Maternal and Post-Weaning High-Fat Diets Produce Distinct DNA Methylation Patterns in Hepatic Metabolic Pathways within Specific Genomic Contexts



Abstract
:1. Introduction
2. Results
2.1. Compared to Maternal HF Diet Exposure, Post-Weaning HF Does Not Alter Body Weight but Induces Greater Hepatic Lipid Accumulation
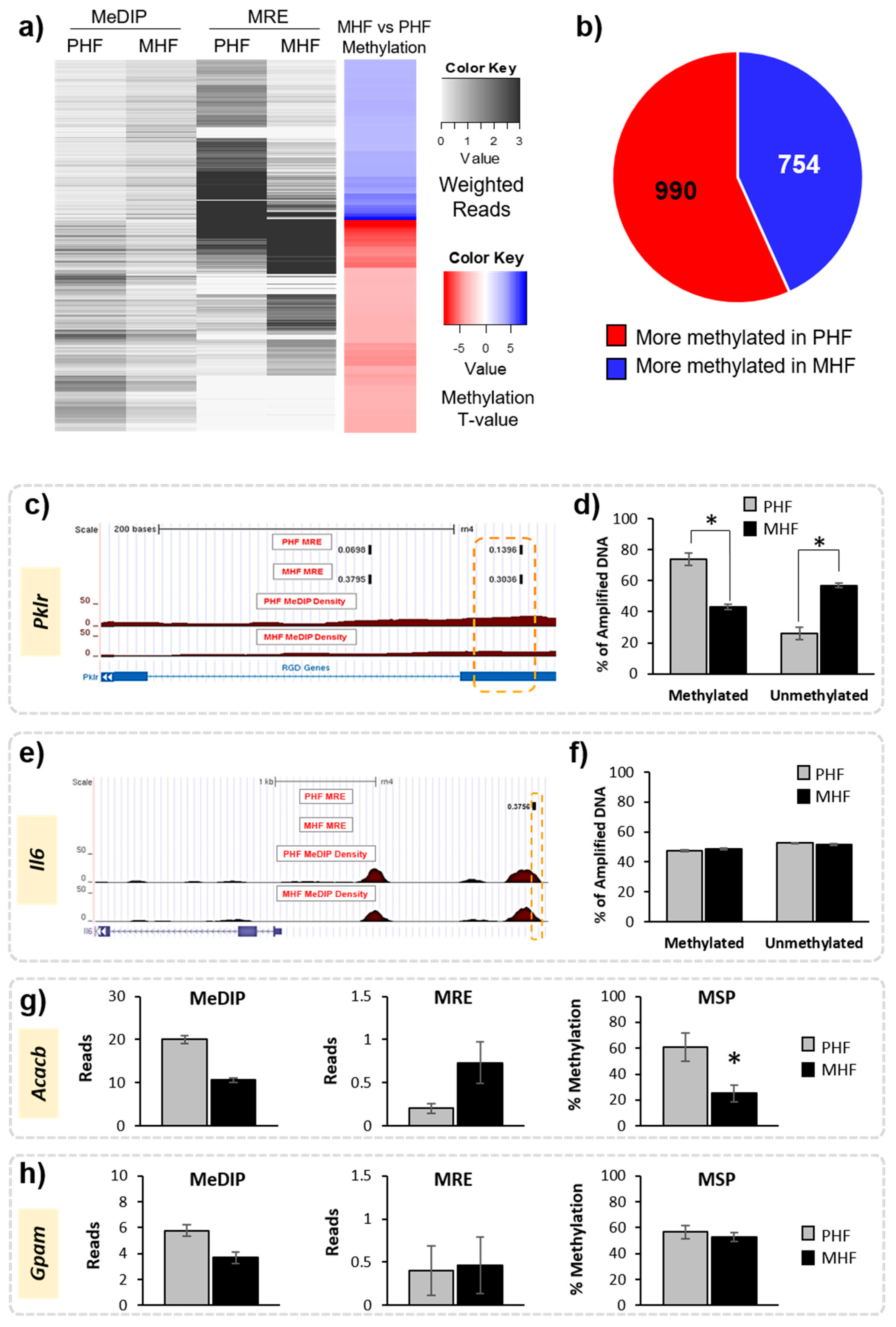
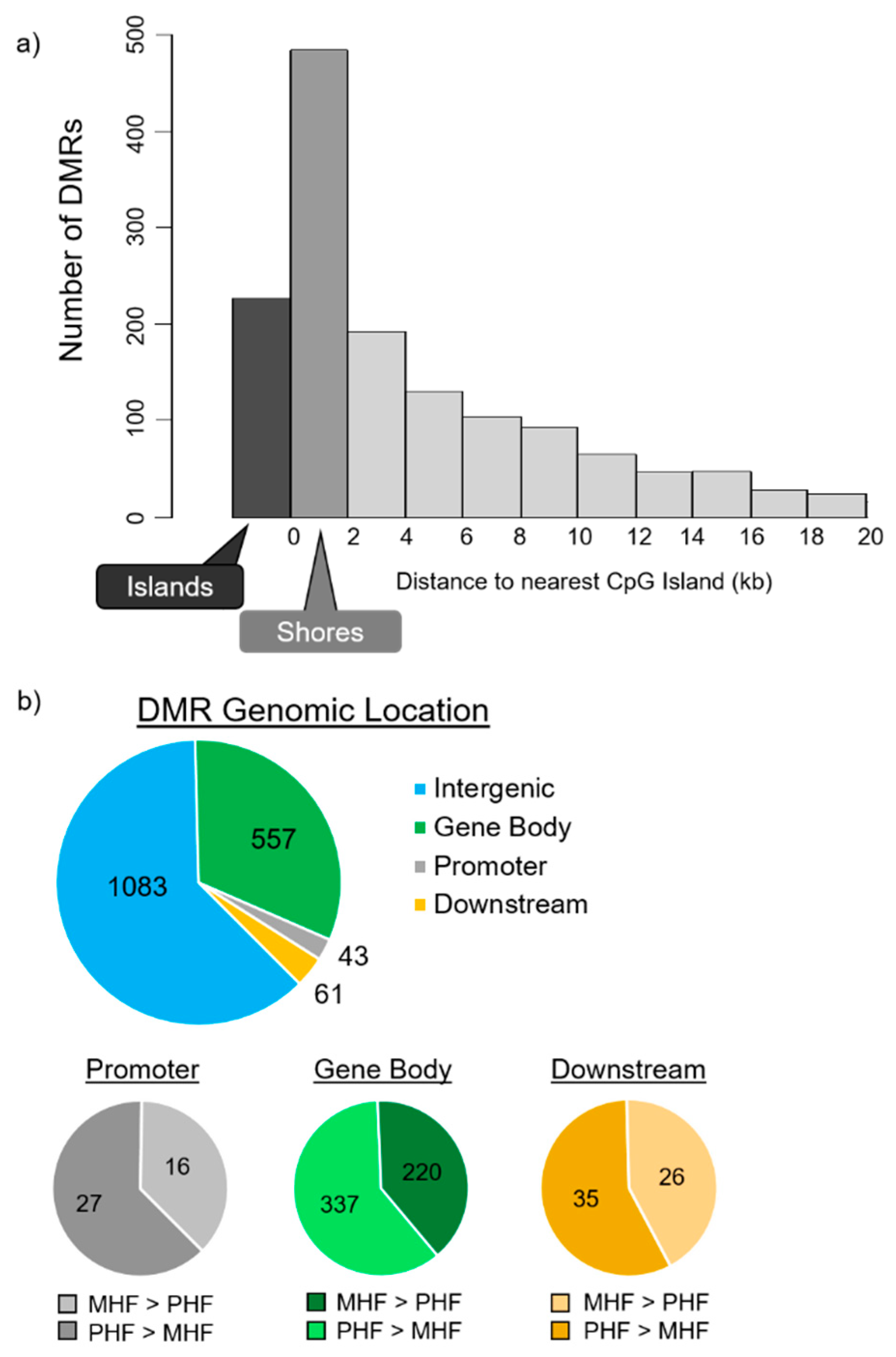
2.2. Timing HF Diet Exposure Determines Distinct Methylation Patterns
2.3. Post-Weaning HF Increases Gene Expression While Maternal HF Decreases Gene Expression in the PI and Insulin Signaling Pathways
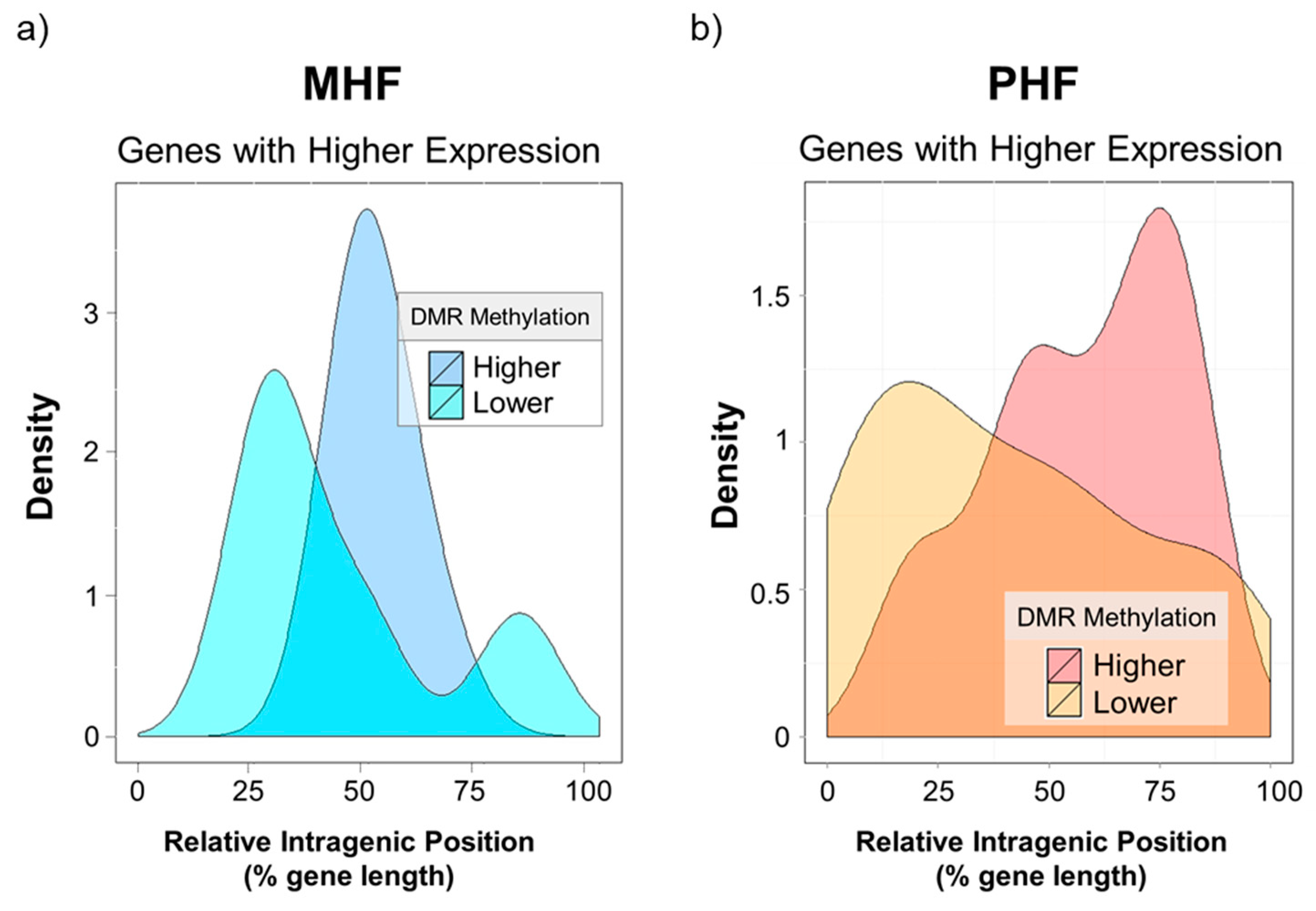
2.4. Genomic Context of DNA Methylation Is Indicative of Gene Expression
3. Discussion
4. Materials and Methods
4.1. Animals and Diets
4.2. Histological Analysis
4.3. Genomic DNA Isolation
4.4. MeDIP-Seq and MRE-seq Sequencing
4.5. Probe-Based Analysis for Validation of DNA Methylation
4.6. Methylation Specific PCR
4.7. Gene Expression Analysis
4.8. DMR Identification
4.9. Genomic Location of DMRs
4.10. Pathway Analysis and Functional Clustering
4.11. General Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HF | high-fat diet |
| MeDIP-seq | methylated DNA immunoprecipitation with high-throughput sequencing |
| MRE-seq | methylation-sensitive restriction enzyme digestion sequencing |
| DMR | differentially methylation regions |
| DMG | differentially methylated genes |
| Mc4r | melanocortin 4 receptor |
| Ppara | peroxisome proliferator-activated receptor α |
| Tlr-1 and Tlr-2 | toll-like receptors 1 and 2 |
| Cdkn1a | cyclin-dependent kinase inhibitor |
| H&E | hematoxylin and eosin |
| QAMA | Quantitative Analysis of Methylated Alleles |
| MSP | methylation specific PCR |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| PCC | Pearson correlation coefficients |
References
- Chang, G.Q.; Gaysinskaya, V.; Karatayev, O.; Leibowitz, S.F. Maternal high-fat diet and fetal programming: Increased proliferation of hypothalamic peptide-producing neurons that increase risk for overeating and obesity. J. Neurosci. 2008, 28, 12107–12119. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Katewa, S.D.; Palaniyappan, A.; Pandya, J.D.; Patel, M.S. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E792–E799. [Google Scholar] [CrossRef] [PubMed]
- Milagro, F.I.; Campion, J.; García-Díaz, D.F.; Goyenechea, E.; Paternain, L.; Martínez, J.A. High fat diet-induced obesity modifies the methylation pattern of leptin promoter in rats. J. Physiol. Biochem. 2009, 65, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Widiker, S.; Kärst, S.; Wagener, A.; Brockmann, G.A. High-fat diet leads to a decreased methylation of theMc4r gene in the obese BFMI and the lean B6 mouse lines. J. Appl. Genet. 2010, 51, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhang, Y.; Liu, M.; Lan, M.S.; Fei, J.; Fan, W.; Gao, X.; Lu, D. Hypermethylation of hepatic glucokinase and L-type pyruvate kinase promoters in high-fat diet-induced obese rats. Endocrinology 2011, 152, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.J.; Luo, S.M.; Lin, F.; Liang, Q.X.; Huang, L.; Wei, Y.C.; Hou, Y.; Han, Z.M.; Schatten, H.; Sun, Q.Y. DNA methylation in oocytes and liver of female mice and their offspring: Effects of high-fat-diet-induced obesity. Environ. Health Perspect. 2014, 122, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, J.; Liu, S.; Zhang, L.; Xiao, H.; Li, J.; Chen, H.; Petersen, R.B.; Huang, K.; Zheng, L. DNA hypomethylation of inflammation-associated genes in adipose tissue of female mice after multigenerational high fat diet feeding. Int. J. Obes. 2014, 38, 198–204. [Google Scholar] [CrossRef]
