Mutations in Prion Protein Gene: Pathogenic Mechanisms in C-Terminal vs. N-Terminal Domain, a Review

Abstract
:1. Introduction
2. The Function of the Prion Protein
3. The PRNP Gene, Mutations, and Inherited Prion Diseases
4. The Structure of the N-Terminal and C-Terminal Domains
4.1. The C-Terminal Domain: Mechanisms Causing a Conformational Change of PrP in Mammalians
4.2. The N-Terminal Domain
4.3. Natural Ligands of the N-Terminal Domain
5. Pathogenic Mechanisms
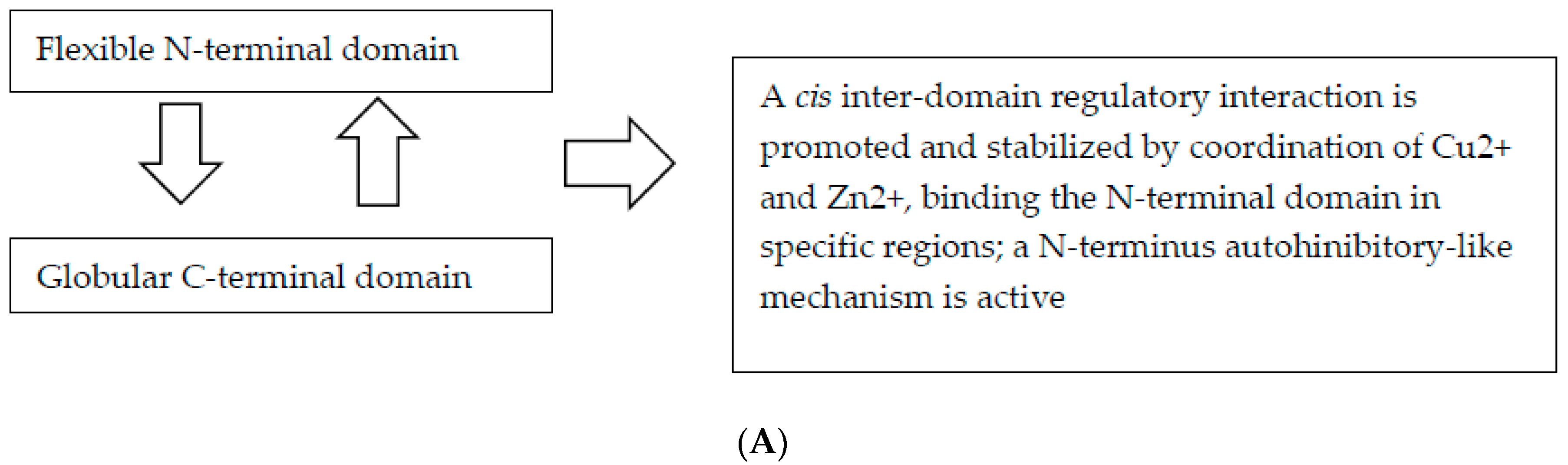
5.1. The N-Terminal Domain Is a Toxic Effector Regulated by the C-Terminus
5.2. Important Key Elements in the Pathogenic Mechanisms: Metal Ions Cu2+ and Zn2+ and the Proline Amino Acid
5.2.1. Cu2+ and Zn2+ Promote Interdomain Interaction in cis
5.2.2. Role of the Proline Amino Acid in the PrP Protein
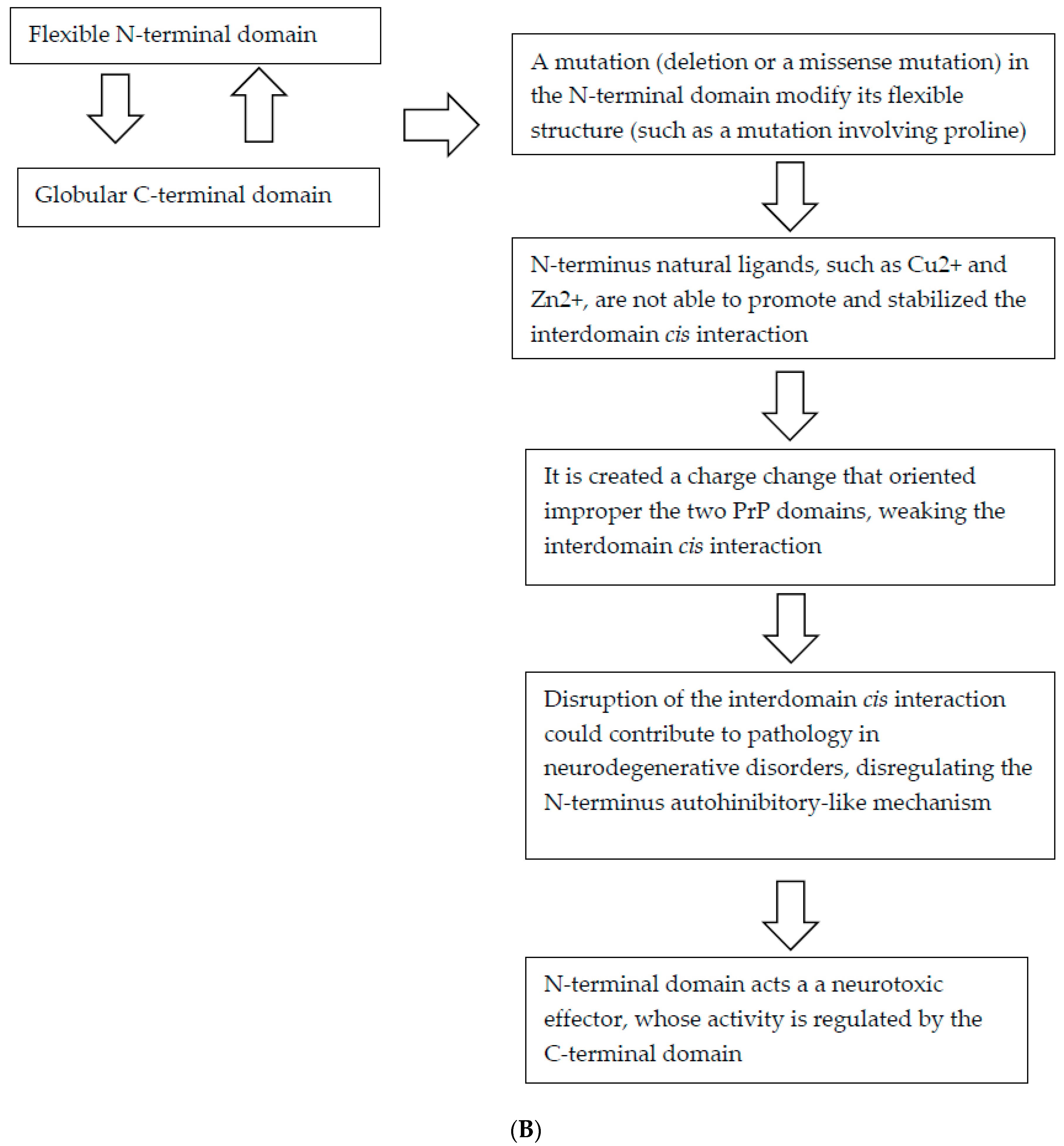
5.3. PRNP Mutations and Disruption of cis Interaction as a Mechanism of Neurotoxicity
5.4. Pathogenic Mutations within the N-terminal Disordered Palindromic Region of PrP Accelerate the Formation of Misfolded Oligomers
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Baiardi, S.; Rossi, M.; Capellari, S.; Parchi, P. Recent advances in the histo-molecular pathology of human prion disease. Brain Pathol. 2019, 29, 278–300. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; Scott, M.R.; DeArmond, S.J.; Cohen, F.E. Prion protein biology. Cell 1998, 93, 337–348. [Google Scholar] [CrossRef]
- Ironside, J.W.; Ritchie, D.L.; Head, M.W. Prion diseases. Handb. Clin. Neurol. 2017, 145, 393–403. [Google Scholar] [PubMed]
