An Ensemble Classifier to Predict Protein–Protein Interactions by Combining PSSM-based Evolutionary Information with Local Binary Pattern Model



Abstract
:1. Introduction
2. Results and Discussion
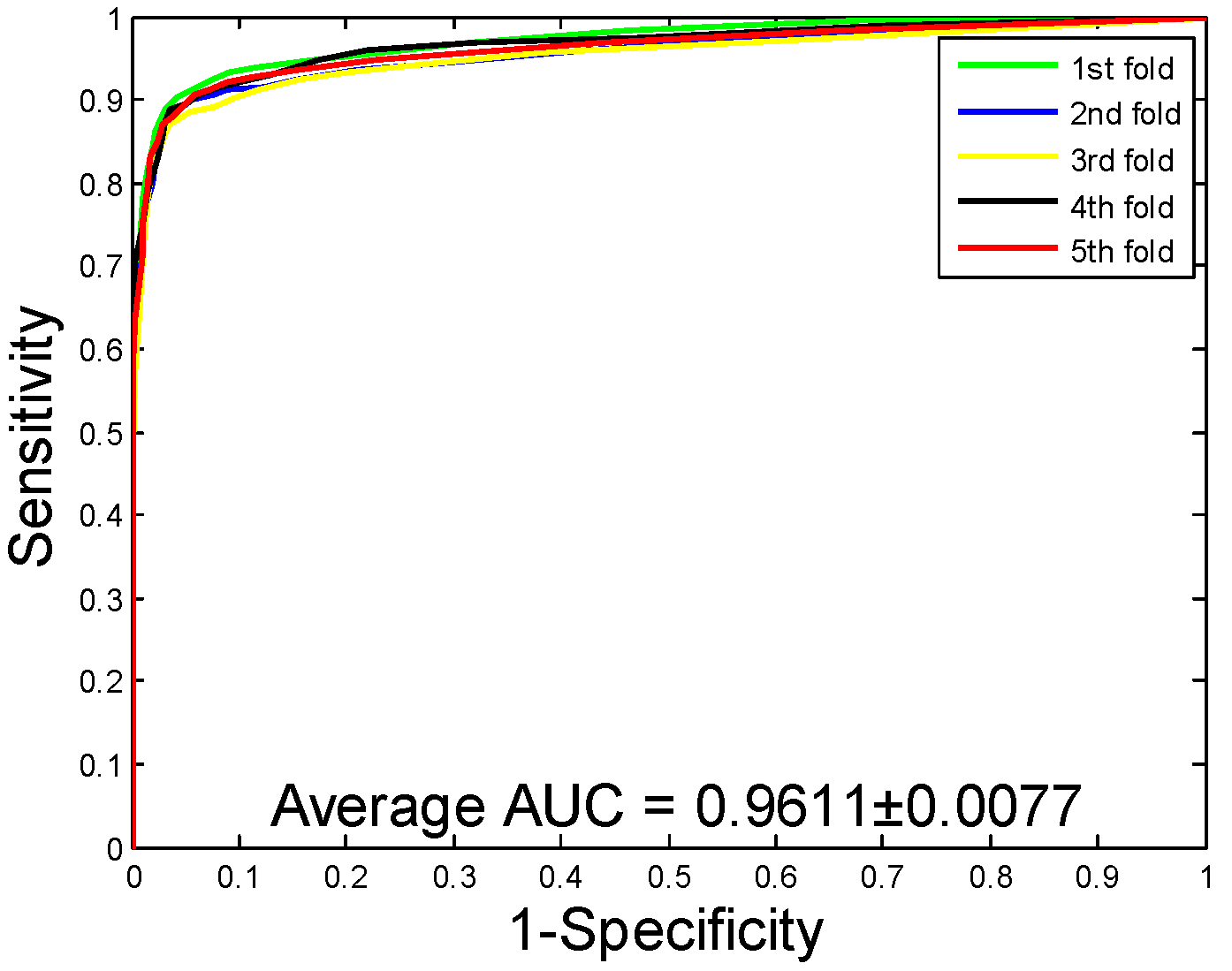
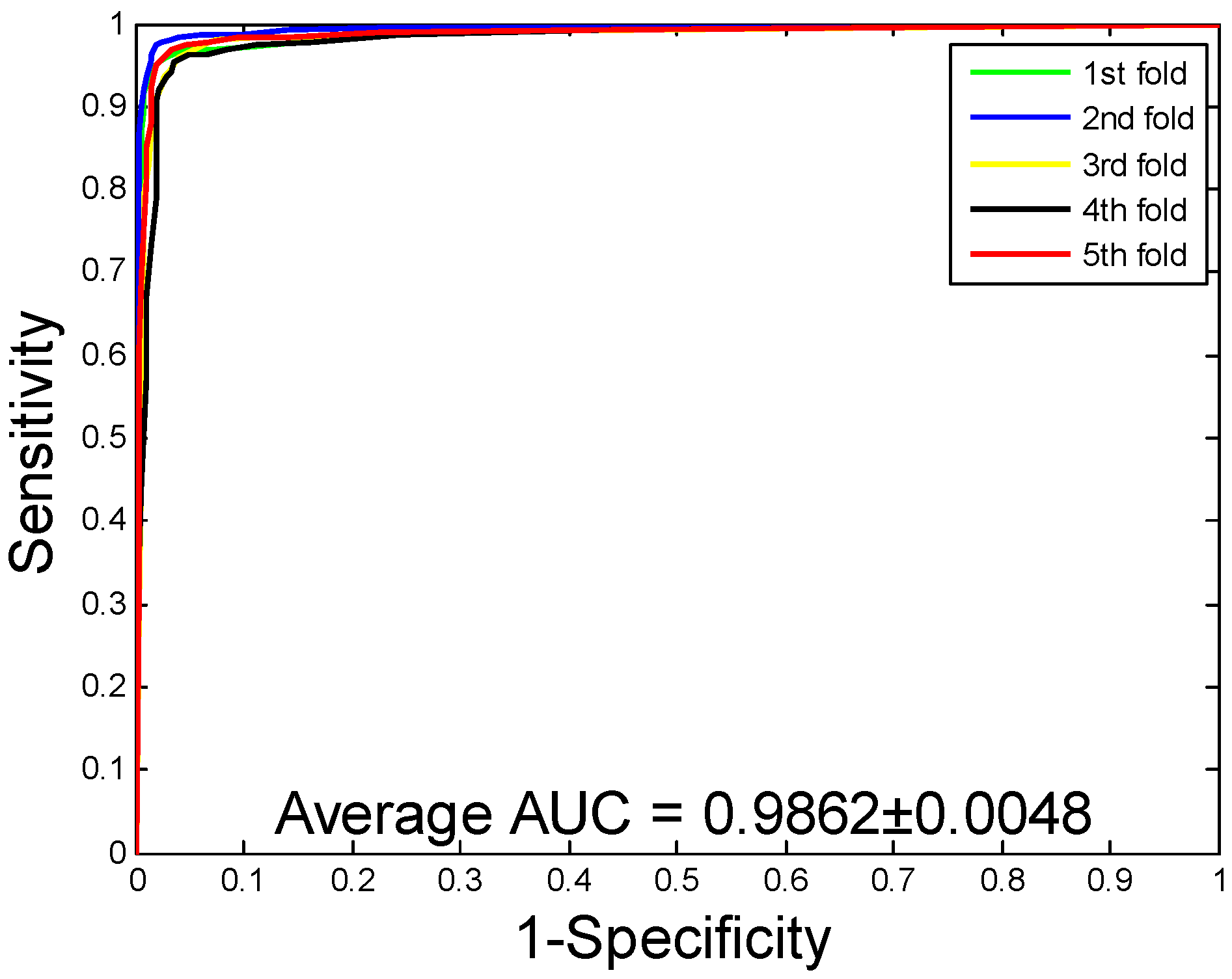
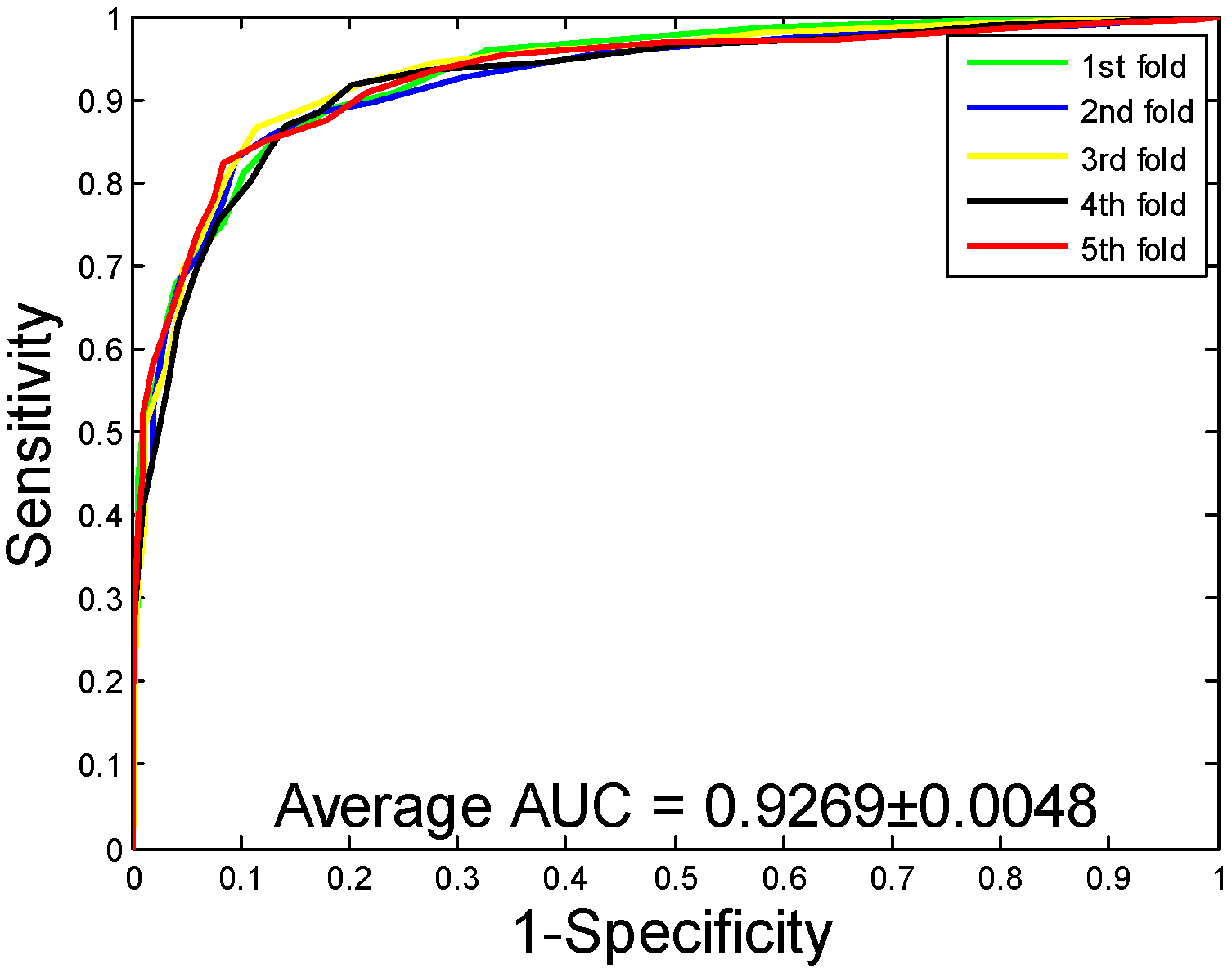
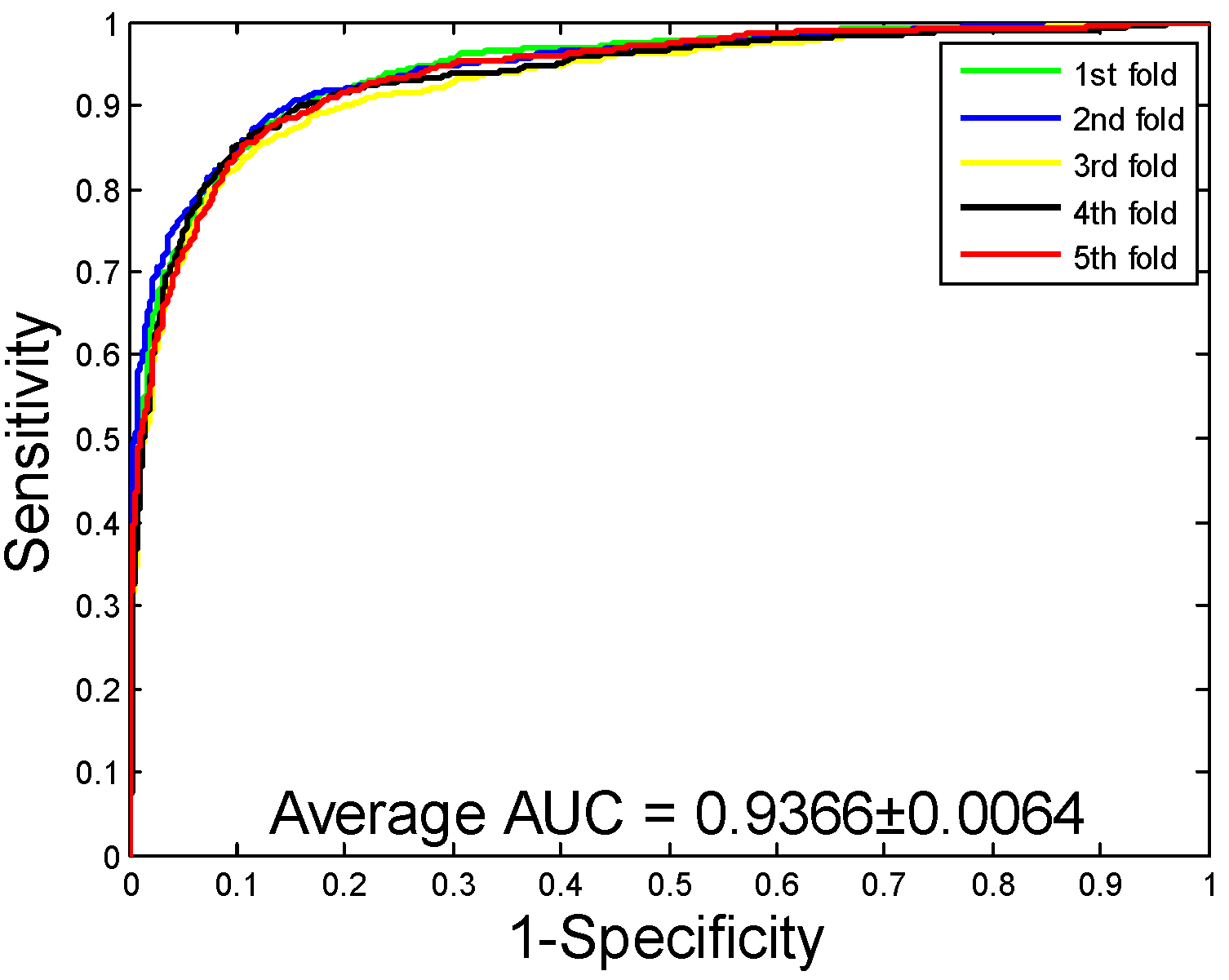
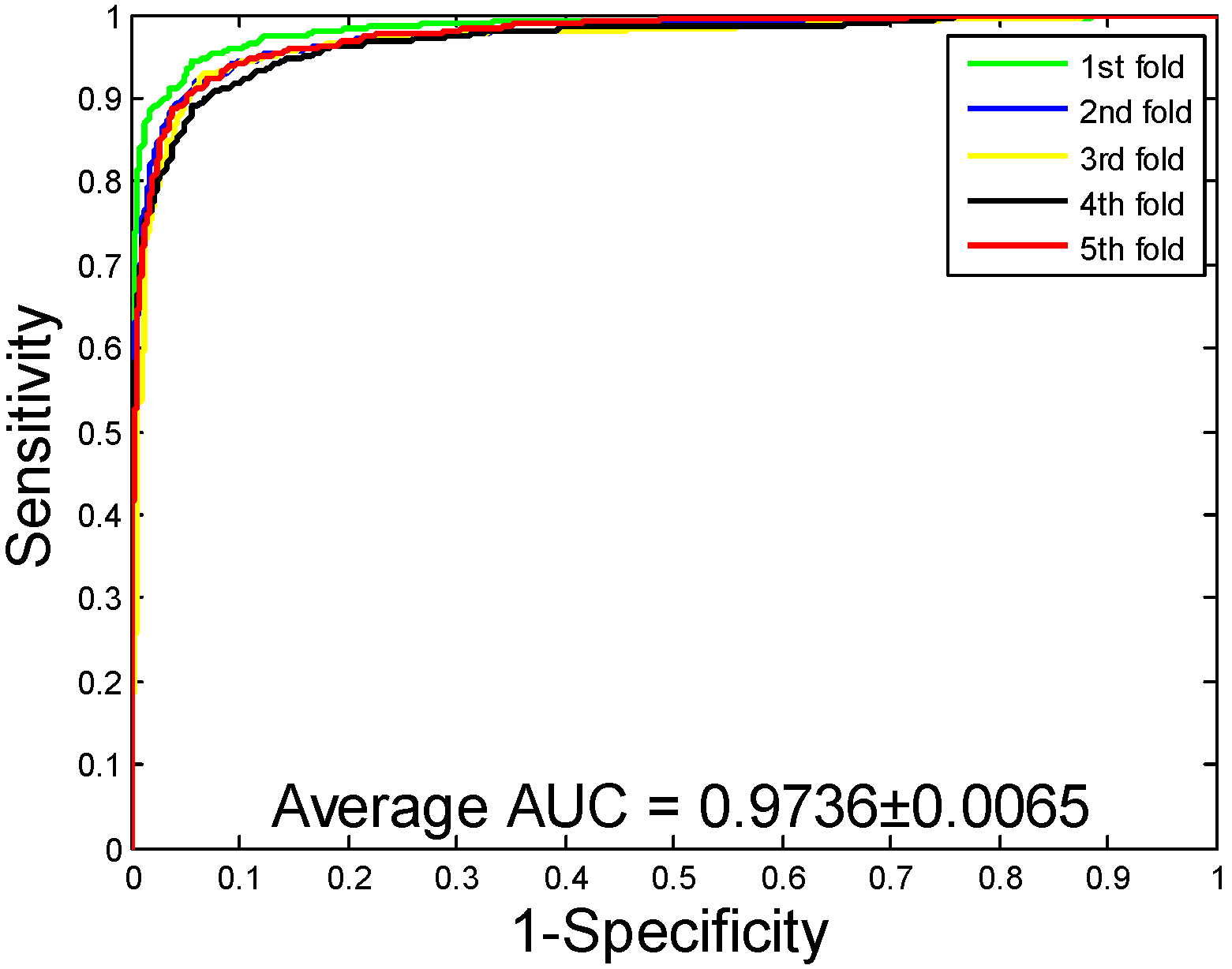
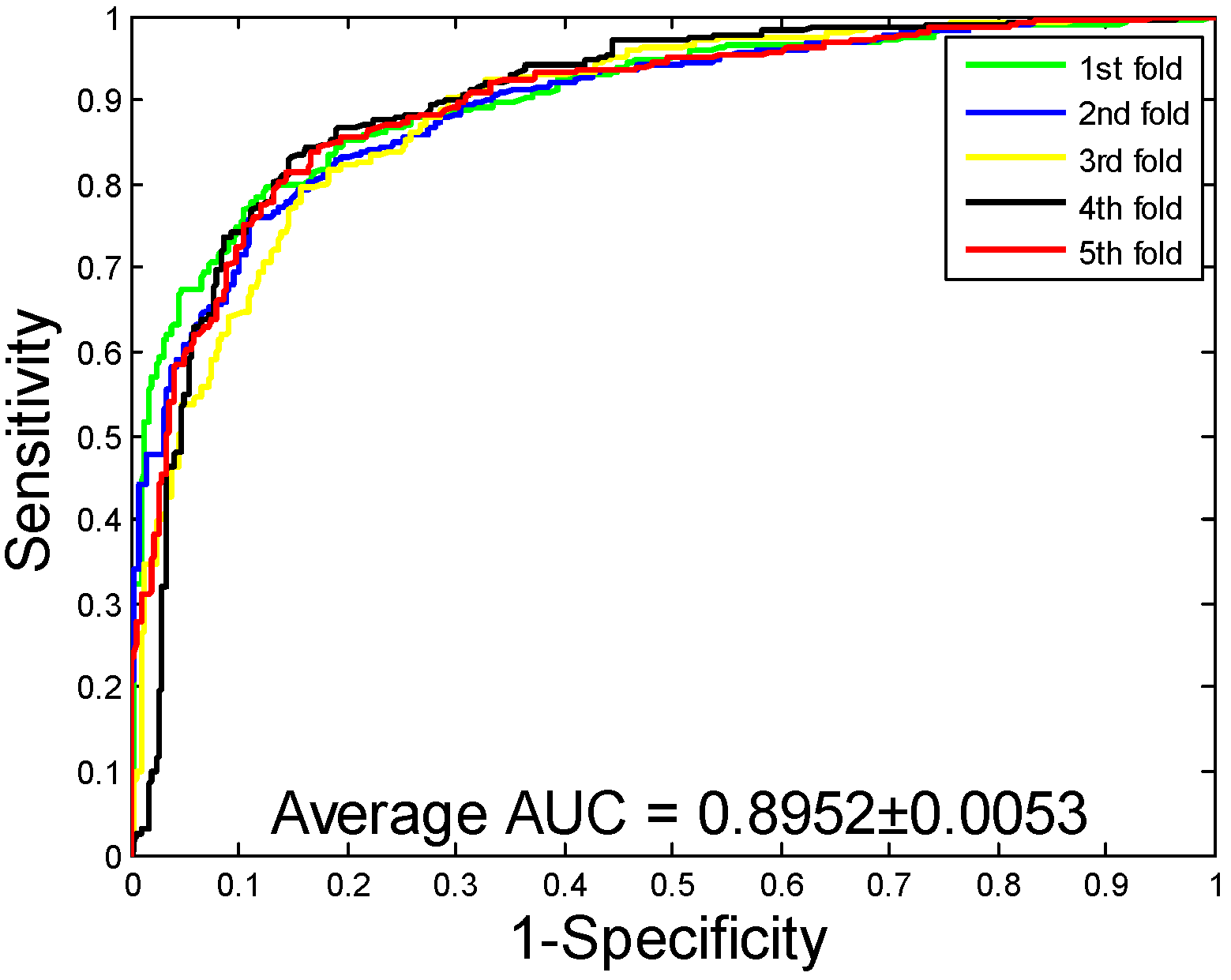
2.1. Performance Evaluation
2.2. Assessment of Prediction Ability
2.3. Comparison with Support Vector Machine (SVM) Classifier
2.4. Comparison with Existing Methods
2.5. Performance on Independent Datasets
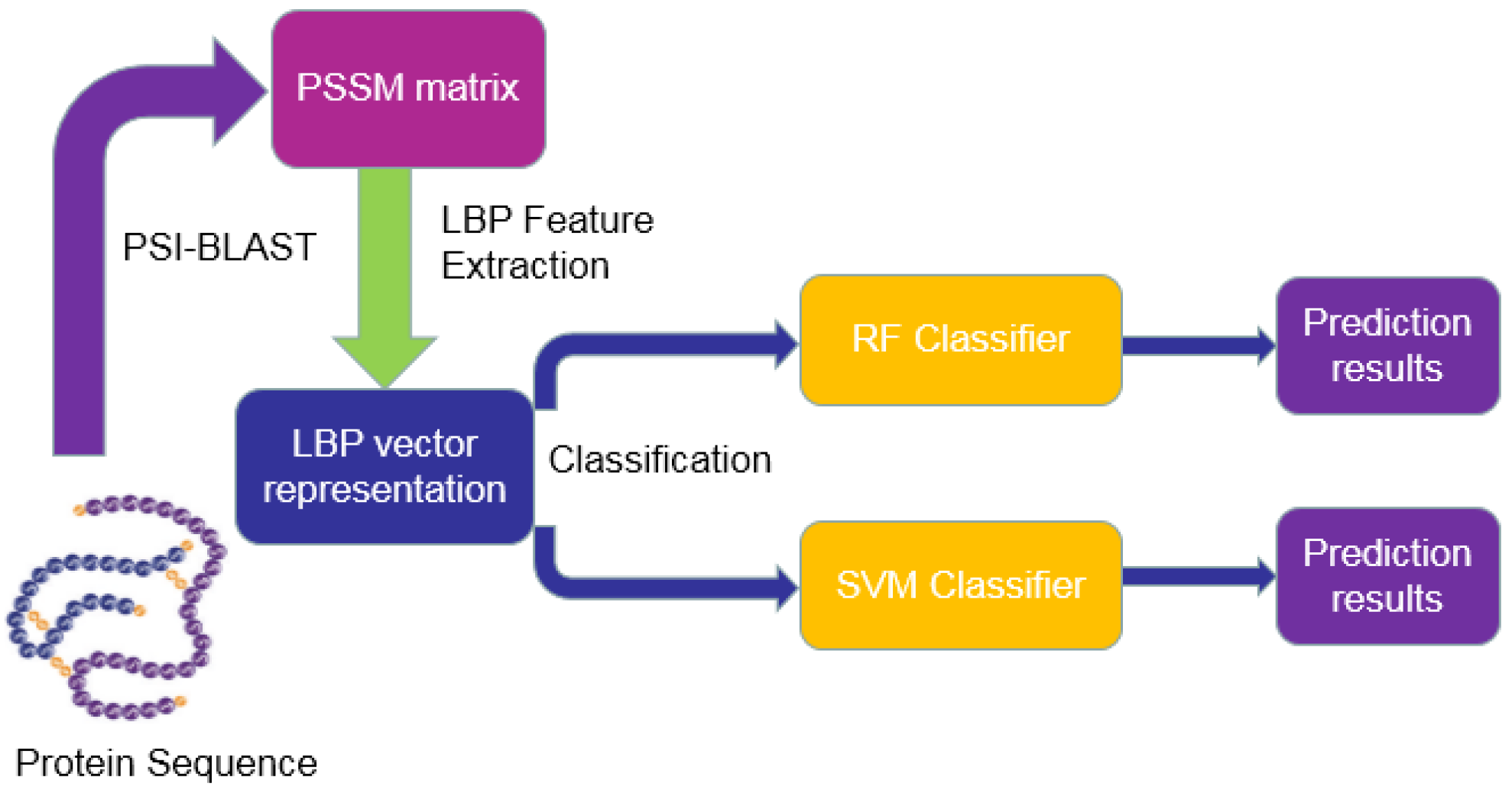
3. Materials and Methodology
3.1. Dataset and Data Collection
3.2. Position-Specific Scoring Matrix (PSSM)
3.3. Local Binary Pattern (LBP)
3.4. Rotation Forest (RF)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Várnai, C.; Burkoff, N.S.; Wild, D.L. Improving protein-protein interaction prediction using evolutionary information from low-quality MSAs. PLoS ONE 2017, 12, 0169356. [Google Scholar]
- Lei, H.; Li, L.; Wu, C.H. Protein-protein interaction prediction based on multiple kernels and partial network with linear programming. BMC Syst. Biol. 2016, 10, 45. [Google Scholar]
- Yi, H.-C.; You, Z.-H.; Huang, D.-S.; Li, X.; Jiang, T.-H.; Li, L.-P. A Deep Learning Framework for Robust and Accurate prediction of ncRNA-Protein Interactions using Evolutionary Information. Mol. Ther. Nucleic Acids 2018, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ivanov, A.A.; Su, R.; Gonzalez-Pecchi, V.; Qi, Q.; Liu, S.; Webber, P.; McMillan, E.; Rusnak, L.; Pham, C.; et al. The OncoPPi network of cancer-focused protein-protein interactions to inform biological insights and therapeutic strategies. Nat. Commun. 2017, 8, 14356. [Google Scholar]
