The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
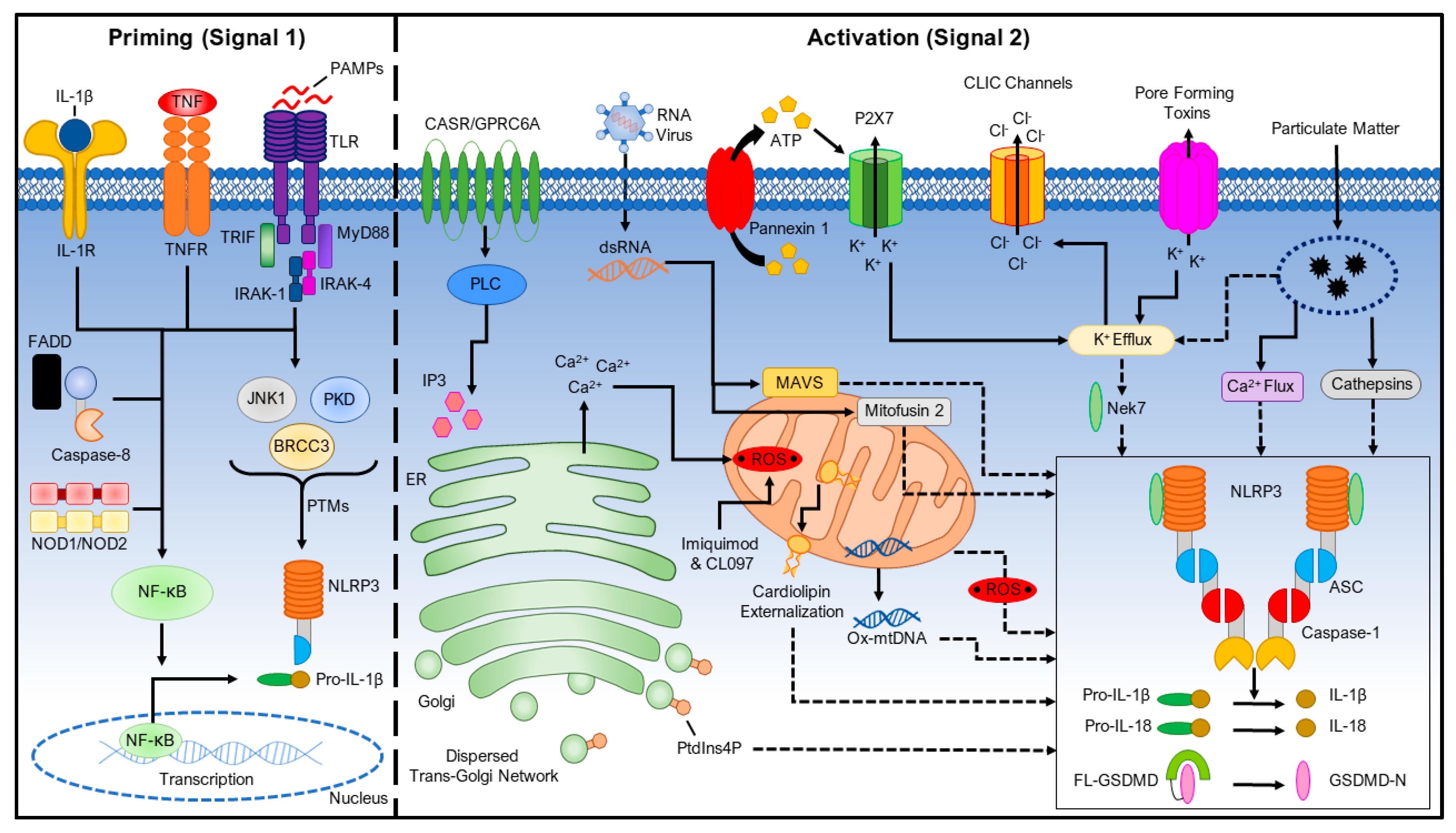
2. Priming the NLRP3 Inflammasome (Signal 1)
3. Activating the NLRP3 Inflammasome (Signal 2)
3.1. Ionic Flux
3.1.1. K+ Efflux
3.1.2. Ca2+ Mobilization
3.1.3. Na+ Influx and Cl− Efflux
3.2. Reactive Oxygen Species (ROS) and Mitochondrial Dysfunction
3.3. Lysosomal Damage
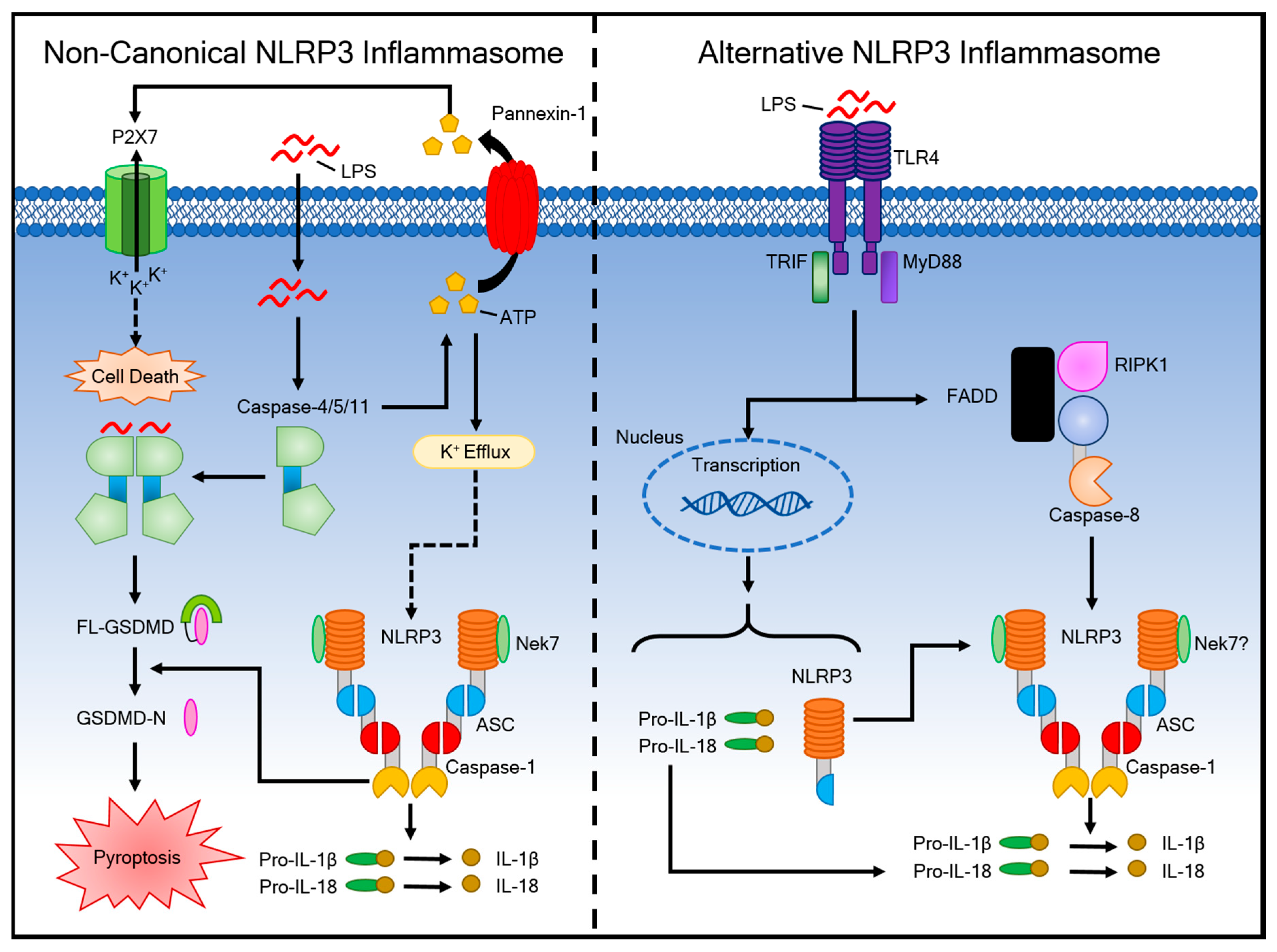
4. Activation of the Non-Canonical Inflammasome Pathway and Alternative Inflammasome Pathway
4.1. The Non-Canonical Inflammasome Pathway
4.2. The Alternative Inflammasome Pathway
5. Regulation of the NLRP3 Inflammasome
5.1. Regulation by Post-Translational Modifications of NLRP3
5.1.1. Ubiquitination
5.1.2. Phosphorylation
5.1.3. Other Post-Translational Modifications
5.2. Regulation by NLRP3 Interacting Partners
6. Concluding Remarks and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PRR | Pattern-recognition receptor |
| PAMP | Pathogen-associated molecular pattern |
| DAMP | Damage-associated molecular pattern |
| PYD | Pyrin domain |
| NOD | Nucleotide-binding oligomerization domain |
| LRR | Leucine-rich repeat |
| NLR | Nod-like receptor |
| NLRP | Nod-like receptor protein |
| NLRP3 | NLR family pyrin domain containing 3 |
| NLRC4 | NLR family CARD domain-containing protein 4 |
| AIM2 | Absent-in-melanoma 2 |
| ASC | Apoptosis-associated speck-like protein containing a caspase-recruitment domain |
| IL | Interleukin |
| IFN | Interferon |
| GSDMD | Gasdermin D |
| CAPS | Cryopyrin-associated periodic syndromes |
| IL-1R | IL-1 receptor |
| MCC950 | 1-(1,2,3,5,6,7-Hexahydro-s-indacen-4-yl)-3-[4-(2-hydroxypropan-2-yl)furan-2-yl]sulfonylurea |
| PtdIns4 | Phosphatidylinositol-4-phosphate |
| ATP | Adenosine triphosphate |
| TLR | Toll-like receptors |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| MYD88 | Myeloid differentiation primary response 88 |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
| FADD | Fas-associated protein with death domain |
| LPS | Lipopolysaccharide |
| IRAK | IL-1 receptor-associated kinase |
| IKK | Inhibitor of NF-κB kinase |
| BRCC | BRCA1-BRCA2-containing complex |
| JNK | c-Jun N-terminal kinase |
| IRF1 | Interferon regulatory transcription factor 1 |
| mtDNA | Mitochondrial DNA |
| PLC | Phospholipase C |
| CL097 | 2-(ethoxymethyl)-3H-imidazo[4,5-c]quinolin-4-amine |
| BAPTA-AM | 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| GPCR | G protein-coupled receptor |
| CaSR | Calcium-sensing receptor |
| GPRC6A | G protein-coupled receptor family C group 6 member A |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| IP3 | Inositol 1,4,5-triphosphate |
| ER | Endoplasmic reticulum |
| IP3R | IP3 receptor |
| 2APB | 2-aminoethoxy diphenylborinate |
| P2RX7 | P2X purinoceptor 7 |
| TRPM | Transient receptor potential ion melastatin |
| TRPV | Transient receptor potential ion vanilloid |
| TXNIP | Thioredoxin interacting protein |
| MSU | Monosodium urate |
| VRAC | Volume-related anion channel |
| CLIC | Chloride intracellular channel |
| ROS | Reactive oxygen species |
| NOX2 | NADPH oxidase 2 |
| NOX4 | NADPH oxidase 4 |
| CPT1A | Carnitine palmitoyltransferase 1A |
| mtROS | Mitochondrial ROS |
| MAVS | Mitochondrial antiviral-signaling protein |
| TRAF3 | TNF receptor associated factor 3 |
| oxPAPC | Oxidized phospholipid 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine |
| GBP2 | Guanylate-binding protein 2 |
| IRGB10 | Immunity-related GTPase family member b10 |
| RIPK1 | Receptor-interacting serine/threonine-protein kinase 1 |
| CASP8 | Caspase-8 |
| DUB | Deubiquitinating enzyme |
| SCF | Skp-Cullin-F box |
| FBXL2 | F-box L2 |
| FBXO3 | F-box O3 |
| MARCH7 | Membrane-associated RING-CH protein VII |
| DRD1 | Dopamine D1 receptor |
| TRIM31 | Tripartite Motif Containing 31 |
| ARIH2 | Ariadne homolog 2 |
| USP | Ubiquitin specific peptidase |
| PKA | Protein kinase A |
| cAMP | Cyclic AMP |
| MAM | Mitochondria-associated membrane |
| DAG | Diacylglycerol |
| PTPN22 | Protein tyrosine phosphatase, non-receptor type 22 |
| PP2A | Phosphotase 2A |
| MAPL | Mitochondrial-anchored protein ligase |
| SENP | Sentrin/SUMO-specific protease |
| HSP90 | Heat shock protein 90 |
| SGT1 | Suppressor of G2 allele of SKP1 |
| TXNIP | Thioredoxin-interacting protein |
| GBP5 | Guanylate-binding protein 5 |
| PKR | Double-stranded RNA-dependent protein kinase |
| MIF | Migration inhibitory factor |
| MARK4 | Microtubule-affinity regulating kinase 4 |