- Dudley, K.J.; Sloboda, D.M.; Connor, K.L.; Beltrand, J.; Vickers, M.H. Offspring of mothers fed a high fat diet display hepatic cell cycle inhibition and associated changes in gene expression and DNA methylation. PLoS ONE 2011, 6, e21662. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2011, 13, 97–109. [Google Scholar] [CrossRef]
- Feil, R.; Walter, J.; Allen, N.D.; Reik, W. Developmental control of allelic methylation in the imprinted mouse Igf2 and H19 genes. Development 1994, 120, 2933–2943. [Google Scholar] [PubMed]
- Lister, R.; Ecker, J.R. Finding the fifth base: Genome-wide sequencing of cytosine methylation. Genome Res. 2009, 19, 959–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lister, R.; Gregory, B.D.; Ecker, J.R. Next is now: New technologies for sequencing of genomes, transcriptomes, and beyond. Curr. Opin. Plant Biol. 2009, 12, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Cheng, J.B.; Li, D.; Xie, M.; Hong, C.; Maire, C.L.; Ligon, K.L.; Hirst, M.; Marra, M.A.; Costello, J.F.; et al. Estimating absolute methylation levels at single CpG resolution from methylation enrichment and restriction enzyme sequencing methods. Genome Res. 2013, 23, 1541–1553. [Google Scholar] [CrossRef] [PubMed]
- Aldoretta, P.W.; Carver, T.D.; Hay, W.W., Jr. Maturation of glucose-stimulated insulin secretion in fetal sheep. Neonatology 1998, 73, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Hay, W.W., Jr. Recent observations on the regulation of fetal metabolism by glucose. J. Physiol. 2006, 572, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Wang, T.; Coarfa, C.; Nagarajan, R.P.; Hong, C.; Downey, S.L.; Johnson, B.E.; Fouse, S.D.; Delaney, A.; Zhao, Y.; et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotechnol. 2010, 28, 1097–1105. [Google Scholar] [CrossRef]
- Moody, L.; Pan, Y.-X.; Chen, H. Postnatal diet remodels hepatic DNA methylation in metabolic pathways established by a maternal high-fat diet. Epigenomics 2017, 9, 1387–1402. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Wu, H.; Feinberg, A.P. A species-generalized probabilistic model-based definition of CpG islands. Mamm. Genome 2009, 20, 674–680. [Google Scholar] [CrossRef] [Green Version]
- Doi, A.; Park, I.-H.; Wen, B.; Murakami, P.; Aryee, M.J.; Irizarry, R.; Herb, B.; Ladd-Acosta, C.; Rho, J.; Loewer, S.; et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009, 41, 1350–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.-H.; Leproust, E.M.; Park, I.-H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Cheng, M.; Zhao, M.; Ge, A.; Guo, F.; Zhang, M.; Yang, Y.; Liu, L.; Yang, N. Differential effects of high-fat-diet rich in lard oil or soybean oil on osteopontin expression and inflammation of adipose tissue in diet-induced obese rats. Eur. J. Nutr. 2013, 52, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ogawa, H.; Kishida, T.; Ebihara, K. The effect of high-amylose cornstarch on lipid metabolism in OVX rats is affected by fructose feeding. J. Nutr. Biochem. 