- Yang, B.; Tang, S.; Ma, C.; Li, S.T.; Shao, G.C.; Dang, B.; Degrado, W.F.; Dong, M.Q.; Wang, P.G.; Ding, S. Spontaneous and specific chemical cross-linking in live cells to capture and identify protein interactions. Nat. Commun. 2017, 8, 2240. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, U.; Liu, Z.; Blundell, J.R.; St Onge, R.P.; Levy, S.F. A scalable double-barcode sequencing platform for characterization of dynamic protein-protein interactions. Nat. Commun. 2017, 8, 15586. [Google Scholar] [CrossRef]
- Li, J.; Bonkowski, M.S.; Moniot, S.; Zhang, D.; Hubbard, B.P.; Ling, A.J.; Rajman, L.A.; Qin, B.; Lou, Z.; Gorbunova, V. A conserved NAD+ binding pocket that regulates protein-protein interactions during aging. Science 2017, 355, 1312. [Google Scholar] [CrossRef]
- Gierer, A. Model for DNA and Protein Interactions and the Function of the Operator. Nature 2017, 212, 1480–1481. [Google Scholar] [CrossRef]
- An, J.Y.; Meng, F.R.; You, Z.H.; Fang, Y.H.; Zhao, Y.J.; Zhang, M. Using the Relevance Vector Machine Model Combined with Local Phase Quantization to Predict Protein-Protein Interactions from Protein Sequences. BioMed Res. Int. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; You, Z.; Zhang, X.; Zhou, Y. Prediction of Protein–Protein Interactions with Clustered Amino Acids and Weighted Sparse Representation. Int. J. Mol. Sci. 2015, 16, 10855–10869. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, X.; You, Z.; Huang, D.; Chan, K. ILNCSIM: Improved lncRNA functional similarity calculation model. Oncotarget 2016, 7, 25902–25914. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-A.; You, Z.-H.; Gao, X.; Wong, L.; Wang, L. Using Weighted Sparse Representation Model Combined with Discrete Cosine Transformation to Predict Protein-Protein Interactions from Protein Sequence. BioMed Res. Int. 2015, 2015, 902198. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Ming, Z.; You, Z.; Li, S.; Xia, Y.; Leung, H. Improving network topology-based protein interactome mapping via collaborative filtering. Knowl. Based Syst. 2015, 90, 23–32. [Google Scholar] [CrossRef]
- Wong, L.; You, Z.-H.; Ming, Z.; Li, J.; Chen, X.; Huang, Y.-A. Detection of Interactions between Proteins through Rotation Forest and Local Phase Quantization Descriptors. Int. J. Mol. Sci. 2015, 17, 21. [Google Scholar] [CrossRef] [PubMed]
- You, Z.H.; Li, S.; Gao, X.; Luo, X.; Ji, Z. Large-Scale Protein-Protein Interactions Detection by Integrating Big Biosensing Data with Computational Model. BioMed Res. Int. 2014, 2014, 598129. [Google Scholar] [CrossRef] [PubMed]
- You, Z.H.; Zhou, M.; Luo, X.; Li, S. Highly Efficient Framework for Predicting Interactions Between Proteins. IEEE Tran. Cybern. 2016, 47, 731–743. [Google Scholar] [CrossRef] [PubMed]
- You, Z.-H.; Lei, Y.-K.; Zhu, L.; Xia, J.; Wang, B. Prediction of protein-protein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis. BMC Bioinform. 2013, 14 (Suppl. 8), S10. [Google Scholar] [CrossRef] [PubMed]
- You, Z.-H.; Li, J.; Gao, X.; He, Z.; Zhu, L.; Lei, Y.-K.; Ji, Z. Detecting protein-protein interactions with a novel matrix-based protein sequence representation and support vector machines. BioMed Res. Int. 2015, 2015, 867516. [Google Scholar] [CrossRef]
- You, Z.-H.; Yin, Z.; Han, K.; Huang, D.-S.; Zhou, X. A semi-supervised learning approach to predict synthetic genetic interactions by combining functional and topological properties of functional gene network. BMC Bioinform. 2010, 11, 343. [Google Scholar] [CrossRef]