- Chen, C.; Dong, X.-P. Epidemiological characteristics of human prion diseases. Infect. Dis. Poverty 2016, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Ghetti, B.; Piccardo, P.; Zanusso, G. Dominantly inherited prion protein cerebral amyloidoses—A modern view of Gerstmann-Sträussler-Scheinker. Handb. Clin. Neurol. 2018, 153, 243–269. [Google Scholar] [PubMed]
- Deleault, N.R.; Walsh, D.J.; Piro, J.R.; Wang, F.; Wang, X.; Ma, J.; Rees, J.R.; Supattapone, S. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. USA 2012, 10, E1938–E1946. [Google Scholar] [CrossRef]
- Zafar, S.; Shafiq, M.; Andreoletti, O.; Zerr, I. Animal TSEs and public health: What remains of past lessons? PLoS Pathog. 2018, 14, e1006759. [Google Scholar] [CrossRef]
- Noble, G.P.; Walsh, D.J.; Miller, M.B.; Jackson, W.S.; Supattapone, S. Requirements for Mutant and Wild-Type Prion Protein Misfolding In Vitro. Biochemistry 2015, 54, 1180–1187. [Google Scholar] [CrossRef] [Green Version]
- Markham, K.A.; Roseman, G.P.; Linsley, R.B.; Lee, H.W.; Millhauser, G.L. Molecular Features of the Zn2+ Binding Site in the Prion Protein Probed by 113Cd NMR. Biophys. J. 2019, 116, 610–620. [Google Scholar] [CrossRef]
- Evans, E.G.B.; Pushie, M.J.; Markham, K.A.; Lee, H.-W.; Millhauser, G.L. Interaction between the Prion Protein’s Copper-Bound Octarepeat Domain and a Charged C-terminal Pocket Suggests a Mechanism for N-terminal Regulation. Structure 2016, 24, 1057–1067. [Google Scholar] [CrossRef]
