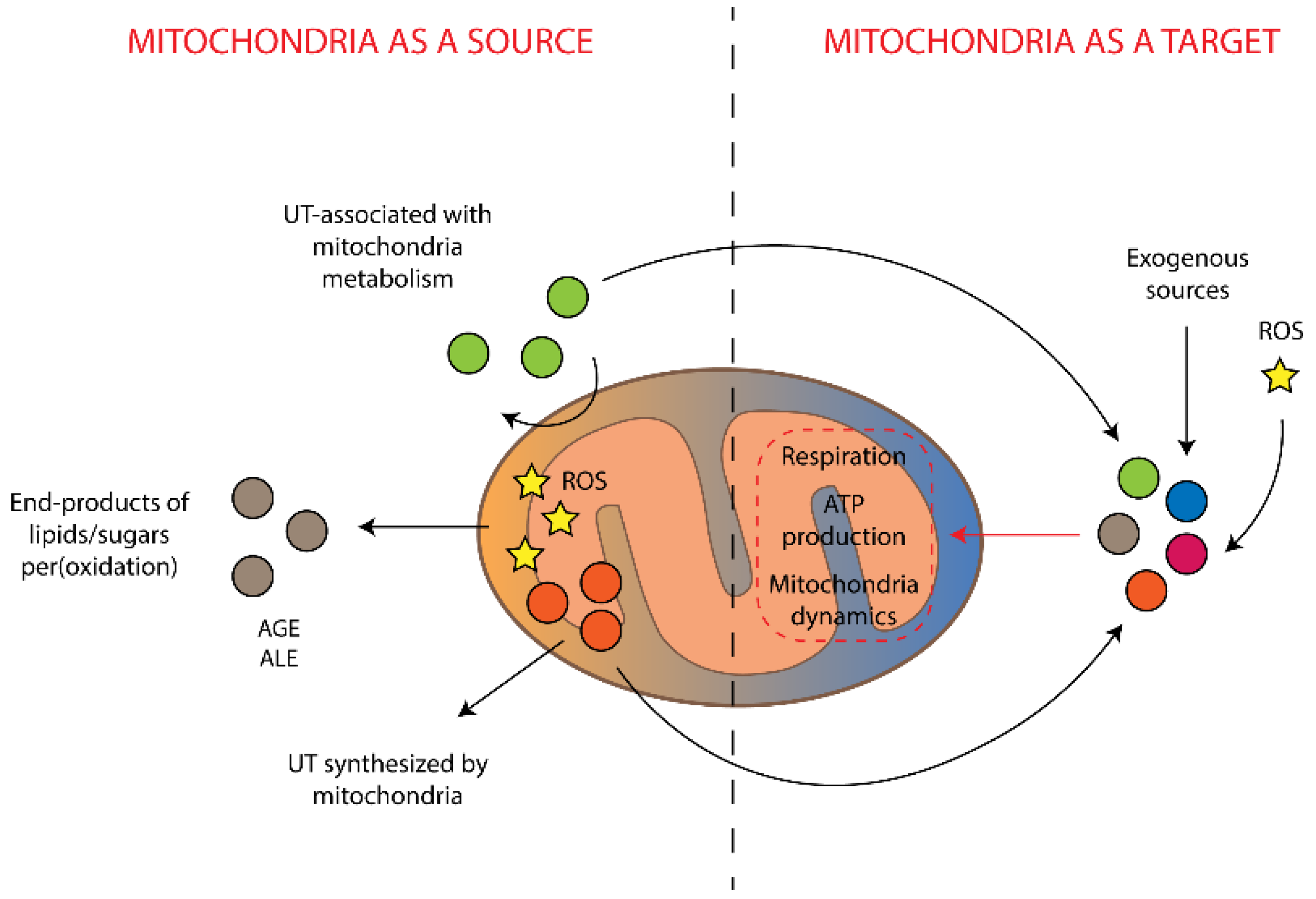
3. Mitochondria as a Source of Damaging and Toxic Molecules
3.1. Enzymatically Produced Compounds
3.2. Oxidation Products: Advanced Glycation and Lipoxidation End Products (AGE and ALE) and Aldehydes
4. Mitochondria as a Target for Uremic Toxins
5. Uremia, Oxidative Stress, and Antioxidant Treatment
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CKD | Chronic kidney disease |
| AKI | Acute kidney injury |
| ROS | Reactive oxygen species |
| EUTox | European Uremic Toxins workgroup |
References
- Meyer, T.W.; Hostetter, T.H. Uremia. N. Engl. J. Med. 2007, 357, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Almeras, C.; Argilés, À. The General Picture of Uremia. Semin. Dial. 2009, 22, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Foley, R.N.; Parfrey, P.S.; Sarnak, M.J. Epidemiology of cardiovascular disease in chronic renal disease. J. Am. Soc. Nephrol. 1998, 9 (Suppl. 12), S16–S23. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. Normal and Pathologic Concentrations of Uremic Toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himmelfarb, J.; Stenvinkel, P.; Ikizler, T.A.; Hakim, R.M. The elephant in uremia: Oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002, 62, 1524–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himmelfarb, J. Uremic toxicity, oxidative stress, and hemodialysis as renal replacement therapy. Semin. Dial. 2009, 22, 636–643. [Google Scholar] [CrossRef]
- Himmelfarb, J.; Hakim, R.M. Oxidative stress in uremia. Curr. Opin. Nephrol. Hypertens. 2003, 12, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Bannikova, S.Y.; Belousov, V.V.; Vyssokikh, M.Y.; Zorova, L.D.; Isaev, N.K.; Krasnikov, B.F.; Plotnikov, E.Y. Reactive oxygen and nitrogen species: Friends or foes? Biochem. (Mosc) 2005, 70, 215–221. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Krata, N.; Zagożdżon, R.; Foroncewicz, B.; Mucha, K. Oxidative Stress in Kidney Diseases: The Cause or the Consequence? Arch. Immunol. Et. Exp. 2018, 66, 211–220. [Google Scholar] [CrossRef]
- Modaresi, A.; Nafar, M.; Sahraei, Z. Oxidative stress in chronic kidney disease. Iran. J. Kidney Dis. 2015, 9, 165–179. [Google Scholar] [PubMed]
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; et al. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The Hallmarks of Mitochondrial Dysfunction in Chronic Kidney Disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Sureshbabu, A.; Ryter, S.W.; Choi, M.E. Oxidative stress and autophagy: Crucial modulators of kidney injury. Redox Biol. 2015, 4, 208–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlakou, P.; Liakopoulos, V.; Eleftheriadis, T.; Mitsis, M.; Dounousi, E. Oxidative Stress and Acute Kidney Injury in Critical Illness: Pathophysiologic Mechanisms—Biomarkers—Interventions, and Future Perspectives. Oxidative Med. Cell. Longev. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gerber, P.A.; Rutter, G.A. The Role of Oxidative Stress and Hypoxia in Pancreatic Beta-Cell Dysfunction in Diabetes Mellitus. Antioxid. Redox Signal. 2017, 26, 501–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, N.; Dabla, P.K. Oxidative stress and antioxidants in hypertension-a current review. Curr. Hypertens. Rev. 2015, 11, 132–142. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Morosanova, M.A.; Pevzner, I.B.; Zorova, L.D.; Manskikh, V.N.; Pulkova, N.V.; Galkina, S.I.; Skulachev, V.P.; Zorov, D.B. Protective effect of mitochondria-targeted antioxidants in an acute bacterial infection. Proc. Natl. Acad. Sci. USA 2013, 110, E3100–E3108. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.H.; Mitch, W.E. Mechanisms of muscle wasting in chronic kidney disease. Nat. Rev. Nephrol. 2014, 10, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Granata, S.; Zaza, G.; Simone, S.; Villani, G.; Latorre, D.; Pontrelli, P.; Carella, M.; Schena, F.; Grandaliano, G.; Pertosa, G. Mitochondrial dysregulation and oxidative stress in patients with chronic kidney disease. Bmc Genom. 2009, 10, 388. [Google Scholar] [CrossRef]