- Zhu, L.; You, Z.-H.; Huang, D.-S. Increasing the reliability of protein–protein interaction networks via non-convex semantic embedding. Neurocomputing 2013, 121, 99–107. [Google Scholar] [CrossRef]
- Zhu, L.; You, Z.-H.; Huang, D.-S. Identifying Spurious Interactions in the Protein-Protein Interaction Networks Using Local Similarity Preserving Embedding. In Bioinformatics Research and Applications; Springer International Publishing: Basel, Switzerland, 2014; pp. 138–148. [Google Scholar]
- Atashin, A.A.; Bagherzadeh, P.; Ghiasishirazi, K. A two-stage learning method for protein-protein interaction prediction. arXiv 2016, arXiv:1606.04561. [Google Scholar]
- Kotlyar, M.; Pastrello, C.; Pivetta, F.; Sardo, A.L.; Cumbaa, C.; Li, H.; Naranian, T.; Niu, Y.; Ding, Z.; Vafaee, F. In silico prediction of physical protein interactions and characterization of interactome orphans. Nat. Methods 2015, 12, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Schoenrock, A.; Samanfar, B.; Pitre, S.; Hooshyar, M.; Jin, K.; Phillips, C.A.; Wang, H.; Phanse, S.; Omidi, K.; Gui, Y. Efficient prediction of human protein-protein interactions at a global scale. BMC Bioinform. 2014, 15, 383. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.-S.; Zhang, L.; Han, K.; Deng, S.; Yang, K.; Zhang, H. Prediction of Protein-Protein Interactions Based on Protein-Protein Correlation Using Least Squares Regression. Curr. Protein Pept. Sci. 2014, 15, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.C.; Petrey, D.; Deng, L.; Qiang, L.; Shi, Y.; Thu, C.A.; Bisikirska, B.; Lefebvre, C.; Accili, D.; Hunter, T.; et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 2012, 490, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Muppirala, U.K.; Honavar, V.G.; Dobbs, D. Predicting RNA-Protein Interactions Using Only Sequence Information. BMC Bioinform. 2011, 12, 489. [Google Scholar] [CrossRef]