| Nek7 | NIMA-related kinase 7 |
References
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The Inflammasome: A Caspase-1 Activation Platform Regulating Immune Responses and Disease Pathogenesis. Nat. Immunol. 2009, 10, 241. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kanneganti, T.-D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and Functions of Inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Dorfleutner, A.; Bryan, N.B.; Yun, C.; Radian, A.D.; de Almeida, L.; Rojanasakul, Y.; Stehlik, C. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 2012, 36, 464–476. [Google Scholar] [CrossRef]
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121. [Google Scholar] [CrossRef]
- Vladimer, G.I.; Weng, D.; Paquette, S.W.M.; Vanaja, S.K.; Rathinam, V.A.K.; Aune, M.H.; Conlon, J.E.; Burbage, J.J.; Proulx, M.K.; Liu, Q.; et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 2012, 37, 96–107. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.-W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef]
- Manji, G.A.; Wang, L.; Geddes, B.J.; Brown, M.; Merriam, S.; Al-Garawi, A.; Mak, S.; Lora, J.M.; Briskin, M.; Jurman, M.; et al. PYPAF1, a PYRIN-containing Apaf1-like protein that assembles with ASC and regulates activation of NF-kappa B. J. Biol. Chem. 2002, 277, 11570–11575. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Warner, N.; Viani, K.; Nuñez, G. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 2009, 227, 106–128. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- He, W.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Miao, E.A.; Leaf, I.A.; Treuting, P.M.; Mao, D.P.; Dors, M.; Sarkar, A.; Warren, S.E.; Wewers, M.D.; Aderem, A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 2010, 11, 1136–1142. [Google Scholar] [CrossRef]
- Thomas, P.G.; Dash, P.; Aldridge, J.R.; Ellebedy, A.H.; Reynolds, C.; Funk, A.J.; Martin, W.J.; Lamkanfi, M.; Webby, R.J.; Boyd, K.L.; et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 2009, 30, 566–575. [Google Scholar] [CrossRef]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P.-Y. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Poeck, H.; Bscheider, M.; Dostert, C.; Hannesschläger, N.; Endres, S.; Hartmann, G.; Tardivel, A.; Schweighoffer, E.; Tybulewicz, V.; et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 2009, 459, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.-D.; Body-Malapel, M.; Amer, A.; Park, J.-H.; Whitfield, J.; Franchi, L.; Taraporewala, Z.F.; Miller, D.; Patton, J.T.; Inohara, N.; et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 2006, 281, 36560–36568. [Google Scholar] [CrossRef] [PubMed]
- Menu, P.; Vince, J.E. The NLRP3 inflammasome in health and disease: The good, the bad and the ugly. Clin. Exp. Immunol. 2011, 166, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Vajjhala, P.R.; Mirams, R.E.; Hill, J.M. Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J. Biol. Chem. 2012, 287, 41732–41743. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.A.; Bergstralh, D.T.; Wang, Y.; Willingham, S.B.; Ye, Z.; Zimmermann, A.G.; Ting, J.P.-Y. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 8041–8046. [Google Scholar] [CrossRef] [Green Version]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Tapia-Abellán, A.; Angosto-Bazarra, D.; Martínez-Banaclocha, H.; Torre-Minguela, C.d.; Cerón-Carrasco, J.P.; Pérez-Sánchez, H.; Arostegui, J.I.; Pelegrin, P. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 2019, 15, 560. [Google Scholar] [CrossRef]
- Hafner-Bratkovič, I.; Sušjan, P.; Lainšček, D.; Tapia-Abellán, A.; Cerović, K.; Kadunc, L.; Angosto-Bazarra, D.; Pelegrίn, P.; Jerala, R. NLRP3 lacking the leucine-rich repeat domain can be fully activated via the canonical inflammasome pathway. Nat. Commun. 2018, 9, 5182. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Kanneganti, T.-D. Nlrp3: An immune sensor of cellular stress and infection. Int. J. Biochem. Cell Biol. 2010, 42, 792–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting Edge: NF-κB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Núñez, G. Cutting Edge: TNF-α Mediates Sensitization to ATP and Silica via the NLRP3 Inflammasome in the Absence of Microbial Stimulation. J. Immunol. 2009, 183, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.S.; Walle, L.V.; Opdenbosch, N.V.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.-D. FADD and Caspase-8 Mediate Priming and Activation of the Canonical and Noncanonical Nlrp3 Inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allam, R.; Lawlor, K.E.; Yu, E.C.-W.; Mildenhall, A.L.; Moujalled, D.M.; Lewis, R.S.; Ke, F.; Mason, K.D.; White, M.J.; Stacey, K.J.; et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 2014, 15, 982–990. [Google Scholar] [CrossRef]