- Oberg, B.P.; McMenamin, E.; Lucas, F.L.E.E.; McMonagle, E.; Morrow, J.; Ikizler, T.A.L.P.; Himmelfarb, J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004, 65, 1009–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, L.F.; Shintani, A.; Ikizler, T.A.; Himmelfarb, J. Oxidative Stress and Inflammation Are Associated with Adiposity in Moderate to Severe CKD. J. Am. Soc. Nephrol. 2008, 19, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Duni, A.; Liakopoulos, V.; Rapsomanikis, K.-P.; Dounousi, E. Chronic Kidney Disease and Disproportionally Increased Cardiovascular Damage: Does Oxidative Stress Explain the Burden? Oxidative Med. Cell. Longev. 2017, 2017, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Bhandari, S.; Seymour, A.-M.L. Mitochondrial dysfunction in uremic cardiomyopathy. Am. J. Physiol. -Ren. Physiol. 2015, 308, F579–F587. [Google Scholar] [CrossRef] [PubMed]
- Hamed, S.A. Neurologic conditions and disorders of uremic syndrome of chronic kidney disease: Presentations, causes, and treatment strategies. Expert Rev. Clin. Pharm. 2019, 12, 61–90. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Vaziri, N.D.; Jabbari, B.; Ni, Z.; Yan, X.X. Increased tyrosine nitration of the brain in chronic renal insufficiency: Reversal by antioxidant therapy and angiotensin-converting enzyme inhibition. J. Am. Soc. Nephrol. 2001, 12, 1892–1899. [Google Scholar] [PubMed]
- Fujisaki, K.; Tsuruya, K.; Yamato, M.; Toyonaga, J.; Noguchi, H.; Nakano, T.; Taniguchi, M.; Tokumoto, M.; Hirakata, H.; Kitazono, T. Cerebral oxidative stress induces spatial working memory dysfunction in uremic mice: Neuroprotective effect of tempol. Nephrol. Dial. Transpl. 2014, 29, 529–538. [Google Scholar] [CrossRef]
- Mutsaers, H.A.M.; Wilmer, M.J.G.; Reijnders, D.; Jansen, J.; van den Broek, P.H.H.; Forkink, M.; Schepers, E.; Glorieux, G.; Vanholder, R.; van den Heuvel, L.P.; et al. Uremic toxins inhibit renal metabolic capacity through interference with glucuronidation and mitochondrial respiration. Biochim. Et Biophys. Acta (Bba) Mol. Basis Dis. 2013, 1832, 142–150. [Google Scholar] [CrossRef]
- Wyss, M.; Kaddurah-Daouk, R. Creatine and Creatinine Metabolism. Physiol. Rev. 2000, 80, 1107–1213. [Google Scholar] [CrossRef]
- Rawls, J.; Knecht, W.; Diekert, K.; Lill, R.; Löffler, M. Requirements for the mitochondrial import and localization of dihydroorotate dehydrogenase. Eur. J. Biochem. 2000, 267, 2079–2087. [Google Scholar] [CrossRef]
- Ellis, S.W.; Rose, M.E.; Grindle, M. Identification of a sterol mutant of Neurospora crassa deficient in delta 14,15-reductase activity. J. Gen. Microbiol. 1991, 137, 2627–2630. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, T.; Tatibana, M. Purification of N-acetyl-L-glutamate synthetase from rat liver mitochondria and substrate and activator specificity of the enzyme. J. Biol. Chem. 1983, 258, 9839–9844. [Google Scholar] [PubMed]
- Humm, A.; Fritsche, E.; Steinbacher, S.; Huber, R. Crystal structure and mechanism of human L-arginine: Glycine amidinotransferase: A mitochondrial enzyme involved in creatine biosynthesis. Embo J. 1997, 16, 3373–3385. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M.; Terai, K.; Kameda, N.; Matsumoto, A.; Kurokawa, Y.; Funase, Y.; Nishikawa, K.; Sugaya, N.; Hiruta, N.; Kishimoto, T. Designation of enzyme activity of glycine-N-acyltransferase family genes and depression of glycine-N-acyltransferase in human hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2012, 420, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Sun, L.; Mochly-Rosen, D. Mitochondrial aldehyde dehydrogenase and cardiac diseases. Cardiovasc. Res. 2010, 88, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sas, K.; Szabó, E.; Vécsei, L. Mitochondria, Oxidative Stress and the Kynurenine System, with a Focus on Ageing and Neuroprotection. Molecules 2018, 23, 191. [Google Scholar] [CrossRef] [PubMed]
- Rus, D.A.; Sastre, J.; Viña, J.; Pallardó, F.V. Induction of mitochondrial xanthine oxidase activity during apoptosis in the rat mammary gland. Front. Biosci. A J. Virtual Libr. 2007, 12, 1184–1189. [Google Scholar] [CrossRef]
- Gladden, J.D.; Zelickson, B.R.; Wei, C.C.; Ulasova, E.; Zheng, J.; Ahmed, M.I.; Chen, Y.; Bamman, M.; Ballinger, S.; Darley-Usmar, V.; et al. Novel insights into interactions between mitochondria and xanthine oxidase in acute cardiac volume overload. Free Radic. Biol. Med. 2011, 51, 1975–1984. [Google Scholar] [CrossRef] [Green Version]