- Wang, L.; You, Z.H.; Chen, X.; Li, J.Q.; Yan, X.; Zhang, W.; Huang, Y.A. An ensemble approach for large-scale identification of protein-protein interactions using the alignments of multiple sequences. Oncotarget 2017, 8, 5149. [Google Scholar] [CrossRef]
- Zhou, C.; Yu, H.; Ding, Y.; Guo, F.; Gong, X.J. Multi-scale encoding of amino acid sequences for predicting protein interactions using gradient boosting decision tree. PLoS ONE 2017, 12, e0181426. [Google Scholar] [CrossRef]
- Juwen, S.; Jian, Z.; Xiaomin, L.; Weiliang, Z.; Kunqian, Y.; Kaixian, C.; Yixue, L.; Hualiang, J. Predicting protein-protein interactions based only on sequences information. Proc. Natl. Acad. Sci. USA 2007, 104, 4337–4341. [Google Scholar]
- Yang, L.; Xia, J.-F.; Gui, J. Prediction of Protein-Protein Interactions from Protein Sequence Using Local Descriptors. Protein Pept. Lett. 2010, 17, 1085–1090. [Google Scholar] [CrossRef]
- Zhou, Y.Z.; Gao, Y.; Zheng, Y.Y. Prediction of Protein-Protein Interactions Using Local Description of Amino Acid Sequence. In Advances in Computer Science and Education Applications, Pt Ii; Zhou, M., Tan, H.H., Eds.; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2011; Volume 202, pp. 254–262. [Google Scholar]
- Nakashima, H.; Nishikawa, K.; Ooi, T. The folding type of a protein is relevant to the amino acid composition. J. Biochem. 1986, 99, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yu, L.; Wen, Z.; Li, M. Using support vector machine combined with auto covariance to predict protein-protein interactions from protein sequences. Nucleic Acids Res. 2008, 36, 3025–3030. [Google Scholar] [CrossRef] [PubMed]
- Zweig, M.H.; Campbell, G. Receiver-operating characteristic (ROC) plots: A fundamental evaluation tool in clinical medicine. Clin. Chem. 1993, 39, 561–577. [Google Scholar] [PubMed]