2010, 21, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Blé-Castillo, J.L.; Aparicio-Trápala, M.A.; Juárez-Rojop, I.E.; Torres-Lopez, J.E.; Méndez, J.D.; Aguilar-Mariscal, H.; Olvera-Hernández, V.; Palma-Cordova, L.C.; Diaz-Zagoya, J.C. Differential Effects of High-Carbohydrate and High-Fat Diet Composition on Metabolic Control and Insulin Resistance in Normal Rats. Int. J. Environ. Res. Public Health 2012, 9, 1663–1676. [Google Scholar] [CrossRef] [Green Version]
- Hallfrisch, J.; Cohen, L.; Reiser, S. Effects of Feeding Rats Sucrose in a High-Fat Diet. J. Nutr. 1981, 111, 531–536. [Google Scholar] [CrossRef]
- Chambers, T.J.G.; Morgan, M.D.; Heger, A.H.; Sharpe, R.M.; Drake, A.J.; Heger, A. High-fat diet disrupts metabolism in two generations of rats in a parent-of-origin specific manner. Sci. Rep. 2016, 6, 31857. [Google Scholar] [CrossRef] [Green Version]
- Ghibaudi, L.; Cook, J.; Farley, C.; Van Heek, M.; Hwa, J.J. Fat intake affects adiposity, comorbidity factors, and energy metabolism of sprague-dawley rats. Obes. Res. 2002, 10, 956–963. [Google Scholar] [CrossRef]
- Loche, E.; Blackmore, H.L.; Carpenter, A.A.; Beeson, J.H.; Pinnock, A.; Ashmore, T.J.; Aiken, C.E.; De Almeida-Faria, J.; Schoonejans, J.M.; Giussani, D.A.; et al. Maternal diet-induced obesity programmes cardiac dysfunction in male mice independently of post-weaning diet. Cardiovasc. Res. 2018, 114, 1372–1384. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez, C.A.; Erthal, R.P.; Ogo, F.M.; Peres, M.N.C.; Vieira, H.R.; Conejo, C.; Tófolo, L.P.; Francisco, F.A.; Silveira, S.D.S.; Malta, A.; et al. A High Fat Diet during Adolescence in Male Rats Negatively Programs Reproductive and Metabolic Function Which is Partially Ameliorated by Exercise. Front. Physiol. 2017, 8, 807. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.M.; Dias-Rocha, C.P.; Souza, A.S.; Muros, M.F.; Mendonca, L.S.; Pazos-Moura, C.C.; Trevenzoli, I.H. Perinatal maternal high-fat diet induces early obesity and sex-specific alterations of the endocannabinoid system in white and brown adipose tissue of weanling rat offspring. Br. J. Nutr. 2017, 118, 788–803. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, L.S.; Souza, L.L.; Souza, A.F.P.; Cordeiro, A.; Kluck, G.E.G.; Atella, G.C.; Trevenzoli, I.H.; Pazos-Moura, C.C. Perinatal maternal high-fat diet promotes alterations in hepatic lipid metabolism and resistance to the hypolipidemic effect of fish oil in adolescent rat offspring. Mol. Nutr. Food Res. 2016, 60, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Keleher, M.R.; Zaidi, R.; Shah, S.; Oakley, M.E.; Pavlatos, C.; El Idrissi, S.; Xing, X.; Li, D.; Wang, T.; Cheverud, J.M. Maternal high-fat diet associated with altered gene expression, DNA methylation, and obesity risk in mouse offspring. PLoS ONE 2018, 13, e0192606. [Google Scholar] [CrossRef] [PubMed]