- Lemmers, B.; Salmena, L.; Bidère, N.; Su, H.; Matysiak-Zablocki, E.; Murakami, K.; Ohashi, P.S.; Jurisicova, A.; Lenardo, M.; Hakem, R.; et al. Essential Role for Caspase-8 in Toll-like Receptors and NFκB Signaling. J. Biol. Chem. 2007, 282, 7416–7423. [Google Scholar] [CrossRef]
- Ranjan, K.; Pathak, C. FADD regulates NF-κB activation and promotes ubiquitination of cFLIPL to induce apoptosis. Sci. Rep. 2016, 6, 22787. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional Priming and Deubiquitination Regulate NLRP3 Inflammasome Activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Sagulenko, V.; Zamoshnikova, A.; Richards, A.A.; Cridland, J.A.; Irvine, K.M.; Stacey, K.J.; Sweet, M.J. Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology 2012, 217, 1325–1329. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.-M.; Hu, W.; Troutman, T.D.; Jennings, M.; Brewer, T.; Li, X.; Nanda, S.; Cohen, P.; Thomas, J.A.; Pasare, C. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc. Natl. Acad. Sci. USA 2014, 111, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Kang, S.; Anderson, C.; Sagara, J.; Fitzgerald, K.A.; Alnemri, E.S. Toll-Like Receptor Signaling Licenses IRAK1 For Rapid Activation Of The NLRP3 Inflammasome. J. Immunol. Baltim. Md 1950 2013, 191, 3995–3999. [Google Scholar]
- Kim, S.-J.; Cha, J.-Y.; Kang, H.S.; Lee, J.-H.; Lee, J.Y.; Park, J.-H.; Bae, J.-H.; Song, D.-K.; Im, S.-S. Corosolic acid ameliorates acute inflammation through inhibition of IRAK-1 phosphorylation in macrophages. BMB Rep. 2016, 49, 276–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Castejon, G.; Luheshi, N.M.; Compan, V.; High, S.; Whitehead, R.C.; Flitsch, S.; Kirov, A.; Prudovsky, I.; Swanton, E.; Brough, D. Deubiquitinases regulate the activity of caspase-1 and interleukin-1β secretion via assembly of the inflammasome. J. Biol. Chem. 2013, 288, 2721–2733. [Google Scholar] [CrossRef] [PubMed]
- Py, B.F.; Kim, M.-S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Liu, Z.-S.; Xue, W.; Bai, Z.-F.; Wang, Q.-Y.; Dai, J.; Liu, X.; Huang, Y.-J.; Cai, H.; Zhan, X.-Y.; et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol. Cell 2017, 68, 185–197.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Silveira, A.A.; Cunningham, C.; Corr, E.; Ferreira, W.A.; Costa, F.F.; Almeida, C.B.; Conran, N.; Dunne, A. Heme Induces NLRP3 Inflammasome Formation in Primary Human Macrophages and May Propagate Hemolytic Inflammatory Processes By Inducing S100A8 Expression. Blood 2016, 128, 1256. [Google Scholar]
- Erdei, J.; Tóth, A.; Balogh, E.; Nyakundi, B.B.; Bányai, E.; Ryffel, B.; Paragh, G.; Cordero, M.D.; Jeney, V. Induction of NLRP3 Inflammasome Activation by Heme in Human Endothelial Cells. Oxid. Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Pétrilli, V.; Bruggen, R.V.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate Immune Activation Through Nalp3 Inflammasome Sensing of Asbestos and Silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eigenbrod, T.; Dalpke, A.H. Bacterial RNA: An Underestimated Stimulus for Innate Immune Responses. J. Immunol. 2015, 195, 411–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Ghosh, S.; Monks, B.; DeOliveira, R.B.; Tzeng, T.-C.; Kalantari, P.; Nandy, A.; Bhattacharjee, B.; Chan, J.; Ferreira, F.; et al. RNA and β-hemolysin of group B Streptococcus induce interleukin-1β (IL-1β) by activating NLRP3 inflammasomes in mouse macrophages. J. Biol. Chem. 2014, 289, 13701–13705. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.-D.; Ozören, N.; Body-Malapel, M.; Amer, A.; Park, J.-H.; Franchi, L.; Whitfield, J.; Barchet, W.; Colonna, M.; Vandenabeele, P.; et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 2006, 440, 233–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, W.; Mitoma, H.; Hanabuchi, S.; Bao, M.; Weng, L.; Sugimoto, N.; Liu, Y.; Zhang, Z.; Zhong, J.; Sun, B.; et al. Human NLRP3 inflammasome senses multiple types of bacterial RNAs. Proc. Natl. Acad. Sci. USA 2014, 111, 16059–16064. [Google Scholar] [CrossRef] [Green Version]
- Greaney, A.J.; Leppla, S.H.; Moayeri, M. Bacterial Exotoxins and the Inflammasome. Front. Immunol. 2015, 6, 570. [Google Scholar] [CrossRef]