- Wang, Y.; You, Z.; Li, X.; Chen, X.; Jiang, T.; Zhang, J. PCVMZM: Using the Probabilistic Classification Vector Machines Model Combined with a Zernike Moments Descriptor to Predict Protein–Protein Interactions from Protein Sequences. Int. J. Mol. Sci. 2017, 18, 1029. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Fang, L.; Wang, X.; Chou, K.C. repDNA: A Python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects. Bioinformatics 2015, 31, 1307. [Google Scholar] [CrossRef] [PubMed]
- Ioannis, X.; Lukasz, S.; Xiaoqun Joyce, D.; Patrick, H.; Sul-Min, K.; David, E. DIP, the Database of Interacting Proteins: A research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002, 30, 303–305. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Bhatti, M.N.A.; Jung, S.K. Local binary pattern variants-based adaptive texture features analysis for posed and nonposed facial expression recognition. J. Electron. Imaging 2017, 26, 053017. [Google Scholar]
- Ahonen, T.; Hadid, A.; Pietikainen, M. Face Description with Local Binary Patterns: Application to Face Recognition. IEEE Trans. Pattern Anal. Mach. Intell. 2006, 28, 2037–2041. [Google Scholar] [CrossRef]
- Huynh, T.; Min, R.; Dugelay, J.L. An Efficient LBP-Based Descriptor for Facial Depth Images Applied to Gender Recognition Using RGB-D Face Data; Springer: Berlin/Heidelberg, Germany, 2013; pp. 133–145. [Google Scholar]
- Ojala, T.; Harwood, I. A Comparative Study of Texture Measures with Classification Based on Feature Distributions. Pattern Recogn. 1996, 29, 51–59. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Kuncheva, L.I. Rotation forest: A new classifier ensemble method. IEEE Trans. Pattern Anal. Mach. Intell. 2006, 28, 1619–1630. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Sets | ACC (%) | PE (%) | SN (%) | MCC (%) | AUC (%) |