- Martínez, D.; Pentinat, T.; Ribó, S.; Daviaud, C.; Bloks, V.W.; Cebria, J.; Villalmanzo, N.; Kalko, S.G.; Ramon-Krauel, M.; Diaz, R.; et al. In utero undernutrition in male mice programs liver lipid metabolism in the second-generation offspring involving altered Lxra DNA methylation. Cell Metab. 2014, 19, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, R.S.; Bird, A.P. CpG islands-’a rough guide’. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Shepherd, P.R.; Withers, D.J.; Siddle, K. Phosphoinositide 3-kinase: The key switch mechanism in insulin signalling. Biochem. J. 1998, 333, 471–490. [Google Scholar] [CrossRef]
- Sleeman, M.W.; Wortley, K.E.; Lai, K.-M.V.; Gowen, L.C.; Kintner, J.; Kline, W.O.; Garcia, K.; Stitt, T.N.; Yancopoulos, G.D.; Wiegand, S.J.; et al. Absence of the lipid phosphatase SHIP2 confers resistance to dietary obesity. Nat. Med. 2005, 11, 199–205. [Google Scholar] [CrossRef]
- Clément, S.; Krause, U.; Desmedt, F.; Tanti, J.-F.; Behrends, J.; Pesesse, X.; Sasaki, T.; Penninger, J.; Doherty, M.; Malaisse, W.; et al. The lipid phosphatase SHIP2 controls insulin sensitivity. Nature 2001, 409, 92–97. [Google Scholar] [CrossRef]
- Gewinner, C.; Wang, Z.C.; Richardson, A.; Teruya-Feldstein, J.; Etemadmoghadam, D.; Bowtell, D.; Barretina, J.; Lin, W.M.; Rameh, L.; Salmena, L.; et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell 2009, 16, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, H.; Ridderstråle, M.; Degerman, E.; Ekholm, D.; Smith, C.J.; Manganiello, V.C.; Belfrage, P.; Tornqvist, H. Evidence for the key role of the adipocyte cGMP-inhibited cAMP phosphodiesterase in the antilipolytic action of insulin. Evidence for the key role of the adipocyte cGMP-inhibited cAMP phosphodiesterase in the antilipolytic action of insulin. Biochim. Biophys. Acta 1995, 1266, 101–107. [Google Scholar] [CrossRef]
- Choi, Y.H.; Park, S.; Hockman, S.; Zmuda-Trzebiatowska, E.; Svennelid, F.; Haluzík, M.; Gavrilova, O.; Ahmad, F.; Pépin, L.; Napolitano, M.; et al. Alterations in regulation of energy homeostasis in cyclic nucleotide phosphodiesterase 3B-null mice. J. Clin. Investig. 2006, 116, 3240–3251. [Google Scholar] [CrossRef] [PubMed]
- Cheverud, J.M.; Ehrich, T.H.; Hrbek, T.; Kenney, J.P.; Pletscher, L.S.; Semenkovich, C.F. Quantitative trait loci for obesity- and diabetes-related traits and their dietary responses to high-fat feeding in LGXSM recombinant inbred mouse strains. Diabetes 2004, 53, 3328–3336. [Google Scholar] [CrossRef] [PubMed]
- Dobrin, R.; Chudin, E.; Suver, C.; Beaulaurier, J.; Zhang, B.; Castro, V.; Zhu, J.; Wang, S.; Molony, C.; Lum, P.Y.; et al. A survey of the genetics of stomach, liver, and adipose gene expression from a morbidly obese cohort. Genome Res. 2011, 21, 1008–1016. [Google Scholar]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42, D1001–D1006. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Bouatia-Naji, N.; Gloyn, A.L.; et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 2010, 42, 105–116. [Google Scholar] [CrossRef]
- Aulchenko, Y.S.; Ripatti, S.; Lindqvist, I.; Boomsma, D.; Heid, I.M.; Pramstaller, P.P.; Penninx, B.W.J.H.; Janssens, A.C.J.W.; Wilson, J.F.; Spector, T.; et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat. Genet. 2009, 41, 47–55. [Google Scholar]