- Lee, M.-S.; Kwon, H.; Lee, E.-Y.; Kim, D.-J.; Park, J.-H.; Tesh, V.L.; Oh, T.-K.; Kim, M.H. Shiga Toxins Activate the NLRP3 Inflammasome Pathway To Promote Both Production of the Proinflammatory Cytokine Interleukin-1β and Apoptotic Cell Death. Infect. Immun. 2016, 84, 172–186. [Google Scholar] [CrossRef]
- Kasper, L.; König, A.; Koenig, P.-A.; Gresnigt, M.S.; Westman, J.; Drummond, R.A.; Lionakis, M.S.; Groß, O.; Ruland, J.; Naglik, J.R.; et al. The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nat. Commun. 2018, 9, 4260. [Google Scholar] [CrossRef]
- Rogiers, O.; Frising, U.C.; Kucharíková, S.; Jabra-Rizk, M.A.; Loo, G.v.; Dijck, P.V.; Wullaert, A. Candidalysin Crucially Contributes to Nlrp3 Inflammasome Activation by Candida albicans Hyphae. mBio 2019, 10, e02221-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skeldon, A.; Saleh, M. The Inflammasomes: Molecular Effectors of Host Resistance Against Bacterial, Viral, Parasitic, and Fungal Infections. Front. Microbiol. 2011, 2, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, A.; Feng, S.; Hayward, J.A.; Ngo, C.; Fox, D.; Atmosukarto, I.I.; Price, J.D.; Schauer, K.; Märtlbauer, E.; Robertson, A.A.B.; et al. A multicomponent toxin from Bacillus cereus incites inflammation and shapes host outcome via the NLRP3 inflammasome. Nat. Microbiol. 2019, 4, 362. [Google Scholar] [CrossRef] [PubMed]
- Perregaux, D.; Gabel, C.A. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem. 1994, 269, 15195–15203. [Google Scholar] [PubMed]
- Walev, I.; Klein, J.; Husmann, M.; Valeva, A.; Strauch, S.; Wirtz, H.; Weichel, O.; Bhakdi, S. Potassium Regulates IL-1β Processing Via Calcium-Independent Phospholipase A2. J. Immunol. 2000, 164, 5120–5124. [Google Scholar] [CrossRef]
- Walev, I.; Reske, K.; Palmer, M.; Valeva, A.; Bhakdi, S. Potassium-inhibited processing of IL-1 beta in human monocytes. EMBO J. 1995, 14, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Pétrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef]
- Rühl, S.; Broz, P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur. J. Immunol. 2015, 45, 2927–2936. [Google Scholar] [CrossRef]
- Schmid-Burgk, J.L.; Gaidt, M.M.; Schmidt, T.; Ebert, T.S.; Bartok, E.; Hornung, V. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur. J. Immunol. 2015, 45, 2911–2917. [Google Scholar] [CrossRef]
- Yang, D.; He, Y.; Muñoz-Planillo, R.; Liu, Q.; Núñez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groß, C.J.; Mishra, R.; Schneider, K.S.; Médard, G.; Wettmarshausen, J.; Dittlein, D.C.; Shi, H.; Gorka, O.; Koenig, P.-A.; Fromm, S.; et al. K + Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 2016, 45, 761–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanman, L.E.; Qian, Y.; Eisele, N.A.; Ng, T.M.; van der Linden, W.A.; Monack, D.M.; Weerapana, E.; Bogyo, M. Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. eLife 2016, 5, e13663. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Zhang, F.; Fuss, I.; Kitani, A.; Strober, W. A Mutation in the Nlrp3 Gene Causing Inflammasome Hyperactivation Potentiates Th17 Cell-Dominant Immune Responses. Immunity 2009, 30, 860–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Brough, D.; Feuvre, R.A.L.; Wheeler, R.D.; Solovyova, N.; Hilfiker, S.; Rothwell, N.J.; Verkhratsky, A. Ca2+ Stores and Ca2+ Entry Differentially Contribute to the Release of IL-1β and IL-1α from Murine Macrophages. J. Immunol. 2003, 170, 3029–3036. [Google Scholar] [CrossRef] [PubMed]
- Feldmeyer, L.; Keller, M.; Niklaus, G.; Hohl, D.; Werner, S.; Beer, H.-D. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr. Biol. CB 2007, 17, 1140–1145. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Thomas, L.M.; Watkins, S.C.; Franchi, L.; Núñez, G.; Salter, R.D. Cholesterol-dependent cytolysins induce rapid release of mature IL-1β from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. J. Leukoc. Biol. 2009, 86, 1227–1238. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.-S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Katsnelson, M.A.; Rucker, L.G.; Russo, H.M.; Dubyak, G.R. K+ Efflux Agonists Induce NLRP3 Inflammasome Activation Independently of Ca2+ Signaling. J. Immunol. 2015, 194, 3937–3952. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.G.; Rivers-Auty, J.; Daniels, M.J.D.; White, C.S.; Schwalbe, C.H.; Schilling, T.; Hammadi, H.; Jaiyong, P.; Spencer, N.G.; England, H.; et al. Boron-Based Inhibitors of the NLRP3 Inflammasome. Cell Chem. Biol. 2017, 24, 1321–1335.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compan, V.; Baroja-Mazo, A.; López-Castejón, G.; Gomez, A.I.; Martínez, C.M.; Angosto, D.; Montero, M.T.; Herranz, A.S.; Bazán, E.; Reimers, D.; et al. Cell Volume Regulation Modulates NLRP3 Inflammasome Activation. Immunity 2012, 37, 487–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Zhai, Y.; Liang, S.; Mori, Y.; Han, R.; Sutterwala, F.S.; Qiao, L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 2013, 4, 1611. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Schilling, J.D. Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J. Biol. Chem. 2014, 289, 9158–9171. [Google Scholar] [CrossRef] [PubMed]
- Schorn, C.; Frey, B.; Lauber, K.; Janko, C.; Strysio, M.; Keppeler, H.; Gaipl, U.S.; Voll, R.E.; Springer, E.; Munoz, L.E.; et al. Sodium overload and water influx activate the NALP3 inflammasome. J. Biol. Chem. 2011, 286, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, P.A.; Kertesy, S.B.; Lundberg, K.; Kahlenberg, J.M.; Dubyak, G.R. Inhibitory effects of chloride on the activation of caspase-1, IL-1beta secretion, and cytolysis by the P2X7 receptor. J. Immunol. Baltim. Md 1950 2005, 175, 7623–7634. [Google Scholar]
- Perregaux, D.G.; Laliberte, R.E.; Gabel, C.A. Human monocyte interleukin-1beta posttranslational processing. Evidence of a volume-regulated response. J. Biol. Chem. 1996, 271, 29830–29838. [Google Scholar] [CrossRef]
- Tang, T.; Lang, X.; Xu, C.; Wang, X.; Gong, T.; Yang, Y.; Cui, J.; Bai, L.; Wang, J.; Jiang, W.; et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat. Commun. 2017, 8, 202. [Google Scholar] [CrossRef]
- Daniels, M.J.D.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 2016, 7, 12504. [Google Scholar] [CrossRef]
- Domingo-Fernández, R.; Coll, R.C.; Kearney, J.; Breit, S.; O’Neill, L.A.J. The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1β transcription and activate the NLRP3 inflammasome. J. Biol. Chem. 2017, 292, 12077–12087. [Google Scholar] [CrossRef] [PubMed]
- Green, J.P.; Yu, S.; Martín-Sánchez, F.; Pelegrin, P.; Lopez-Castejon, G.; Lawrence, C.B.; Brough, D. Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc. Natl. Acad. Sci. USA 2018, 115, E9371–E9380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.M.; Persechini, P.M.; Ojcius, D.M. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef] [PubMed]
- van Bruggen, R.; Köker, M.Y.; Jansen, M.; van Houdt, M.; Roos, D.; Kuijpers, T.W.; van den Berg, T.K. Human NLRP3 inflammasome activation is Nox1-4 independent. Blood 2010, 115, 5398–5400. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.W.; Wang, J.; Dhandapani, K.M.; Brann, D.W. NADPH Oxidase 2 Regulates NLRP3 Inflammasome Activation in the Brain after Traumatic Brain Injury. Oxid. Med. Cell. Longev. 2017, 2017, 6057609. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-S.; Nakahira, K.; Chung, K.-P.; DeNicola, G.M.; Koo, M.J.; Pabón, M.A.; Rooney, K.T.; Yoon, J.-H.; Ryter, S.W.; Stout-Delgado, H.; et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat. Med. 2016, 22, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.; Bartok, E.; Rieger, A.; Franchi, L.; Núñez, G.; Hornung, V. Cutting edge: Reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. Baltim. Md 1950 2011, 187, 613–617. [Google Scholar] [CrossRef]
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; Yu, J.-W.; Alnemri, E.S. The mitochondrial anti-viral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. Baltim. Md 1950 2013, 191, 4358–4366. [Google Scholar]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The Adaptor MAVS Promotes NLRP3 Mitochondrial Localization and Inflammasome Activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ermler, M.E.; Traylor, Z.; Patel, K.; Schattgen, S.A.; Vanaja, S.K.; Fitzgerald, K.A.; Hise, A.G. Rift Valley fever virus infection induces activation of the NLRP3 inflammasome. Virology 2014, 449, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Ozkurede, U.; Kim, Y.-G.; Chakrabarti, A.; Gale, M.; Silverman, R.H.; Colonna, M.; Akira, S.; et al. Cytosolic Double-Stranded RNA Activates the NLRP3 Inflammasome via MAVS-Induced Membrane Permeabilization and K+ Efflux. J. Immunol. 2014, 193, 4214–4222. [Google Scholar] [CrossRef] [PubMed]
- Guan, K.; Wei, C.; Zheng, Z.; Song, T.; Wu, F.; Zhang, Y.; Cao, Y.; Ma, S.; Chen, W.; Xu, Q.; et al. MAVS Promotes Inflammasome Activation by Targeting ASC for K63-Linked Ubiquitination via the E3 Ligase TRAF3. J. Immunol. 2015, 194, 4880–4890. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Yamazaki, T.; Koshiba, T.; Yanagi, Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. USA 2013, 110, 17963–17968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Elliott, E.I.; Miller, A.N.; Banoth, B.; Iyer, S.S.; Stotland, A.; Weiss, J.P.; Gottlieb, R.A.; Sutterwala, F.S.; Cassel, S.L. Cutting Edge: Mitochondrial Assembly of the NLRP3 Inflammasome Complex Is Initiated at Priming. J. Immunol. 2018, 200, 3047–3052. [Google Scholar] [CrossRef] [Green Version]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, C.; Mao, K.; Chen, S.; Meng, G.; Sun, B. Cellular localization of NLRP3 inflammasome. Protein Cell 2013, 4, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Chen, Z.J. PtdIns4P on dispersed trans -Golgi network mediates NLRP3 inflammasome activation. Nature 2018, 564, 71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Meszaros, G.; He, W.; Xu, Y.; Magliarelli, H.d.F.; Mailly, L.; Mihlan, M.; Liu, Y.; Gámez, M.P.; Goginashvili, A.; et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med. 2017, 214, 2671–2693. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassel, S.L.; Eisenbarth, S.C.; Iyer, S.S.; Sadler, J.J.; Colegio, O.R.; Tephly, L.A.; Carter, A.B.; Rothman, P.B.; Flavell, R.A.; Sutterwala, F.S. The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. USA 2008, 105, 9035–9040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Pétrilli, V.; De Smedt, T.; Rolaz, A.; Hammad, H.; van Nimwegen, M.; Bergen, I.M.; Castillo, R.; Lambrecht, B.N.; Tschopp, J. Cutting edge: Alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J. Immunol. Baltim. Md 1950 2008, 181, 3755–3759. [Google Scholar] [CrossRef] [PubMed]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of Inflammasome by Aggregated α–Synuclein, an Inflammatory Response in Synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Guarda, G.; Romero, J.F.; Menu, P.; Gross, O.; Tardivel, A.; Suva, M.-L.; Stehle, J.-C.; Kopf, M.; Stamenkovic, I.; et al. Malarial Hemozoin Is a Nalp3 Inflammasome Activating Danger Signal. PLoS ONE 2009, 4, e6510. [Google Scholar] [CrossRef]
- Orlowski, G.M.; Colbert, J.D.; Sharma, S.; Bogyo, M.; Robertson, S.A.; Rock, K.L. Multiple Cathepsins Promote Pro-IL-1β Synthesis and NLRP3-Mediated IL-1β Activation. J. Immunol. Baltim. Md 1950 2015, 195, 1685–1697. [Google Scholar] [CrossRef]
- Barlan, A.U.; Griffin, T.M.; McGuire, K.A.; Wiethoff, C.M. Adenovirus membrane penetration activates the NLRP3 inflammasome. J. Virol. 2011, 85, 146–155. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, A.; Karki, R.; Berwin, B.; Yamamoto, M.; Kanneganti, T.-D. Guanylate binding proteins facilitate caspase-11-dependent pyroptosis in response to type 3 secretion system-negative Pseudomonas aeruginosa. Cell Death Discov. 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.J.; Boucher, D.; Bierschenk, D.; Tebartz, C.; Whitney, P.G.; D’Silva, D.B.; Tanzer, M.C.; Monteleone, M.; Robertson, A.A.B.; Cooper, M.A.; et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur. J. Immunol. 2015, 45, 2918–2926. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Broz, P.; Ruby, T.; Belhocine, K.; Bouley, D.M.; Kayagaki, N.; Dixit, V.M.; Monack, D.M. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 2012, 490, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Pelegrin, P.; Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006, 25, 5071–5082. [Google Scholar] [CrossRef]
- Piccini, A.; Carta, S.; Tassi, S.; Lasiglié, D.; Fossati, G.; Rubartelli, A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1β and IL-18 secretion in an autocrine way. Proc. Natl. Acad. Sci. USA 2008, 105, 8067–8072. [Google Scholar] [CrossRef]
- Alves, L.A.; de Melo Reis, R.A.; de Souza, C.A.M.; de Freitas, M.S.; Teixeira, P.C.N.; Neto Moreira Ferreira, D.; Xavier, R.F. The P2X7 receptor: Shifting from a low- to a high-conductance channel—An enigmatic phenomenon? Biochim. Biophys. Acta BBA-Biomembr. 2014, 1838, 2578–2587. [Google Scholar] [CrossRef]