|---|---|---|---|---|---|
| Yeast | 92.12 ± 0.54 | 94.20 ± 0.78 | 89.76 ± 0.96 | 85.46 ± 0.92 | 96.11 ± 0.77 |
| Human | 96.21 ± 0.76 | 97.23 ± 1.19 | 94.77 ± 1.09 | 92.70 ± 1.42 | 98.62 ± 0.48 |
| H. pylori | 86.59 ± 0.48 | 87.70 ± 1.89 | 85.17 ± 2.20 | 76.73 ± 0.74 | 92.69 ± 0.48 |
| Dataset | Classifier | ACC (%) | PE (%) | SN (%) | MCC (%) | AUC (%) |
|---|---|---|---|---|---|---|
| Yeast | RF | 92.12 ± 0.54 | 94.20 ± 0.78 | 89.76 ± 0.96 | 85.46 ± 0.92 | 96.11 ± 0.77 |
| SVM | 86.99 ± 0.43 | 88.05 ± 0.88 | 85.62 ± 1.23 | 77.36 ± 0.64 | 93.66 ± 0.64 | |
| Human | RF | 96.21 ± 0.76 | 97.23 ± 1.19 | 94.77 ± 1.09 | 92.70 ± 1.42 | 98.62 ± 0.48 |
| SVM | 92.56 ± 0.70 | 93.71 ± 1.06 | 90.47 ± 0.82 | 86.18 ± 1.23 | 97.36 ± 0.65 | |
| H. pylori | RF | 86.59 ± 0.48 | 87.70 ± 1.89 | 85.17 ± 2.20 | 76.73 ± 0.74 | 92.69 ± 0.48 |
| SVM | 81.62 ± 1.22 | 80.73 ± 3.79 | 83.40 ± 3.56 | 69.93 ± 1.56 | 89.52 ± 0.53 |
| Author | Model | ACC (%) | PE (%) | SN (%) | MCC (%) |
|---|---|---|---|---|---|
| Guos’ work [34] | ACC | 89.33 ± 2.67 | 88.87 ± 6.16 | 89.93 ± 3.68 | N/A |
| AC | 87.36 ± 1.38 | 87.82 ± 4.33 | 87.30 ± 4.68 | N/A | |
| You et al.’s work [17] | PCA-EELM | 87.00 ± 0.29 | 87.59 ± 0.32 | 86.15 ± 0.43 | 77.36 ± 0.44 |
| Yang et al.’s work [31] | Cod1 | 75.08 ± 1.13 | 74.75 ± 1.23 | 75.81 ± 1.20 | N/A |
| Cod2 | 80.04 ± 1.06 | 82.17 ± 1.35 | 76.77 ± 0.69 | N/A | |
| Cod3 | 80.41 ± 0.47 | 81.86 ± 0.99 | 78.14 ± 0.90 | N/A | |
| Cod4 | 86.15 ± 1.17 | 90.24 ± 1.34 | 81.03 ± 1.74 | N/A | |
| Zhou et al.’s work [32] | SVM + LD | 88.56 ± 0.33 | 89.50 ± 0.60 | 87.37 ± 0.22 | 77.15 ± 0.68 |
| Wang et al.’s work [36] | PCVM + ZM | 94.48 ± 1.2 | 93.92 ± 2.4 | 95.13 ± 2.0 | 89.58 ± 2.2 |
| Our method | SVM + PSSM | 86.99 ± 0.43 | 88.05 ± 0.88 | 85.62 ± 1.23 | 77.36 ± 0.64 |
| RF + PSSM | 92.12 ± 0.54 | 94.20 ± 0.78 | 89.76 ± 0.96 | 85.46 ± 0.92 |
| Model | ACC (%) | SN (%) | MCC (%) |
|---|---|---|---|
| LDA + RF [37] | 96.4 | 94.2 | 92.8 |
| LDA + RoF | 95.7 | 97.6 | 91.8 |
| LDA + SVM | 90.7 | 89.7 | 81.3 |
| AC + RF | 95.5 | 94.0 | 91.4 |
| AC + RoF | 95.1 | 93.3 | 91.0 |
| AC + SVM | 89.3 | 94.0 | 79.2 |
| Our method | 96.21 | 94.77 | 92.70 |
| Species | Test Pairs | ACC (%) |
|---|---|---|
| C. elegans | 4013 | 94.82 |
| H. pylori | 1420 | 94.79 |
| H. sapiens | 1412 | 95.11 |
| M. musculus | 313 | 93.93 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Li, L.-P.; Wang, L.; Yu, C.-Q.; Wang, Z.; You, Z.-H. An Ensemble Classifier to Predict Protein–Protein Interactions by Combining PSSM-based Evolutionary Information with Local Binary Pattern Model. Int. J. Mol. Sci. 2019, 20, 3511. https://doi.org/10.3390/ijms20143511
Li Y, Li L-P, Wang L, Yu C-Q, Wang Z, You Z-H. An Ensemble Classifier to Predict Protein–Protein Interactions by Combining PSSM-based Evolutionary Information with Local Binary Pattern Model. International Journal of Molecular Sciences. 2019; 20(14):3511. https://doi.org/10.3390/ijms20143511
Chicago/Turabian StyleLi, Yang, Li-Ping Li, Lei Wang, Chang-Qing Yu, Zheng Wang, and Zhu-Hong You. 2019. "An Ensemble Classifier to Predict Protein–Protein Interactions by Combining PSSM-based Evolutionary Information with Local Binary Pattern Model" International Journal of Molecular Sciences 20, no. 14: 3511. https://doi.org/10.3390/ijms20143511
APA StyleLi, Y., Li, L. -P., Wang, L., Yu, C. -Q., Wang, Z., & You, Z. -H. (2019). An Ensemble Classifier to Predict Protein–Protein Interactions by Combining PSSM-based Evolutionary Information with Local Binary Pattern Model. International Journal of Molecular Sciences, 20(14), 3511. https://doi.org/10.3390/ijms20143511