- Levillain, O.; Balvay, S.; Peyrol, S. Mitochondrial expression of arginase II in male and female rat inner medullary collecting ducts. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2005, 53, 533–541. [Google Scholar] [CrossRef]
- Rose, A.M.; Belford, H.G.; Shen, W.C.; Greer, C.L.; Hopper, A.K.; Martin, N.C. Location of N2, N2-dimethylguanosine-specific tRNA methyltransferase. Biochimie 1995, 77, 45–53. [Google Scholar] [CrossRef]
- Zaganelli, S.; Rebelo-Guiomar, P.; Maundrell, K.; Rozanska, A.; Pierredon, S.; Powell, C.A.; Jourdain, A.A.; Hulo, N.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.; et al. The pseudouridine synthase RPUSD4 is an essential component of mitochondrial RNA granules. J. Biol. Chem. 2017, 292, 4519–4532. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.A.; Kopajtich, R.; D’Souza, A.R.; Rorbach, J.; Kremer, L.S.; Husain, R.A.; Dallabona, C.; Donnini, C.; Alston, C.L.; Griffin, H.; et al. TRMT5 Mutations Cause a Defect in Post-transcriptional Modification of Mitochondrial tRNA Associated with Multiple Respiratory-Chain Deficiencies. Am. J. Hum. Genet. 2015, 97, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broch, O.J.; Ueland, P.M. Regional and subcellular distribution of S-adenosylhomocysteine hydrolase in the adult rat brain. J. Neurochem. 1980, 35, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N. Toxic dimethylarginines: Asymmetric dimethylarginine (ADMA) and symmetric dimethylarginine (SDMA). Toxins 2017, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47, 3–27. [Google Scholar] [CrossRef]
- Miyata, T.; van Ypersele de Strihou, C.; Kurokawa, K.; Baynes, J.W. Alterations in nonenzymatic biochemistry in uremia: Origin and significance of “carbonyl stress” in long-term uremic complications. Kidney Int. 1999, 55, 389–399. [Google Scholar] [CrossRef]
- Miyata, T.; Fu, M.X.; Kurokawa, K.; Van Ypersele De Strihou, C.; Thorpe, S.R.; Baynes, J.W. Autoxidation products of both carbohydrates and lipids are increased in uremic plasma: Is there oxidative stress in uremia? Kidney Int. 1998, 54, 1290–1295. [Google Scholar] [CrossRef] [Green Version]
- Niwa, T.; Takeda, N.; Yoshizumi, H.; Tatematsu, A.; Ohara, M.; Tomiyama, S.; Niimura, K. Presence of 3-Deoxyglucosone, a Potent Protein Crosslinking Intermediate of Maillard Reaction, in Diabetic Serum. Biochem. Biophys. Res. Commun. 1993, 196, 837–843. [Google Scholar] [CrossRef]
- Ueda, Y.; Miyata, T.; Hashimoto, T.; Yamada, H.; Izuhara, Y.; Sakai, H.; Kurokawa, K. Implication of altered redox regulation by antioxidant enzymes in the increased plasma pentosidine, an advanced glycation end product, in uremia. Biochem. Biophys. Res. Commun. 1998, 245, 785–790. [Google Scholar] [CrossRef]
- Odani, H.; Shinzato, T.; Matsumoto, Y.; Usami, J.; Maeda, K. Increase in three α, β-dicarbonyl compound levels in human uremic plasma: Specific in vivo determination of intermediates in advanced Maillard reaction. Biochem. Biophys. Res. Commun. 1999, 256, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Wada, Y.; Cai, Z.; Iida, Y.; Horie, K.; Yasuda, Y.; Maeda, K.; Kurokawa, K.; Van Ypersele De Strihou, C. Implication of an increased oxidative stress in the formation of advanced glycation end products in patients with end-stage renal failure. Kidney Int. 1997, 51, 1170–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnaitman, C.; Erwin, V.G.; Greenawalt, J.W. The submitochondrial localization of monoamine oxidase. An enzymatic marker for the outer membrane of rat liver mitochondria. J. Cell Biol. 1967, 32, 719–735. [Google Scholar] [CrossRef] [PubMed]
- Pun, P.B.L.; Logan, A.; Darley-Usmar, V.; Chacko, B.; Johnson, M.S.; Huang, G.W.; Rogatti, S.; Prime, T.A.; Methner, C.; Krieg, T.; et al. A mitochondria-targeted mass spectrometry probe to detect glyoxals: Implications for diabetes. Free Radic. Biol. Med. 2014, 67, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Awe, S.O.; Vladykovskaya, E.; Ahmed, Y.; Liu, S.-Q.; Bhatnagar, A.; Srivastava, S. Myocardial ischaemia inhibits mitochondrial metabolism of 4-hydroxy-trans-2-nonenal. Biochem. J. 2009, 417, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Huszti, Z. Comparative studies on mitochondrial and soluble diamine oxidases. Enzymologia 1967, 33, 179–191. [Google Scholar] [PubMed]
- Hirai, K.; Kuroyanagi, H.; Tatebayashi, Y.; Hayashi, Y.; Hirabayashi-Takahashi, K.; Saito, K.; Haga, S.; Uemura, T.; Izumi, S. Dual role of the carboxyl-terminal region of pig liver l-kynurenine 3-monooxygenase: Mitochondrial-targeting signal and enzymatic activity. J. Biochem. 2010, 148, 639–650. [Google Scholar] [CrossRef] [PubMed]
- MACKENZIE, C.G.; FRISELL, W.R. The metabolism of dimethylglycine by liver mitochondria. J. Biol. Chem. 1958, 232, 417–427. [Google Scholar]
- Luka, Z.; Pakhomova, S.; Loukachevitch, L.V.; Newcomer, M.E.; Wagner, C. Folate in demethylation: The crystal structure of the rat dimethylglycine dehydrogenase complexed with tetrahydrofolate. Biochem. Biophys. Res. Commun. 2014, 449, 392–398. [Google Scholar] [CrossRef] [Green Version]
- Breitzig, M.; Bhimineni, C.; Lockey, R.; Kolliputi, N. 4-Hydroxy-2-nonenal: A critical target in oxidative stress? Am. J. Physiol. Cell Physiol. 2016, 311, C537–C543. [Google Scholar] [CrossRef]
- Dalleau, S.; Baradat, M.; Guéraud, F.; Huc, L. Cell death and diseases related to oxidative stress:4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013, 20, 1615–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzu, V.; Parker, N.; Brand, M.D. High membrane potential promotes alkenal-induced mitochondrial uncoupling and influences adenine nucleotide translocase conformation. Biochem. J. 2008, 413, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, H.J.; Fukuto, J.M.; Miller, T.; Zhang, H.; Rinna, A.; Levy, S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch. Biochem. Biophys. 2008, 15, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Shoeb, M.; Ansari, N.; Srivastava, S.; Ramana, K. 4-Hydroxynonenal in the Pathogenesis and Progression of Human Diseases. Curr. Med. Chem. 2013, 21, 230–237. [Google Scholar] [CrossRef]
- Jinsmaa, Y.; Florang, V.R.; Rees, J.N.; Anderson, D.G.; Strack, S.; Doorn, J.A. Products of oxidative stress inhibit aldehyde oxidation and reduction pathways in dopamine catabolism yielding elevated levels of a reactive intermediate. Chem. Res. Toxicol. 2009, 22, 835–841. [Google Scholar] [CrossRef]
- Neuzil, J.; Wang, X.-F.; Dong, L.-F.; Low, P.; Ralph, S.J. Molecular mechanism of ’mitocan’-induced apoptosis in cancer cells epitomizes the multiple roles of reactive oxygen species and Bcl-2 family proteins. Febs Lett. 2006, 580, 5125–5229. [Google Scholar] [CrossRef]
- Forman, H.J. Reactive oxygen species and α, β-unsaturated aldehydes as second messengers in signal transduction. Ann. N. Y. Acad. Sci. 2010, 1203, 35–44. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell. Longev. 2014. [Google Scholar] [CrossRef]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199. [Google Scholar] [CrossRef]
- Bradley, M.A.; Markesbery, W.R.; Lovell, M.A. Increased levels of 4-hydroxynonenal and acrolein in the brain in preclinical Alzheimer disease. Free Radic. Biol. Med. 2010, 48, 1570–1576. [Google Scholar] [CrossRef] [Green Version]
- Selley, M.L. (E)-4-Hydroxy-2-nonenal may be involved in the pathogenesis of Parkinson’s disease. Free Radic. Biol. Med. 1998, 25, 169–174. [Google Scholar] [CrossRef]
- Pashkow, F.J. Oxidative Stress and Inflammation in Heart Disease: Do Antioxidants Have a Role in Treatment and/or Prevention? Int. J. Inflamm. 2011, 2011, 514623. [Google Scholar] [CrossRef] [PubMed]
- Shinmura, K.; Bolli, R.; Liu, S.Q.; Tang, X.L.; Kodani, E.; Xuan, Y.T.; Srivastava, S.; Bhatnagar, A. Aldose reductase is an obligatory mediator of the late phase of ischemic preconditioning. Circ. Res. 2002, 91, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.; Riahi, Y.; Sunda, V.; Deplano, S.; Chatgilialoglu, C.; Ferreri, C.; Kaiser, N.; Sasson, S. Signaling properties of 4-hydroxyalkenals formed by lipid peroxidation in diabetes. Free Radic. Biol. Med. 2013, 65, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Galam, L.; Failla, A.; Soundararajan, R.; Lockey, R.F.; Kolliputi, N. 4-Hydroxynonenal regulates mitochondrial function in human small airway epithelial cells. Oncotarget 2015, 6, 41508–41521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkowski, P.; Malgorzewicz, S.; Slominska, E.; Renke, M.; Lysiak-Szydlowska, W.; Swierczynski, J.; Rutkowski, B. Interrelationship Between Uremic Toxicity and Oxidative Stress. J. Ren. Nutr. 2006, 16, 190–193. [Google Scholar] [CrossRef]
- Leong, S.C.; Sirich, T.L. Indoxyl Sulfate-Review of Toxicity and Therapeutic Strategies. Toxins 2016, 8, 358. [Google Scholar] [CrossRef]
- Wu, I.-W.; Hsu, K.-H.; Lee, C.-C.; Sun, C.-Y.; Hsu, H.-J.; Tsai, C.-J.; Tzen, C.-Y.; Wang, Y.-C.; Lin, C.-Y.; Wu, M.-S. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol. Dial. Transpl. 2011, 26, 938–947. [Google Scholar] [CrossRef]
- Ellis, R.J.; Small, D.M.; Vesey, D.A.; Johnson, D.W.; Francis, R.; Vitetta, L.; Gobe, G.C.; Morais, C. Indoxyl sulphate and kidney disease: Causes, consequences and interventions. Nephrol. (Carlton) 2016, 21, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Deguchi, T.; Ohtsuki, S.; Otagiri, M.; Takanaga, H.; Asaba, H.; Mori, S.; Terasaki, T. Major role of organic anion transporter 3 in the transport of indoxyl sulfate in the kidney. Kidney Int. 2002, 61, 1760–1768. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.-J.; Hsu, S.-C.; Chang, Y.-L.; Huang, S.-M.; Shih, C.-C.; Tsai, C.-S.; Lin, C.-Y. Indoxyl sulfate upregulates the cannabinoid type 1 receptor gene via an ATF3/c-Jun complex-mediated signaling pathway in the model of uremic cardiomyopathy. Int. J. Cardiol. 2018, 252, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Watanabe, T.; Nakayama, M. Cerebro-renal interactions: Impact of uremic toxins on cognitive function. NeuroToxicology 2014, 44, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, N.; Sagata, M.; Saigo, C.; Yoneda, G.; Yamamoto, Y.; Nomura, Y.; Nishi, K.; Fujino, R.; Jono, H.; Saito, H. Indoxyl Sulfate as a Mediator Involved in Dysregulation of Pulmonary Aquaporin-5 in Acute Lung Injury Caused by Acute Kidney Injury. Int. J. Mol. Sci. 2016, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-F.; Liang, S.-S.; Thanasekaran, P.; Chang, H.-W.; Wen, L.-L.; Chen, C.-H.; Liou, J.-C.; Yeh, J.-C.; Liu, S.-H.; Dai, H.-M.; et al. Translational Medicine in Pulmonary-Renal Crosstalk: Therapeutic Targeting of p-Cresyl Sulfate Triggered Nonspecific ROS and Chemoattractants in Dyspneic Patients with Uremic Lung Injury. J. Clin. Med. 2018, 7, 266. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Mao, X.; Guo, H.; Wang, L.; Li, Z.; Zhang, Y.; Wang, Y.; Wang, H.; Zhang, X.; Peng, W. Indoxyl sulfate potentiates endothelial dysfunction via reciprocal role for reactive oxygen species and RhoA/ROCK signaling in 5/6 nephrectomized rats. Free Radic. Res. 2017, 51, 237–252. [Google Scholar] [CrossRef]
- Lin, C.-J.; Wu, C.-J.; Wu, P.-C.; Pan, C.-F.; Wang, T.-J.; Sun, F.-J.; Liu, H.-L.; Chen, H.-H.; Yeh, H.-I. Indoxyl Sulfate Impairs Endothelial Progenitor Cells and Might Contribute to Vascular Dysfunction in Patients with Chronic Kidney Disease. Kidney Blood Press. Res. 2016, 41, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Enoki, Y.; Watanabe, H.; Arake, R.; Fujimura, R.; Ishiodori, K.; Imafuku, T.; Nishida, K.; Sugimoto, R.; Nagao, S.; Miyamura, S.; et al. Potential therapeutic interventions for chronic kidney disease-associated sarcopenia via indoxyl sulfate-induced mitochondrial dysfunction. J. Cachexiasarcopenia Muscle 2017, 8, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Ishimori, N.; Takada, S.; Saito, A.; Kadoguchi, T.; Furihata, T.; Fukushima, A.; Matsushima, S.; Yokota, T.; Kinugawa, S.; et al. AST-120 ameliorates lowered exercise capacity and mitochondrial biogenesis in the skeletal muscle from mice with chronic kidney disease via reducing oxidative stress. Nephrol. Dial. Transpl. 2015, 30, 934–942. [Google Scholar] [CrossRef] [Green Version]
- Fujii, H.; Nakai, K.; Fukagawa, M. Role of Oxidative Stress and Indoxyl Sulfate in Progression of Cardiovascular Disease in Chronic Kidney Disease. Apher. Dial. 2011, 15, 125–128. [Google Scholar] [CrossRef]
- Gelasco, A.K.; Raymond, J.R. Indoxyl sulfate induces complex redox alterations in mesangial cells. Am. J. Physiol. Ren. Physiol. 2006, 290, F1551–F1558. [Google Scholar] [CrossRef] [Green Version]
- Owada, S.; Goto, S.; Bannai, K.; Hayashi, H.; Nishijima, F.; Niwa, T. Indoxyl Sulfate Reduces Superoxide Scavenging Activity in the Kidneys of Normal and Uremic Rats. Am. J. Nephrol. 2008, 28, 446–454. [Google Scholar] [CrossRef]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Li, Y.; Trush, M.A. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem. Biophys. Res. Commun. 1998, 253, 295–299. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Vasileva, A.K.; Arkhangelskaya, A.A.; Pevzner, I.B.; Skulachev, V.P.; Zorov, D.B. Interrelations of mitochondrial fragmentation and cell death under ischemia/reoxygenation and UV-irradiation: Protective effects of SkQ1, lithium ions and insulin. Febs Lett. 2008, 582, 3117–3124. [Google Scholar] [CrossRef] [Green Version]
- Ježek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Cheng, M.-L.; Pan, H.-C.; Lee, J.-H.; Lee, C.-C. Protein-bound uremic toxins impaired mitochondrial dynamics and functions. Oncotarget 2017, 8, 77722–77733. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, A. EFFECTS OF CRESOLS (O-, M-, AND P-ISOMERS) ON THE BIOENERGETIC SYSTEM IN ISOLATED RAT LIVER MITOCHONDRIA. Drug Chem. Toxicol. 2001, 24, 39–47. [Google Scholar] [CrossRef]
- Adam-Vizi, V. Production of Reactive Oxygen Species in Brain Mitochondria: Contribution by Electron Transport Chain and Non–Electron Transport Chain Sources. Antioxid. Redox Signal. 2005, 7, 1140–1149. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef]
- Meyer, J.N.; Leung, M.C.K.; Rooney, J.P.; Sendoel, A.; Hengartner, M.O.; Kisby, G.E.; Bess, A.S. Mitochondria as a Target of Environmental Toxicants. Toxicol. Sci. 2013, 134, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ellis, R.J.; Small, D.M.; Ng, K.L.; Vesey, D.A.; Vitetta, L.; Francis, R.S.; Gobe, G.C.; Morais, C. Indoxyl Sulfate Induces Apoptosis and Hypertrophy in Human Kidney Proximal Tubular Cells. Toxicol. Pathol. 2018, 46, 449–459. [Google Scholar] [CrossRef] [Green Version]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [Green Version]
- Shang, F.; Wang, S.-C.; Hsu, C.-Y.; Miao, Y.; Martin, M.; Yin, Y.; Wu, C.-C.; Wang, Y.-T.; Wu, G.; Chien, S.; et al. MicroRNA-92a Mediates Endothelial Dysfunction in CKD. J. Am. Soc. Nephrol. 2017, 28, 3251–3261. [Google Scholar] [CrossRef] [Green Version]
- Dzúrik, R.; Spustová, V.; Krivošíková, Z.; Gazdíková, K. Hippurate participates in the correction of metabolic acidosis. Kidney Int. 2001, 59, S278–S281. [Google Scholar] [CrossRef] [Green Version]
- Baran, H.; Staniek, K.; Kepplinger, B.; Gille, L.; Stolze, K.; Nohl, H. Kynurenic Acid Influences the Respiratory Parameters of Rat Heart Mitochondria. Pharmacology 2001, 62, 119–123. [Google Scholar] [CrossRef]
- Baran, H.; Staniek, K.; Kepplinger, B.; Stur, J.; Draxler, M.; Nohl, H. Kynurenines and the respiratory parameters on rat heart mitochondria. Life Sci. 2003, 72, 1103–1115. [Google Scholar] [CrossRef]
- Bosch-Panadero, E.; Mas, S.; Civantos, E.; Abaigar, P.; Camarero, V.; Ruiz-Priego, A.; Ortiz, A.; Egido, J.; González-Parra, E. Bisphenol A is an exogenous toxin that promotes mitochondrial injury and death in tubular cells. Environ. Toxicol. 2018, 33, 325–332. [Google Scholar] [CrossRef]
- Khan, S.; Beigh, S.; Chaudhari, B.P.; Sharma, S.; Aliul Hasan Abdi, S.; Ahmad, S.; Ahmad, F.; Parvez, S.; Raisuddin, S. Mitochondrial dysfunction induced by Bisphenol A is a factor of its hepatotoxicity in rats. Environ. Toxicol. 2016, 31, 1922–1934. [Google Scholar] [CrossRef]
- Ceballos-Picot, I.; Witko-Sarsat, V.; Merad-Boudia, M.; Nguyen, A.T.; Thévenin, M.; Jaudon, M.C.; Zingraff, J.; Verger, C.; Jungers, P.; Descamps-Latscha, B. Glutathione antioxidant system as a marker of oxidative stress in chronic renal failure. Free Radic. Biol. Med. 1996, 21, 845–853. [Google Scholar] [CrossRef]
- Kaya, Y.; Ari, E.; Demir, H.; Soylemez, N.; Cebi, A.; Alp, H.; Bakan, E.; Gecit, I.; Asicioglu, E.; Beytur, A. Accelerated atherosclerosis in haemodialysis patients; correlation of endothelial function with oxidative DNA damage. Nephrol. Dial. Transpl. 2012, 27, 1164–1169. [Google Scholar] [CrossRef]
- Lim, P.S.; Chan, E.C.; Lu, T.C.; Yu, Y.L.; Kuo, S.Y.; Wang, T.H.; Wei, Y.H. Lipophilic antioxidants and iron status in ESRD patients on hemodialysis. Nephron 2000, 86, 428–435. [Google Scholar] [CrossRef]
- Wang, S.; Eide, T.C.; Sogn, E.M.; Berg, K.J.; Sund, R.B. Plasma ascorbic acid in patients undergoing chronic haemodialysis. Eur. J. Clin. Pharm. 1999, 55, 527–532. [Google Scholar] [CrossRef]
- Roehrs, M.; Valentini, J.; Paniz, C.; Moro, A.; Charão, M.; Bulcão, R.; Freitas, F.; Brucker, N.; Duarte, M.; Leal, M.; et al. The relationships between exogenous and endogenous antioxidants with the lipid profile and oxidative damage in hemodialysis patients. Bmc Nephrol. 2011, 12, 59. [Google Scholar] [CrossRef]
- Aminzadeh, M.A.; Vaziri, N.D. Downregulation of the renal and hepatic hydrogen sulfide (H2S)-producing enzymes and capacity in chronic kidney disease. Nephrol. Dial. Transpl. 2012, 27, 498–504. [Google Scholar] [CrossRef]
- Drüeke, T.; Witko-Sarsat, V.; Massy, Z.; Descamps-Latscha, B.; Guerin, A.P.; Marchais, S.J.; Gausson, V.; London, G.M. Iron therapy, advanced oxidation protein products, and carotid artery intima-media thickness in end-stage renal disease. Circulation 2002, 106, 2212–2217. [Google Scholar] [CrossRef]
- Fumeron, C.; Nguyen-Khoa, T.; Saltiel, C.; Kebede, M.; Buisson, C.; Drüeke, T.B.; Lacour, B.; Massy, Z.A. Effects of oral vitamin C supplementation on oxidative stress and inflammation status in haemodialysis patients. Nephrol. Dial. Transpl. 2005, 20, 1874–1879. [Google Scholar] [CrossRef] [Green Version]
- Boudouris, G.; Verginadis, I.I.; Simos, Y.V.; Zouridakis, A.; Ragos, V.; Karkabounas, S.C.; Evangelou, A.M. Oxidative stress in patients treated with continuous ambulatory peritoneal dialysis (CAPD) and the significant role of vitamin C and E supplementation. Int Urol Nephrol 2013, 45, 1137–1144. [Google Scholar] [CrossRef]
- Fatouros, I.G.; Douroudos, I.; Panagoutsos, S.; Pasadakis, P.; Nikolaidis, M.G.; Chatzinikolaou, A.; Sovatzidis, A.; Michailidis, Y.; Jamurtas, A.Z.; Mandalidis, D.; et al. Effects of L-carnitine on oxidative stress responses in patients with renal disease. Med. Sci. Sports Exerc. 2010, 42, 1809–1818. [Google Scholar] [CrossRef]
- Sener, G.; Paskaloğlu, K.; Satiroglu, H.; Alican, I.; Kaçmaz, A.; Sakarcan, A. L-carnitine ameliorates oxidative damage due to chronic renal failure in rats. J. Cardiovasc. Pharm. 2004, 43, 698–705. [Google Scholar] [CrossRef]
- Bouzidi, N.; Mekki, K.; Boukaddoum, A.; Dida, N.; Kaddous, A.; Bouchenak, M. Effects of omega-3 polyunsaturated fatty-acid supplementation on redox status in chronic renal failure patients with dyslipidemia. J. Ren. Nutr. 2010, 20, 321–328. [Google Scholar] [CrossRef]
- Sakata, T.; Furuya, R.; Shimazu, T.; Odamaki, M.; Ohkawa, S.; Kumagai, H. Coenzyme Q10 administration suppresses both oxidative and antioxidative markers in hemodialysis patients. Blood Purif. 2008, 26, 371–378. [Google Scholar] [CrossRef]
- Ivanovski, O.; Szumilak, D.; Nguyen-Khoa, T.; Ruellan, N.; Phan, O.; Lacour, B.; Descamps-Latscha, B.; Drüeke, T.B.; Massy, Z.A. The antioxidant N-acetylcysteine prevents accelerated atherosclerosis in uremic apolipoprotein E knockout mice. Kidney Int. 2005, 67, 2288–2294. [Google Scholar] [CrossRef] [Green Version]
- Zachwieja, J.; Zaniew, M.; Bobkowski, W.; Stefaniak, E.; Warzywoda, A.; Ostalska-Nowicka, D.; Dobrowolska-Zachwieja, A.; Lewandowska-Stachowiak, M.; Siwińska, A. Beneficial in vitro effect of N-acetyl-cysteine on oxidative stress and apoptosis. Pediatr. Nephrol. 2005, 20, 725–731. [Google Scholar] [CrossRef]
- Hsu, S.-P.; Chiang, C.-K.; Yang, S.-Y.; Chien, C.-T. N-acetylcysteine for the management of anemia and oxidative stress in hemodialysis patients. Nephron Clin. Pract. 2010, 116, c207–c216. [Google Scholar] [CrossRef]
- Tepel, M.; van der Giet, M.; Statz, M.; Jankowski, J.; Zidek, W. The antioxidant acetylcysteine reduces cardiovascular events in patients with end-stage renal failure: A randomized, controlled trial. Circulation 2003, 107, 992–995. [Google Scholar] [CrossRef]
- Jun, M.; Venkataraman, V.; Razavian, M.; Cooper, B.; Zoungas, S.; Ninomiya, T.; Webster, A.C.; Perkovic, V. Antioxidants for chronic kidney disease. Cochrane Database Syst. Rev. 2012, 10, CD008176. [Google Scholar] [CrossRef]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. Available online: https://www.ncbi.nlm.nih.gov/pubmed/28033563 (accessed on 29 April 2019). [CrossRef]
- Plotnikov, E.Y.; Chupyrkina, A.A.; Jankauskas, S.S.; Pevzner, I.B.; Silachev, D.N.; Skulachev, V.P.; Zorov, D.B. Mechanisms of nephroprotective effect of mitochondria-targeted antioxidants under rhabdomyolysis and ischemia/reperfusion. Biochim. Biophys. Acta 2011, 1812, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, E.Y.; Silachev, D.N.; Zorova, L.D.; Pevzner, I.B.; Jankauskas, S.S.; Zorov, S.D.; Babenko, V.A.; Skulachev, M.V.; Zorov, D.B. Lithium salts—Simple but magic. Biochem. Mosc. 2014, 79, 740–749. [Google Scholar] [CrossRef]
- Lemoine, S.; Pillot, B.; Rognant, N.; Augeul, L.; Rayberin, M.; Varennes, A.; Laville, M.; Ovize, M.; Juillard, L. Postconditioning with cyclosporine a reduces early renal dysfunction by inhibiting mitochondrial permeability transition. Transplantation 2015, 99, 717–723. [Google Scholar] [CrossRef]
- Reddy, P.H. Inhibitors of mitochondrial fission as a therapeutic strategy for diseases with oxidative stress and mitochondrial dysfunction. J. Alzheimers Dis. 2014, 40, 245–256. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Toxins | Metabolic Pathway |
|---|---|
| Creatine | Phosphocreatine ← Creatine kinase, mitochondrial (CKMT1B/CKMT1A/CKMT2) → Creatine |
| Orotic acid | Dihydroorotate ← Dihydroorotate dehydrogenase (DHODH) → Orotic acid |
| Hippuric acid | Benzoyl-CoA← Glycine-N-acyltransferase → Hippuric acid |
| Methylglyoxal | Aminoacetone ← Monoamine oxidase A/B (MAOA/MAOB) → Methylglyoxal |
| α-Keto-δ-guanidinovaleric acid | Arginine ← D-amino acid oxidase (DAO) → α-keto-δ-Guanidinovaleric acid |
| Urea | Arginine ← Arginase 1 (ARG1) → Urea |
| γ-Guanidinobutyric acid | 4-Aminobutanoate ← Glycine amidinotransferase, mitochondrial (GATM) → γ-Guanidinobutyric acid |
| Indole-3-acetic acid | Indole-3-acetaldehyde ← Aldehyde dehydrogenase (ALDH3A2) → Indole-3-acetic acid |
| Inosine | Adenosine ← Adenosine deaminase (ADA) → Inosine |
| Hypoxanthine | Inosine ← Purine-nucleoside phosphorylase (PNP) → Hypoxanthine |
| Xanthine | Hypoxanthine ← Xanthine oxidoreductase (XOR) → Xanthine |
| Urea | Xanthine ← Xanthine oxidoreductase (XOR) → Urea |
| Xanthosine | Xanthine ← Purine-nucleoside phosphorylase (PNP) → Xanthosine |
| 1-Methylinosine | 1-Methyladenosine ← Adenosine deaminase (ADA) → 1-Methylinosine |
| Cytidine | Cytidine-5′-monophosphate ← 5′,3′-Nucleotidase (NT5M) → Cytidine |
| Uridine | Uridine-5′-monophosphate ← 5′,3′-Nucleotidase (NT5M) → Uridine |
| Putrescine | Arginine ← Arginase 1 (ARG1) → Ornithine← Ornithine decarboxylase (ODC1)→ Putrescine |
| Phenylacetic acid | Phenylacetaldehyde ← Aldehyde Dehydrogenase (ALDH3A2) → Phenylacetic acid |
| Melatonin | Serotonin ← Arylalkylamine N-acetyltransferase (AANAT)→ N-Acetylserotonin ← Acetylserotonin O-methyltransferase(ASMT) → Melatonin |
| Phenylacetylglutamine | L-glutamine ← Glutamine N-phenylacetyltransferase → Phenylacetylglutamine |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popkov, V.A.; Silachev, D.N.; Zalevsky, A.O.; Zorov, D.B.; Plotnikov, E.Y. Mitochondria as a Source and a Target for Uremic Toxins. Int. J. Mol. Sci. 2019, 20, 3094. https://doi.org/10.3390/ijms20123094
Popkov VA, Silachev DN, Zalevsky AO, Zorov DB, Plotnikov EY. Mitochondria as a Source and a Target for Uremic Toxins. International Journal of Molecular Sciences. 2019; 20(12):3094. https://doi.org/10.3390/ijms20123094
Chicago/Turabian StylePopkov, Vasily A., Denis N. Silachev, Arthur O. Zalevsky, Dmitry B. Zorov, and Egor Y. Plotnikov. 2019. "Mitochondria as a Source and a Target for Uremic Toxins" International Journal of Molecular Sciences 20, no. 12: 3094. https://doi.org/10.3390/ijms20123094
APA StylePopkov, V. A., Silachev, D. N., Zalevsky, A. O., Zorov, D. B., & Plotnikov, E. Y. (2019). Mitochondria as a Source and a Target for Uremic Toxins. International Journal of Molecular Sciences, 20(12), 3094. https://doi.org/10.3390/ijms20123094