- Lau, A.; McDonald, A.; Daude, N.; Mays, C.E.; Walter, E.D.; Aglietti, R.; Mercer, R.C.; Wohlgemuth, S.; van der Merwe, J.; Yang, J.; et al. Octarepeat region flexibility impacts prion function, endoprioteolysis and disease manifestation. EMBO Mol. Med. 2015, 7, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Sabareesan, A.; Udgaonkar, J.B. Pathogenic Mutations within the Disordered Palindromic Region of the Prion Protein Induce Structure Therein and Accelerate the Formation of Misfolded Oligomers. J. Mol. Biol. 2016, 428, 3935–3947. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, L.; Cupidi, C.; Frangipane, F.; Anfossi, M.; Gallo, M.; Conidi, M.E.; Vasso, F.; Colao, R.; Puccio, G.; Curcio, S.A.; et al. Novel N-terminal domain mutation in prion protein detected in 2 patients diagnosed with frontotemporal lobar degeneration syndrome. Neurobiol. Aging 2014, 35, 2657.e7–2657.e11. [Google Scholar] [CrossRef] [PubMed]
- Oldoni, E.; Fumagalli, G.G.; Serpente, M.; Fenoglio, C.; Scarioni, M.; Arighi, A.; Bruno, G.; Talarico, G.; Confaloni, A.; Piscopo, P.; et al. PRNP P39L Variant is a Rare Cause of Frontotemporal Dementia in Italian Population. J. Alzheimers Dis. 2016, 50, 353–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, L.; Cupidi, C.; Bruni, A.C. Pathogenic mechanisms of the prion protein gene mutations: A review and speculative hypotheses for pathogenic potential of the Pro39Leu mutation in the associated FTD-like phenotype. J. Neurol. Neurosci. 2017, 8, 208. [Google Scholar] [CrossRef]
- Weissmann, C.; Raeber, A.J.; Montrasio, F.; Hegyi, I.; Frigg, R.; Klein, M.A.; