The Significance of IL-36 Hyperactivation and IL-36R Targeting in Psoriasis




Abstract
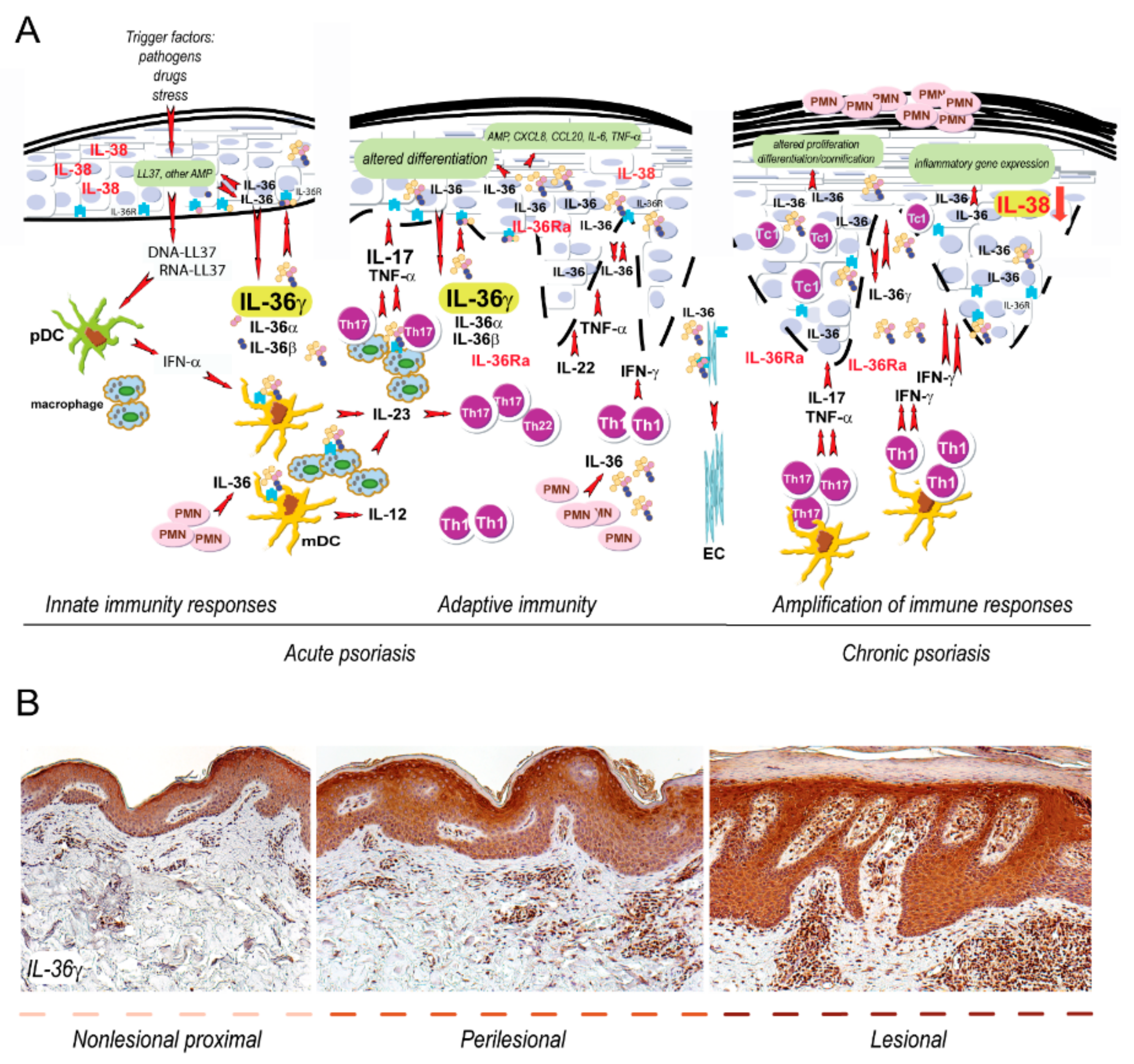
:1. Introduction
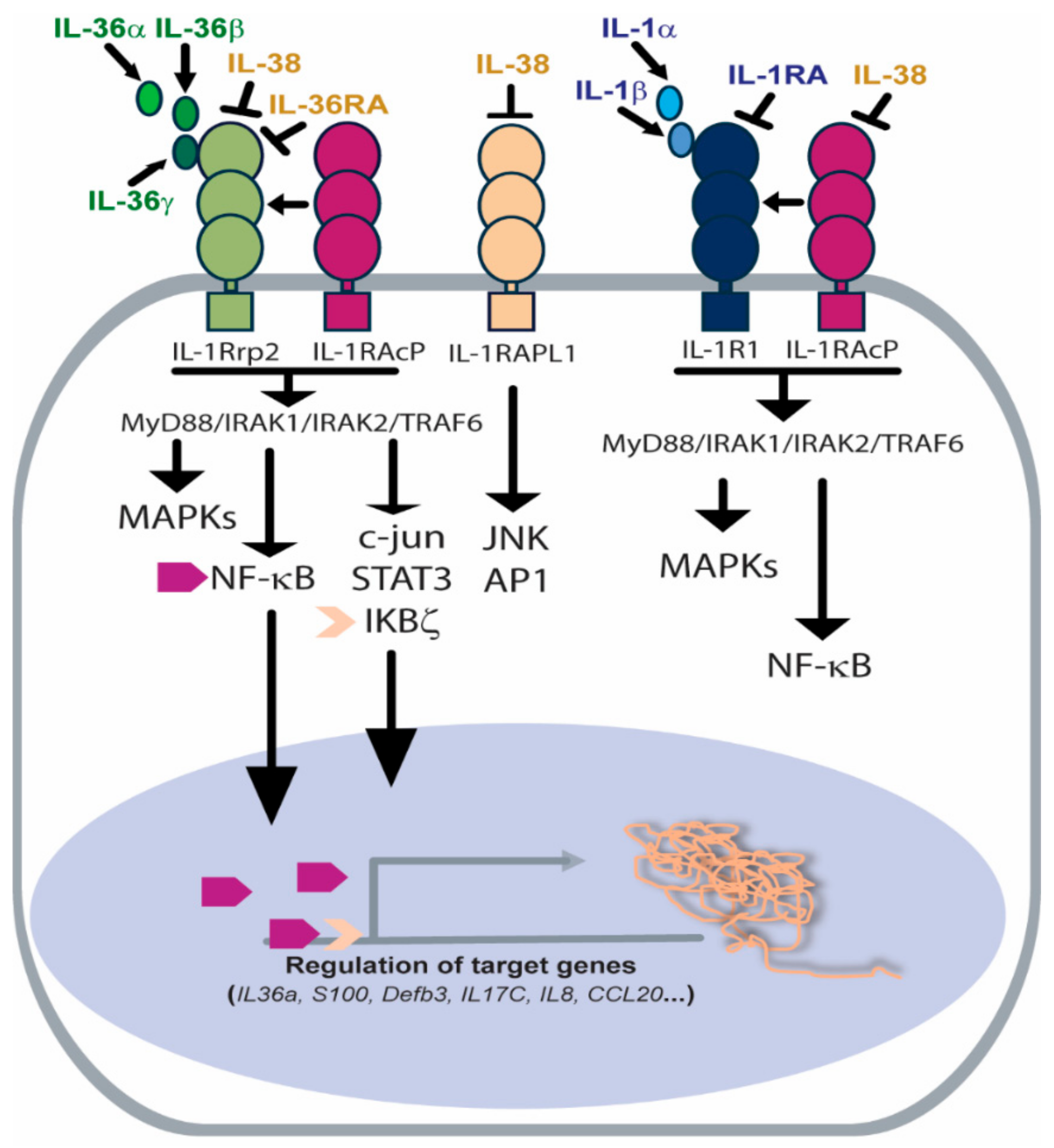
2. Structural and Functional Characterization of Il-36 Receptor
2.1. Organization of IL-36R Molecular Complex
2.2. IL-36R-Dependent Intracellular Signaling
3. IL36R-Dependent Responses in Psoriatic Resident Skin Cells
3.1. Epidermal Keratinocytes
3.2. Dermal Endothelial Cells and Fibroblasts
4. IL36R Function in Psoriasis Immune Cells
5. Impact of IL-36R Manipulation in Preclinical Models of Skin Inflammation
6. Therapeutic Potential of IL36R-Targeting in Psoriasis
7. Conclusions
Acknowledgements
Conflicts of Interest
Abbreviations
| IL-1R | IL-1 Receptor |
| IL-1Ra | IL-1 Receptor antagonist |
| IL1RAPL1 | X-linked interleukin-1 receptor accessory protein-like 1 |
| JNK | Jun N-terminal Kinase |
| MyD88 | Myeloid Differentiation Primary Response Protein 88 |
| MAPK | Mitogen-Activated Protein Kinases |
| IkBz | Nuclear Factor-kappa B inhibitor zeta |
| NFkB | Nuclear Factor-kappa B |
| TRAF | TNF Receptor-Associated Factor |
| IRAK | Interleukin 1 Receptor Associated Kinase 1 |
| STAT | Signal Transducer and Activator of Transcription |
References
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar] [CrossRef]
- Lowes, M.A.; Suárez-Fariñas, M.; Krueger, J.G. Immunology of psoriasis. Annu. Rev. Immunol. 2014, 32, 227–255. [Google Scholar] [CrossRef] [PubMed]
- Kuida, K.; Lippke, J.A.; Ku, G.; Harding, M.W.; Livingston, D.J.; Su, M.S.; Flavell, R.A. Altered cytokine export and apoptosis in mice deficient in interleukin-1β converting enzyme. Science 1995, 267, 2000–2003. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Towne, J.E.; Renshaw, B.R.; Douangpanya, J.; Lipsky, B.P.; Shen, M.; Gabel, C.A.; Sims, J.E. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J. Biol. Chem. 2011, 286, 42594–42602. [Google Scholar] [CrossRef] [PubMed]
- Balato, A.; Mattii, M.; Caiazzo, G.; Raimondo, A.; Patruno, C.; Balato, N.; Ayala, F.; Lembo, S. IL-36gamma is involved in psoriasis and allergic contact dermatitis. J. Investig. Dermatol. 2016, 136, 1520–1523. [Google Scholar] [CrossRef]
- Di Caprio, R.; Balato, A.; Caiazzo, G.; Lembo, S.; Raimondo, A.; Fabbrocini, G.; Manfrecola, G. IL-36 cytokines are increased in acne and hidradenitis suppurativa. Arch. Dermatol. Res. 2017, 309, 673–678. [Google Scholar] [CrossRef]
- Girolomoni, G.; Strohal, R.; Puig, L.; Bachelez, H.; Barker, J.; Boehncke, W.H.; Prinz, J.C. The role of IL-23 and the IL-23/TH17 immune axis in the pathogenesis and treatment of psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1616–1626. [Google Scholar] [CrossRef]
- Lowes, M.A.; Russell, C.B.; Martin, D.A.; Towne, J.E.; Krueger, J.G. The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol. 2013, 34, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, A.; Vollmer, S.; Besgen, P.; Galinski, A.; Summer, B.; Kawakami, Y.; Wollenberg, A.; Dornmair, K.; Spannagl, M.; Ruzicka, T.; et al. Unopposed IL-36 Activity Promotes Clonal CD4+ T-Cell Responses with IL-17A Production in Generalized Pustular Psoriasis. J. Invest. Dermatol. 2018, 138, 1338–1347. [Google Scholar] [CrossRef]
- Mahil, S.K.; Catapano, M.; Di Meglio, P.; Dand, N.; Ahlfors, H.; Carr, I.M.; Smith, C.H.; Trembath, R.C.; Peakman, M.; Wright, J.; et al. An analysis of IL-36 signature genes and individuals with IL1RL2 knockout mutations validates IL-36 as a psoriasis therapeutic target. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, K.; Takemoto, A.; Yamaguchi, M.; Takahashi, H.; Shoda, Y.; Mitsuma, T.; Tsuda, K.; Nishida, E.; Togawa, Y.; Nakajima, K. The majority of generalized pustular psoriasis without psoriasis vulgaris is caused by deficiency of interleukin-36 receptor antagonist. J. Investig. Dermatol. 2013, 133, 2514–2521. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, L.; Morelli, M.; Scarponi, C.; Eisenmesser, E.Z.; Doti, N.; Pagnanelli, G.; Gubinelli, E.; Mazzanti, C.; Cavani, A.; Ruvo, M.; et al. IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment. Cell Death Dis. 2018, 9, 1104. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Scheiermann, P.; Härdle, L.; Pfeilschifter, J.; Mühl, H. IL-36γ/IL-1F9, an innate T-bet target in myeloid cells. J. Biol. Chem. 2012, 287, 41684–41696. [Google Scholar] [CrossRef] [PubMed]
- Towne, J.E.; Sims, J.E. IL-36 in psoriasis. Curr. Opin. Pharmacol. 2012, 12, 486–490. [Google Scholar] [CrossRef] [PubMed]
- D’Erme, A.M.; Wilsmann-Theis, D.; Wagenpfeil, J.; Holzel, M.; Ferring-Schmitt, S.; Sternberg, S. IL-36[gamma] (IL-1F9) is a biomarker for psoriasis skin lesions. J. Investig. Dermatol. 2015, 135, 1025–1032. [Google Scholar] [CrossRef]
- Boutet, M.A.; Bart, G.; Penhoat, M.; Amiaud, J.; Brulin, B.; Charrier, C.; Morel, F.; Lecron, J.C.; Rolli-Derkinderen, M.; Bourreille, A. Distinct expression of interleukin (IL)-36alpha, beta and gamma, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn’s disease. Clin. Exp. Immunol. 2016, 184, 159–173. [Google Scholar] [CrossRef]
- Albanesi, C.; Madonna, S.; Gisondi, P.; Girolomoni, G. The Interplay between keratinocytes and immune cells in the pathogenesis of psoriasis. Front. Immunol. 2018, 9, 1549. [Google Scholar] [CrossRef]
- Foster, A.M.; Baliwag, J.; Chen, C.S.; Guzman, A.M.; Stoll, S.W.; Gudjonsson, J.E.; Ward, N.L.; Johnston, A. IL-36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J. Immunol. 2014, 192, 6053–6061. [Google Scholar] [CrossRef]
- Li, N.; Yamasaki, K.; Saito, R.; Fukushi-Takahashi, S.; Shimada-Omori, R.; Asano, M.; Aiba, S. Alarmin function of cathelicidin antimicrobial peptide LL37 through IL-36gamma induction in human epidermal keratinocytes. J. Immunol. 2014, 193, 5140–5148. [Google Scholar] [CrossRef]
- Bridgewood, C.; Stacey, M.; Alase, A.; Lagos, D.; Graham, A.; Wittmann, M. IL-36gamma has proinflammatory effects on human endothelial cells. Exp. Dermatol. 2017, 26, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Scheibe, K.; Backert, I.; Wirtz, S.; Hueber, A.; Schett, G.; Vieth, M.; Probst, H.C.; Bopp, T.; Neurath, M.F.; Neufert, C. IL-36R signaling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut 2017, 66, 823–838. [Google Scholar] [CrossRef] [PubMed]
- Bridgewood, C.; Fearnley, G.W.; Berekmeri, A.; Laws, P.; Macleod, T.; Ponnambalam, S.; Stacey, M.; Graham, A.; Wittmann, M. IL-36γ Is a Strong Inducer of IL-23 in Psoriatic Cells and Activates Angiogenesis. Front. Immunol. 2018, 26, 200. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.; Xing, X.; Wolterink, L.; Barnes, D.H.; Yin, Z.; Reingold, L.; Kahlenberg, J.M.; Harms, P.W.; Gudjonsson, J.E. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J. Allergy Clin. Immunol. 2017, 140, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Traks, T.; Keermann, M.; Prans, E.; Karelson, M.; Loite, U.; Kõks, G.; Silm, H.; Kõks, S.; Kingo, K. Polymorphisms in IL36G gene are associated with plaque psoriasis. BMC Med. Genet. 2019, 20, 10. [Google Scholar] [CrossRef]
- Marrakchi, S.; Guigue, P.; Renshaw, B.R.; Puel, A.; Pei, X.Y.; Fraitag, S.; Zribi, J.; Bal, E.; Cluzeau, C.; Chrabieh, M. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 2011, 365, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Onoufriadis, A.; Simpson, M.A.; Pink, A.E.; Di Meglio, P.; Smith, C.H.; Pullabhatla, V.; Knight, J.; Spain, S.L.; Nestle, F.O.; Burden, A.D. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am. J. Hum. Genet. 2011, 89, 432–437. [Google Scholar] [CrossRef]
- Boehner, A.; Navarini, A.A.; Eyerich, K. Generalized pustular psoriasis—A model disease for specific targeted immunotherapy, systematic review. Exp. Dermatol. 2018, 27, 1067–1077. [Google Scholar] [CrossRef]
- Twelves, S.; Mostafa, A.; Dand, N.; Burri, E.; Farkas, K.; Wilson, R.; Cooper, H.L.; Irvine, A.D.; Oon, H.H.; Kingo, K.; et al. Clinical and genetic differences between pustular psoriasis subtypes. J. Allergy Clin. Immunol. 2019, 143, 1021–1026. [Google Scholar] [CrossRef]
- Blumberg, H.; Dinh, H.; Dean, C., Jr.; Trueblood, E.S.; Bailey, K.; Shows, D.; Bhagavathula, N.; Aslam, M.N.; Varani, J.; Towne, J.E.; et al. IL-1RL2 and its ligands contribute to the cytokine network in psoriasis. J. Immunol. 2010, 185, 4354–4362. [Google Scholar] [CrossRef]
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The family of the interleukin-1 receptors. Immunol. Rev. 2018, 281, 197–232. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tu, J.; Hu, Y.; Song, G.; Yin, Z. Cathepsin G cleaves and activates IL-36γ and promotes the inflammation of psoriasis. Drug Des. Devel. Ther. 2019, 8, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.; Schlemmer, A.; Wittig, I.; Richter, F.; Putyrski, M.; Frank, A.C.; Han, Y.; Jung, M.; Ernst, A.; Weigert, A.; et al. Interleukin-38 is released from apoptotic cells to limit inflammatory macrophage responses. J. Mol. Cell Biol. 2016, 8, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Mora, J.; Huard, A.; da Silva, P.; Wiechmann, S.; Putyrski, M.; Schuster, C.; Elwakeel, E.; Lang, G.; Scholz, A.; et al. IL-38 Ameliorates Skin Inflammation and Limits IL-17 Production from γδ T Cells. Cell Rep. 2019, 27, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.M.; Sullivan, G.P.; Clancy, D.M.; Afonina, I.S.; Kulms, D.; Martin, S.J. Neutrophil-Derived Proteases Escalate Inflammation through Activation of IL-36 Family Cytokines. Cell Rep. 2016, 14, 708–722. [Google Scholar] [CrossRef] [Green Version]
- Gresnigt, M.S.; van de Veerdonk, F.L. Biology of IL-36 cytokines and their role in disease. Semin. Immunol. 2013, 25, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Yi, G.; Ybe, J.A.; Saha, S.S.; Caviness, G.; Raymond, E.; Ganesan, R.; Mbow, M.L.; Kao, C.C. Structural and Functional Attributes of the Interleukin-36 Receptor. J. Biol. Chem. 2016, 291, 16597–16609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Towne, J.E.; Garka, K.E.; Renshaw, B.R.; Virca, G.D.; Sims, J.E. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J. Biol. Chem. 2004, 279, 13677–13688. [Google Scholar] [CrossRef]
- Saha, S.S.; Singh, D.; Raymond, E.L.; Ganesan, R.; Caviness, G.; Grimaldi, C.; Woska, J.R., Jr.; Mennerich, D. Signal Transduction and Intracellular Trafficking by the Interleukin 36 Receptor. J. Biol. Chem. 2015, 290, 23997–24006. [Google Scholar] [CrossRef] [Green Version]
- Ohko, K.; Nakajima, K.; Kataoka, S.; Takaishi, M.; Sano, S. IL-36 Signaling Is Essential for Psoriatic Inflammation through the Augmentation of Innate Immune Responses. J. Investig. Dermatol. 2019, 139, 1400–1404. [Google Scholar] [CrossRef] [Green Version]
- Takaishi, M.; Satoh, T.; Akira, S.; Sano, S. Regnase-1, an Immunomodulator, Limits the IL-36/IL-36R Autostimulatory Loop in Keratinocytes to Suppress Skin Inflammation. J. Investig. Dermatol. 2018, 138, 1439–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohani, M.G.; DiJulio, D.H.; An, J.Y.; Hacker, B.M.; Dale, B.A.; Chung, W.O. PAR1-and PAR2-induced innate immune markers are negatively regulated by PI3K/Akt signaling pathway in oral keratinocytes. BMC Immunol. 2010, 11, 53. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Hennig, A.; Lorscheid, S.; Grondona, P.; Schulze-Osthoff, K.; Hailfinger, S.; Kramer, D. IκBζ is a key transcriptional regulator of IL-36-driven psoriasis-related gene expression in keratinocytes. Proc. Natl. Acad. Sci. USA 2018, 115, 10088–10093. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, V.; Hahn, M.; Kästele, V.; Wagner, O.; Wiendl, M.; Derer, A.; Taddeo, A.; Hahne, S.; Radbruch, A.; Jäck, H.M.; et al. Interleukin-36 receptor mediates the crosstalk between plasma cells and synovial fibroblasts. Eur. J. Immunol. 2017, 47, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, F.L.; de Graaf, D.M.; Joosten, L.A.; Dinarello, C.A. Biology of IL-38 and its role in disease. Immunol. Rev. 2018, 281, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, F.L.; Stoeckman, A.K.; Wu, G.; Boeckermann, A.N.; Azam, T.; Netea, M.G.; Joosten, L.A.; van der Meer, J.W.; Hao, R.; Kalabokis, V.; et al. IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc. Natl. Acad. Sci. USA 2012, 109, 3001–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, A.; Xing, X.; Guzman, A.M.; Riblett, M.; Loyd, C.M.; Ward, N.L.; Wohn, C.; Prens, E.P.; Wang, F.; Maier, L.E.; et al. IL-1F5, -F6, -F8, and -F9: A novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J. Immunol. 2011, 186, 2613–2622. [Google Scholar] [CrossRef]
- Bassoy, E.Y.; Towne, J.E.; Gabay, C. Regulation and function of interleukin-36 cytokines. Immunol. Rev. 2018, 281, 169–178. [Google Scholar] [CrossRef]
- Boutet, M.A.; Nerviani, A.; Pitzalis, C. IL-36, IL-37, and IL-38 Cytokines in Skin and Joint Inflammation: A Comprehensive Review of Their Therapeutic Potential. Int. J. Mol. Sci. 2019, 20, 1257. [Google Scholar] [CrossRef]
- Shao, S.; Fang, H.; Dang, E.; Xue, K.; Zhang, J.; Li, B.; Qiao, H.; Cao, T.; Zhuang, Y.; Shen, S.; et al. Neutrophil Extracellular Traps Promote Inflammatory Responses in Psoriasis via Activating Epidermal TLR4/IL-36R Crosstalk. Front. Immunol. 2019, 10, 746. [Google Scholar] [CrossRef] [Green Version]
- Carrier, Y.; Ma, H.L.; Ramon, H.E.; Napierata, L.; Small, C.; O’Toole, M.; Young, D.A.; Fouser, L.A.; Nickerson-Nutter, C.; Collins, M.; et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: Implications in psoriasis pathogenesis. J. Investig. Dermatol. 2011, 131, 2428–2437. [Google Scholar] [CrossRef]
- Pfaff, C.M.; Marquardt, Y.; Fietkau, K.; Baron, J.M.; Lüscher, B. The psoriasis-associated IL-17A induces and cooperates with IL-36 cytokines to control keratinocyte differentiation and function. Sci. Rep. 2017, 7, 15631. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, D.; Martin, P.; Flacher, V.; Sun, Y.; Jarrossay, D.; Brembilla, N.; Mueller, C.; Arnett, H.A.; Palmer, G.; Towne, J.; et al. Interleukin-36 potently stimulates human M2 macrophages, Langerhans cells and keratinocytes to produce pro-inflammatory cytokines. Cytokine 2016, 84, 88–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigne, S.; Palmer, G.; Lamacchia, C.; Martin, P.; Talabot-Ayer, D.; Rodriguez, E.; Ronchi, F.; Sallusto, F.; Dinh, H.; Sims, J.E.; et al. IL-36R ligands are potent regulators of dendritic and T cells. Blood 2011, 118, 5813–5823. [Google Scholar] [CrossRef] [PubMed]
- Vigne, S.; Palmer, G.; Martin, P.; Lamacchia, C.; Strebel, D.; Rodriguez, E.; Olleros, M.L.; Vesin, D.; Garcia, I.; Ronchi, F. IL-36 signaling amplifies Th1 responses by enhancing proliferation and Th1 polarization of naive CD4+ T cells. Blood 2012, 120, 3478–8347. [Google Scholar] [CrossRef]
- Clark, R.A.; Kupper, T.S. Misbehaving macrophages in the pathogenesis of psoriasis. J. Clin. Investig. 2006, 116, 2084–2087. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Ganesan, R.; Raymond, E.L.; Mennerich, D.; Woska, J.R., Jr.; Caviness, G.; Grimaldi, C.; Ahlberg, J.; Perez, R.; Roberts, S.; Yang, D.; et al. Generation and functional characterization of anti-human and anti-mouse IL-36R antagonist monoclonal antibodies. MAbs 2017, 9, 1143–1154. [Google Scholar] [CrossRef]
- Tortola, L.; Rosenwald, E.; Abel, B.; Blumberg, H.; Schäfer, M.; Coyle, A.J.; Renauld, J.C.; Werner, S.; Kisielow, J.; Kopf, M. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J. Clin. Investig. 2012, 122, 3965–3976. [Google Scholar] [CrossRef]
- Su, Z.; Paulsboe, S.; Wetter, J.; Salte, K.; Kannan, A.; Mathew, S.; Horowitz, A.; Gerstein, C.; Namovic, M.; Todorović, V.; et al. IL-36 receptor antagonistic antibodies inhibit inflammatory responses in preclinical models of psoriasiform dermatitis. Exp. Dermatol. 2019, 28, 113–120. [Google Scholar] [CrossRef]
- Campbell, J.J.; Ebsworth, K.; Ertl, L.S.; McMahon, J.P.; Wang, Y.; Yau, S.; Mali, V.R.; Chhina, V.; Kumamoto, A.; Liu, S.; et al. Efficacy of Chemokine Receptor Inhibition in Treating IL-36α-Induced Psoriasiform Inflammation. J. Immunol. 2019, 202, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, H.; Dinh, H.; Trueblood, E.S.; Pretorius, J.; Kugler, D.; Weng, N.; Kanaly, S.T.; Towne, J.E.; Willis, C.R.; Kuechle, M.K.; et al. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J. Exp. Med. 2007, 204, 2603–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milora, K.A.; Fu, H.; Dubaz, O.; Jensen, L.E. Unprocessed Interleukin-36α Regulates Psoriasis-Like Skin Inflammation in Cooperation with Interleukin-1. J. Investig. Dermatol. 2015, 135, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Ahn, R.; Yan, D.; Chang, H.W.; Lee, K.; Bhattarai, S.; Huang, Z.M.; Nakamura, M.; Singh, R.; Afifi, L.; Taravati, K.; et al. RNA-seq and flow-cytometry of conventional, scalp, and palmoplantar psoriasis reveal shared and distinct molecular pathways. Sci. Rep. 2018, 27, 11368. [Google Scholar] [CrossRef]
- Astorri, E.; Nerviani, A.; Bombardieri, M.; Pitzalis, C. Towards a stratified targeted approach with biologic treatments in rheumatoid arthritis: Role of synovial pathobiology. Curr. Pharm. Des. 2015, 21, 2216–2224. [Google Scholar] [CrossRef]
- Germán, B.; Wei, R.; Hener, P.; Martins, C.; Ye, T.; Gottwick, C.; Yang, J.; Seneschal, J.; Boniface, K.; Li, M. Disrupting the IL-36 and IL-23/IL-17 loop underlies the efficacy of calcipotriol and corticosteroid therapy for psoriasis. JCI Insight 2019, 24, 4. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, G.P.; Henry, C.M.; Clancy, D.M.; Mametnabiev, T.; Belotcerkovskaya, E.; Davidovich, P.; Sura-Trueba, S.; Garabadzhiu, A.V.; Martin, S.J. Suppressing IL-36-driven inflammation using peptide pseudosubstrates for neutrophil proteases. Cell Death Dis. 2018, 9, 378. [Google Scholar] [CrossRef]
- Gresnigt, M.S.; Rösler, B.; Jacobs, C.W.; Becker, K.L.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Dinarello, C.A.; van de Veerdonk, F.L. The IL-36 receptor pathway regulates Aspergillus fumigatus-induced Th1 and Th17 responses. Eur. J. Immunol. 2013, 43, 416–426. [Google Scholar] [CrossRef]
- Lockwood, S.J.; Prens, L.M.; Kimball, A.B. Book Adverse Reactions to Biologics in Psoriasis; Karger Publishers: Basel, Switzerland, 2018; Volume 53, pp. 1–14. [Google Scholar]



{kind=link}
{kind=link}
| IL-36 Members | Expression | Induction | Dow-Regulation | Refs |
|---|---|---|---|---|
| IL-36α (IL-1F6) | Keratinocytes, endothelial cells, macrophages, dendritic cells, Langerhans cells | IL-17A, TNF-α, IL-22 | [13,17,18,49] | |
| IL-36β (IL-1F8) | Endothelial cells | IL-36β, IL-17A, TNF-α, IL-22, IL-1β, LPS | [13,17,18,49] | |
| IL-36γ (IL-1F9) | Keratinocytes, endothelial cells, macrophages, dendritic cells, Langerhans cells | IL-17A, TNF-α, IL-36γ, TLR3, LPS, NETs | [13,17,18,49,50] | |
| IL-36Ra (IL-1F10) | Keratinocytes | IL-17A, TNF-α, IL.36γ | [13,17,18,49] | |
| IL-38 (IL-1F5) | Keratinocytes | IL-17A, TNF-α IL-22, IL-36γ, IFN-γ | [13,34] | |
| IL-36R complex | Keratinocyte, fibroblasts, endothelial cells, macrophages, dendritic cells, Langerhans cells | [13,14,19,21,23,53,54,55] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madonna, S.; Girolomoni, G.; Dinarello, C.A.; Albanesi, C. The Significance of IL-36 Hyperactivation and IL-36R Targeting in Psoriasis. Int. J. Mol. Sci. 2019, 20, 3318. https://doi.org/10.3390/ijms20133318
Madonna S, Girolomoni G, Dinarello CA, Albanesi C. The Significance of IL-36 Hyperactivation and IL-36R Targeting in Psoriasis. International Journal of Molecular Sciences. 2019; 20(13):3318. https://doi.org/10.3390/ijms20133318
Chicago/Turabian StyleMadonna, Stefania, Giampiero Girolomoni, Charles A. Dinarello, and Cristina Albanesi. 2019. "The Significance of IL-36 Hyperactivation and IL-36R Targeting in Psoriasis" International Journal of Molecular Sciences 20, no. 13: 3318. https://doi.org/10.3390/ijms20133318
APA StyleMadonna, S., Girolomoni, G., Dinarello, C. A., & Albanesi, C. (2019). The Significance of IL-36 Hyperactivation and IL-36R Targeting in Psoriasis. International Journal of Molecular Sciences, 20(13), 3318. https://doi.org/10.3390/ijms20133318