- Chu, L.H.; Indramohan, M.; Ratsimandresy, R.A.; Gangopadhyay, A.; Morris, E.P.; Monack, D.M.; Dorfleutner, A.; Stehlik, C. The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nat. Commun. 2018, 9, 996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, S.M.; Karki, R.; Sasai, M.; Place, D.E.; Kesavardhana, S.; Temirov, J.; Frase, S.; Zhu, Q.; Malireddi, R.K.S.; Kuriakose, T.; et al. IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell 2016, 167, 382–396.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meunier, E.; Dick, M.S.; Dreier, R.F.; Schürmann, N.; Kenzelmann Broz, D.; Warming, S.; Roose-Girma, M.; Bumann, D.; Kayagaki, N.; Takeda, K.; et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 2014, 509, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Nold-Petry, C.A.; Nold, M.F.; Joosten, L.A.B.; Opitz, B.; van der Meer, J.H.M.; van de Veerdonk, F.L.; Ferwerda, G.; Heinhuis, B.; Devesa, I.; et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 2009, 113, 2324–2335. [Google Scholar] [CrossRef] [PubMed]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.B.; Cooper, M.A.; Graf, T.; Hornung, V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Franchi, L.; Núñez, G. TLR Agonists Stimulate Nlrp3-Dependent IL-1β Production Independently of the Purinergic P2X7 Receptor in Dendritic Cells and In Vivo. J. Immunol. 2013, 190, 334–339. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Z.; Xiao, T.S. Post-translational regulation of inflammasomes. Cell. Mol. Immunol. 2017, 14, 65–79. [Google Scholar] [CrossRef]
- Han, S.; Lear, T.B.; Jerome, J.A.; Rajbhandari, S.; Snavely, C.A.; Gulick, D.L.; Gibson, K.F.; Zou, C.; Chen, B.B.; Mallampalli, R.K. Lipopolysaccharide Primes the NALP3 Inflammasome by Inhibiting Its Ubiquitination and Degradation Mediated by the SCFFBXL2 E3 Ligase. J. Biol. Chem. 2015, 290, 18124–18133. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine Controls Systemic Inflammation through Inhibition of NLRP3 Inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Liu, B.; Huai, W.; Yu, Z.; Wang, W.; Zhao, J.; Han, L.; Jiang, G.; Zhang, L.; Gao, C.; et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 2016, 7, 13727. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, A.; Karasawa, T.; Tago, K.; Kimura, H.; Kamata, R.; Usui-Kawanishi, F.; Watanabe, S.; Ohta, S.; Funakoshi-Tago, M.; Yanagisawa, K.; et al. ARIH2 Ubiquitinates NLRP3 and Negatively Regulates NLRP3 Inflammasome Activation in Macrophages. J. Immunol. 2017, 199, 3614–3622. [Google Scholar] [CrossRef] [PubMed]
- Humphries, F.; Bergin, R.; Jackson, R.; Delagic, N.; Wang, B.; Yang, S.; Dubois, A.V.; Ingram, R.J.; Moynagh, P.N. The E3 ubiquitin ligase Pellino2 mediates priming of the NLRP3 inflammasome. Nat. Commun. 2018, 9, 1560. [Google Scholar] [CrossRef] [PubMed]
- Palazón-Riquelme, P.; Worboys, J.D.; Green, J.; Valera, A.; Martín-Sánchez, F.; Pellegrini, C.; Brough, D.; López-Castejón, G. USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation. EMBO Rep. 2018, 19, e44766. [Google Scholar] [CrossRef] [PubMed]
- Sandall, C.F.; MacDonald, J.A. Effects of phosphorylation on the NLRP3 inflammasome. Arch. Biochem. Biophys. 2019. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, L.; Moreau, F.; MacDonald, J.A.; Chadee, K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat. Immunol. 2016, 17, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Spalinger, M.R.; Kasper, S.; Gottier, C.; Lang, S.; Atrott, K.; Vavricka, S.R.; Scharl, S.; Gutte, P.M.; Grütter, M.G.; Beer, H.-D.; et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J. Clin. Investig. 2016, 126, 1783–1800. [Google Scholar] [CrossRef] [Green Version]
- Stutz, A.; Kolbe, C.-C.; Stahl, R.; Horvath, G.L.; Franklin, B.S.; van Ray, O.; Brinkschulte, R.; Geyer, M.; Meissner, F.; Latz, E. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med. 2017, 214, 1725–1736. [Google Scholar] [CrossRef]
- Hernandez-Cuellar, E.; Tsuchiya, K.; Hara, H.; Fang, R.; Sakai, S.; Kawamura, I.; Akira, S.; Mitsuyama, M. Cutting edge: Nitric oxide inhibits the NLRP3 inflammasome. J. Immunol. Baltim. Md 1950 2012, 189, 5113–5117. [Google Scholar] [CrossRef]
- Mao, K.; Chen, S.; Chen, M.; Ma, Y.; Wang, Y.; Huang, B.; He, Z.; Zeng, Y.; Hu, Y.; Sun, S.; et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013, 23, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.B.; Rathinam, V.A.K.; Martens, G.W.; Martinot, A.J.; Kornfeld, H.; Fitzgerald, K.A.; Sassetti, C.M. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nat. Immunol. 2013, 14, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Barry, R.; John, S.W.; Liccardi, G.; Tenev, T.; Jaco, I.; Chen, C.-H.; Choi, J.; Kasperkiewicz, P.; Fernandes-Alnemri, T.; Alnemri, E.; et al. SUMO-mediated regulation of NLRP3 modulates inflammasome activity. Nat. Commun. 2018, 9, 3001. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Segovia, J.A.; Somarajan, S.R.; Chang, T.-H.; Kannan, T.R.; Baseman, J.B. ADP-Ribosylation of NLRP3 by Mycoplasma pneumoniae CARDS Toxin Regulates Inflammasome Activity. mBio 2014, 5, e02186-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayor, A.; Martinon, F.; De Smedt, T.; Pétrilli, V.; Tschopp, J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol. 2007, 8, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Piippo, N.; Korhonen, E.; Hytti, M.; Skottman, H.; Kinnunen, K.; Josifovska, N.; Petrovski, G.; Kaarniranta, K.; Kauppinen, A. Hsp90 inhibition as a means to inhibit activation of the NLRP3 inflammasome. Sci. Rep. 2018, 8, 6720. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Song, X.; Su, G.; Wang, Y.; Wang, Z.; Qing, S.; Jia, J.; Wang, Y.; Huang, L.; Zheng, K.; et al. AT-533, a Hsp90 inhibitor, attenuates HSV-1-induced inflammation. Biochem. Pharmacol. 2019, 166, 82–92. [Google Scholar] [CrossRef]
- Zuo, Y.; Wang, J.; Liao, F.; Yan, X.; Li, J.; Huang, L.; Liu, F. Inhibition of Heat Shock Protein 90 by 17-AAG Reduces Inflammation via P2X7 Receptor/NLRP3 Inflammasome Pathway and Increases Neurogenesis After Subarachnoid Hemorrhage in Mice. Front. Mol. Neurosci. 2018, 11, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef]
- Shenoy, A.R.; Wellington, D.A.; Kumar, P.; Kassa, H.; Booth, C.J.; Cresswell, P.; MacMicking, J.D. GBP5 Promotes NLRP3 Inflammasome Assembly and Immunity in Mammals. Science 2012, 336, 481–485. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Malireddi, R.K.S.; Neale, G.; Vogel, P.; Yamamoto, M.; Lamkanfi, M.; Kanneganti, T.-D. The transcription factor IRF1 and guanylate-binding proteins target AIM2 inflammasome activation by Francisella infection. Nat. Immunol. 2015, 16, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Meunier, E.; Wallet, P.; Dreier, R.F.; Costanzo, S.; Anton, L.; Rühl, S.; Dussurgey, S.; Dick, M.S.; Kistner, A.; Rigard, M.; et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat. Immunol. 2015, 16, 476–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundbäck, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Okamura, H.; Hiroshima, Y.; Abe, K.; Kido, J.-I.; Shinohara, Y.; Ozaki, K. PKR induces the expression of NLRP3 by regulating the NF-κB pathway in Porphyromonas gingivalis-infected osteoblasts. Exp. Cell Res. 2017, 354, 57–64. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Franchi, L.; Núñez, G. The protein kinase PKR is critical for LPS-induced iNOS production but dispensable for inflammasome activation in macrophages. Eur. J. Immunol. 2013, 43, 1147–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Thome, S.; Ma, X.; Amrute-Nayak, M.; Finigan, A.; Kitt, L.; Masters, L.; James, J.R.; Shi, Y.; Meng, G.; et al. MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule-dependent mechanism. Nat. Commun. 2017, 8, 15986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, T.; Lee, J.P.W.; Elgass, K.; Pinar, A.A.; Tate, M.D.; Aitken, E.H.; Fan, H.; Creed, S.J.; Deen, N.S.; Traore, D.A.K.; et al. Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat. Commun. 2018, 9, 2223. [Google Scholar] [CrossRef]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Núñez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [Green Version]
- Schmid-Burgk, J.L.; Chauhan, D.; Schmidt, T.; Ebert, T.S.; Reinhardt, J.; Endl, E.; Hornung, V. A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J. Biol. Chem. 2016, 291, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef]
- Salem, H.; Rachmin, I.; Yissachar, N.; Cohen, S.; Amiel, A.; Haffner, R.; Lavi, L.; Motro, B. Nek7 kinase targeting leads to early mortality, cytokinesis disturbance and polyploidy. Oncogene 2010, 29, 4046–4057. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. https://doi.org/10.3390/ijms20133328
Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. International Journal of Molecular Sciences. 2019; 20(13):3328. https://doi.org/10.3390/ijms20133328
Chicago/Turabian StyleKelley, Nathan, Devon Jeltema, Yanhui Duan, and Yuan He. 2019. "The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation" International Journal of Molecular Sciences 20, no. 13: 3328. https://doi.org/10.3390/ijms20133328
APA StyleKelley, N., Jeltema, D., Duan, Y., & He, Y. (2019). The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. International Journal of Molecular Sciences, 20(13), 3328. https://doi.org/10.3390/ijms20133328