- Kathiresan, S.; Willer, C.J.; Peloso, G.M.; Demissie, S.; Musunuru, K.; Schadt, E.E.; Kaplan, L.; Bennett, D.; Li, Y.; Tanaka, T.; et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 2009, 41, 56–65. [Google Scholar] [CrossRef]
- Sabatti, C.; Service, S.K.; Hartikainen, A.-L.; Pouta, A.; Ripatti, S.; Brodsky, J.; Jones, C.G.; Zaitlen, N.A.; Varilo, T.; Kaakinen, M.; et al. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat. Genet. 2009, 41, 35–46. [Google Scholar] [CrossRef]
- DiStefano, J.K.; Kingsley, C.; Wood, G.C.; Chu, X.; Argyropoulos, G.; Still, C.D.; Doné, S.C.; Legendre, C.; Tembe, W.; Gerhard, G.S. Genome-wide analysis of hepatic lipid content in extreme obesity. Acta Diabetol. 2015, 52, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, Y.; Fridman, M.V.; Oparina, N.J.; Malko, D.B.; Ermakova, E.O.; Kulakovskiy, I.V.; Heinzel, A.; Makeev, V.J. Intergenic, gene terminal, and intragenic CpG islands in the human genome. BMC Genom. 2010, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Pérez, R.F.; Santamarina, P.; Tejedor, J.R.; Urdinguio, R.G.; Álvarez-Pitti, J.; Redon, P.; Fernández, A.F.; Fraga, M.F.; Lurbe, E. Longitudinal genome-wide DNA methylation analysis uncovers persistent early-life DNA methylation changes. J. Transl. Med. 2019, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Frigola, J.; Vendrell, E.; Risques, R.-A.; Fraga, M.F.; Morales, C.; Moreno, V.; Esteller, M.; Capellà, G.; Ribas, M.; et al. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res. 2006, 66, 8462–9468. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.; Heyn, H.A.; Serra-Musach, J.; Bibikova, M.; Moran, S.; Pujana, M.A.; Esteller, M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011, 6, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Timp, W.; Bravo, H.C.; McDonald, O.G.; Goggins, M.; Umbricht, C.; Zeiger, M.; Feinberg, A.P.; Irizarry, R.A. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med. 2014, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, N.; Reinius, L.E.; Vitezic, M.; Fortino, V.; Söderhäll, C.; Honkanen, H.; Veijola, R.; Simell, O.; Toppari, J.; Ilonen, J.; et al. Age-associated DNA methylation changes in immune genes, histone modifiers and chromatin remodeling factors within 5 years after birth in human blood leukocytes. Clin. Epigenet. 2015, 7, 34. [Google Scholar] [CrossRef]
- Alisch, R.S.; Barwick, B.G.; Chopra, P.; Myrick, L.K.; Satten, G.A.; Conneely, K.N.; Warren, S.T. Age-associated DNA methylation in pediatric populations. Genome Res. 2012, 22, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Christensen, B.C.; Houseman, E.A.; Marsit, C.J.; Zheng, S.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Padbury, J.F.; Bueno, R.; et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009, 5, e1000602. [Google Scholar] [CrossRef]
- Lurbe, E.; Urdinguio, R.G.; Torró, M.I.; Bayón, G.F.; Álvarez-Pitti, J.; Fernández, A.F.; Redón, P.; Fraga, M.F. Longitudinal study of DNA methylation during the first 5 years of life. J. Transl. Med. 2016, 14, 160. [Google Scholar]
- Day, K.; Waite, L.L.; Thalacker-Mercer, A.; West, A.; Bamman, M.M.; Brooks, J.D.; Myers, R.M.; Absher, D. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 2013, 14, R102. [Google Scholar] [CrossRef] [PubMed]
- Gelfman, S.; Ast, G. When epigenetics meets alternative splicing: The roles of DNA methylation and GC architecture. Epigenomics 2013, 5, 351–353. [Google Scholar] [CrossRef] [PubMed]
- Gelfman, S.; Cohen, N.; Yearim, A.; Ast, G. DNA-methylation effect on cotranscriptional splicing is dependent on GC architecture of the exon-intron structure. Genome Res. 2013, 23, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Hoivik, E.A.; Bjanesoy, T.E.; Mai, O.; Okamoto, S.; Minokoshi, Y.; Shima, Y.; Morohashi, K.-I.; Boehm, U.; Bakke, M. DNA methylation of intronic enhancers directs tissue-specific expression of steroidogenic factor 1/adrenal 4 binding protein (SF-1/Ad4BP). Endocrinology 2011, 152, 2100–2112. [Google Scholar] [CrossRef] [PubMed]
- Unoki, M.; Nakamura, Y. Methylation at CpG islands in intron 1 of EGR2 confers enhancer-like activity. FEBS Lett. 2003, 554, 67–72. [Google Scholar] [CrossRef]
- Portella, G.; Battistini, F.; Orozco, M. Understanding the connection between epigenetic DNA methylation and nucleosome positioning from computer simulations. PLoS Comput. Biol. 2013, 9, e1003354. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, G.; Kelly, T.K.; Lay, F.D.; Berman, B.P.; Jones, P.A. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012, 22, 2497–2506. [Google Scholar] [Green Version]
- Chodavarapu, R.K.; Feng, S.; Bernatavichute, Y.V.; Chen, P.-Y.; Stroud, H.; Yu, Y.; Hetzel, J.A.; Kuo, F.; Kim, J.; Cokus, S.J.; et al. Relationship between nucleosome positioning and DNA methylation. Nature 2010, 466, 388–392. [Google Scholar] [CrossRef] [Green Version]
- Pennings, S.; Allan, J.; Davey, C.S. DNA methylation, nucleosome formation and positioning. Brief. Funct. Genom. 2005, 3, 351–361. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Rauch, T.A.; Pfeifer, G.P. The MIRA method for DNA methylation analysis. Methods Mol. Biol. 2009, 507, 65–75. [Google Scholar] [PubMed]
- Yang, Y.; Yang, C.; Zhu, Y.; Chen, H.; Zhao, R.; He, X.; Tao, L.; Wang, P.; Zhou, L.; Zhao, L.; et al. Intragenic and extragenic disruptions of FOXL2 mapped by whole genome low-coverage sequencing in two BPES families with chromosome reciprocal translocation. Genomics 2014, 104, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Hanson, M.A.; Slater-Jefferies, J.L.; Lillycrop, K.A. Epigenetic regulation of transcription: A mechanism for inducing variations in phenotype (fetal programming) by differences in nutrition during early life? Br. J. Nutr. 2007, 97, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Waldecker, M.; Kautenburger, T.; Daumann, H.; Busch, C.; Schrenk, D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J. Nutr. Biochem. 2008, 19, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; McCaffery, J.M.; Irizarry, R.A.; Boeke, J.D. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol. Cell 2006, 23, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Bacolla, A.; Wells, R.D.; Roberts, R. Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem. 1999, 274, 33002–33010. [Google Scholar] [CrossRef]
- Pogribny, I.P.; Tryndyak, V.P.; Bagnyukova, T.V.; Melnyk, S.; Montgomery, B.; Ross, S.A.; Latendresse, J.R.; Rusyn, I.; Beland, F.A. Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. J. Hepatol. 2009, 51, 176–186. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.B.; He, J.L.; Liu, X.Q.; Chen, X.M.; Long, C.L.; Wang, Y.X. Expression of DNA methyltransferases in the mouse uterus during early pregnancy and susceptibility to dietary folate deficiency. Reproduction 2012, 144, 91–100. [Google Scholar] [CrossRef]
- Ghoshal, K.; Li, X.; Datta, J.; Bai, S.; Pogribny, I.; Pogribny, M.; Huang, Y.; Young, D.; Jacob, S.T. A folate- and methyl-deficient diet alters the expression of DNA methyltransferases and methyl CpG binding proteins involved in epigenetic gene silencing in livers of F344 rats. J. Nutr. 2006, 136, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Kovacheva, V.P.; Mellott, T.J.; Davison, J.M.; Wagner, N.; Lopez-Coviella, I.; Schnitzler, A.C.; Blusztajn, J.K. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J. Biol. Chem. 2007, 282, 31777–31788. [Google Scholar] [CrossRef]
- Xia, L.; Wang, C.; Lu, Y.; Fan, C.; Ding, X.; Fu, H.; Qi, K. Time-specific changes in DNA methyltransferases associated with the leptin promoter during the development of obesity. Nutr. Hosp. 2014, 30, 1248–1255. [Google Scholar] [PubMed]
- Strakovsky, R.S.; Wang, H.; Engeseth, N.J.; Flaws, J.A.; Helferich, W.G.; Pan, Y.-X.; Lezmi, S. Developmental bisphenol A (BPA) exposure leads to sex-specific modification of hepatic gene expression and epigenome at birth that may exacerbate high-fat diet-induced hepatic steatosis. Toxicol. Appl. Pharmacol. 2015, 284, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhou, D.; Pan, Y.-X.; Strakovsky, R.S.; Pan, Y. Gestational high fat diet programs hepatic phosphoenolpyruvate carboxykinase gene expression and histone modification in neonatal offspring rats. J. Physiol. 2011, 589, 2707–2717. [Google Scholar]
- Zhou, D.; Wang, H.; Cui, H.; Chen, H.; Pan, Y.-X. Early-life exposure to high-fat diet may predispose rats to gender-specific hepatic fat accumulation by programming Pepck expression. J. Nutr. Biochem. 2015, 26, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Strakovsky, R.; Zhou, D.; Zhang, Y.; Pan, Y.-X. A maternal high-fat diet represses the expression of antioxidant defense genes and induces the cellular senescence pathway in the liver of male offspring rats. J. Nutr. 2011, 141, 1254–1259. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Zhou, D.; Moody, L.; Lezmi, S.; Chen, H.; Pan, Y.-X. High-fat diet caused widespread epigenomic differences on hepatic methylome in rat. Physiol. Genom. 2015, 47, 514–523. [Google Scholar] [CrossRef]
- Li, D.; Zhang, B.; Xing, X.; Wang, T. Combining MeDIP-seq and MRE-seq to investigate genome-wide CpG methylation. Methods 2015, 72, 29–40. [Google Scholar] [CrossRef]
- Claussen, U.; Michel, S.; Boehringer, S.; Gillessen-Kaesbach, G.; Buiting, K.; Horsthemke, B.; Nazlican, H.; Zeschnigk, M. Somatic mosaicism in patients with Angelman syndrome and an imprinting defect. Hum. Mol. Genet. 2004, 13, 2547–2555. [Google Scholar]
- Suzuki, T.; Higgins, P.J.; Crawford, D.R. Control Selection for RNA Quantitation. Biotechniques 2000, 29, 332–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wilson, G.J.; Zhou, D.; Lezmi, S.; Chen, X.; Layman, D.K.; Pan, Y.-X. Induction of autophagy through the activating transcription factor 4 (ATF4)-dependent amino acid response pathway in maternal skeletal muscle may function as the molecular memory in response to gestational protein restriction to alert offspring to maternal nutrition. Br. J. Nutr. 2015, 114, 519–532. [Google Scholar] [PubMed] [Green Version]
- Bass, J.I.F.; Diallo, A.; Nelson, J.; Soto, J.M.; Myers, C.L.; Walhout, A.J.M. Using networks to measure similarity between genes: Association index selection. Nat. Methods 2013, 10, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Position | Sequence |
|---|---|---|
| Pklr (+ chr2) | Forward Primer + 4333 | 5′-TGGTGTTATTTAGATGTTGGAGAGTATGA-3′ |
| Reverse Primer + 4557 | 5′-AACATAATACAATCAACCCCATCCA-3′ | |
| Methylated Probe + 4467 | 5′-VIC-AGGTTCGATTAATTCGGGCG-MGB-3′ | |
| Unmethylated Probe + 4470 | 5′-FAM-TGATTAATTTGGGTGGAGATAA-MGB-3′ | |
| IL-6 | Forward Primer − 2516 | GTGAGTAAGGGATTTAGTTTGAGTATGGT |
| Reverse Primer − 2441 | CTTATTCCTAAATATCTAATACCCTCTTATAACCTC | |
| Methylated Probe − 2480 | 5′-VIC-CGTGTGTGAATGTGCGTTA-3′ | |
| Unmethylated Probe − 2483 | 5′-FAM-TTTGTGTGTGAATGTGTGTT-MGB-3′ |
| Pathway | DMGs | Fold Enrichment | p-Value |
|---|---|---|---|
| Phosphatidylinositol signaling system | 11 | 5.2 | 0.00004 |
| Insulin signaling pathway | 11 | 2.8 | 0.006 |
| Axon guidance | 10 | 2.6 | 0.01 |
| Pathways in cancer | 18 | 1.9 | 0.01 |
| Purine metabolism | 10 | 2.1 | 0.04 |
| Glycerolipid metabolism | 5 | 3.8 | 0.04 |
| Fc gamma R-mediated phagocytosis | 7 | 2.6 | 0.05 |
| Gene | Position | Sequence |
|---|---|---|
| Acacb | Forward M + 75,096 | 5′-TTGGGTTCGGTTTTTAGTTTCG-3′ |
| Reverse M + 75,222 | 5′-ACGTATATCCCTATAATCCAACTCGC-3′ | |
| Forward UM + 75,095 | 5′-TTTGGGTTTGGTTTTTAGTTTTGAA-3′ | |
| Reverse UM + 75,225 | 5′-CACATATATCCCTATAATCCAACTCACTCT -3′ | |
| Gpam | Forward M + 7289 | 5′-AGTCGTAGTGGTCGGGTAATCG-3′ |
| Reverse M + 7357 | 5′-CCGCTTATTTTAAACAACATCGAA-3′ | |
| Forward UM + 7288 | 5′-AAGTTAAGTTGTAGTGGTTGGGTAATTG-3′ | |
| Reverse UM + 7360 | 5′-CCCACTTATTTTAAACAACATCAAACC-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moody, L.; Wang, H.; Jung, P.M.; Chen, H.; Pan, Y.-X. Maternal and Post-Weaning High-Fat Diets Produce Distinct DNA Methylation Patterns in Hepatic Metabolic Pathways within Specific Genomic Contexts. Int. J. Mol. Sci. 2019, 20, 3229. https://doi.org/10.3390/ijms20133229
Moody L, Wang H, Jung PM, Chen H, Pan Y-X. Maternal and Post-Weaning High-Fat Diets Produce Distinct DNA Methylation Patterns in Hepatic Metabolic Pathways within Specific Genomic Contexts. International Journal of Molecular Sciences. 2019; 20(13):3229. https://doi.org/10.3390/ijms20133229
Chicago/Turabian StyleMoody, Laura, Huan Wang, Paul M. Jung, Hong Chen, and Yuan-Xiang Pan. 2019. "Maternal and Post-Weaning High-Fat Diets Produce Distinct DNA Methylation Patterns in Hepatic Metabolic Pathways within Specific Genomic Contexts" International Journal of Molecular Sciences 20, no. 13: 3229. https://doi.org/10.3390/ijms20133229
APA StyleMoody, L., Wang, H., Jung, P. M., Chen, H., & Pan, Y. -X. (2019). Maternal and Post-Weaning High-Fat Diets Produce Distinct DNA Methylation Patterns in Hepatic Metabolic Pathways within Specific Genomic Contexts. International Journal of Molecular Sciences, 20(13), 3229. https://doi.org/10.3390/ijms20133229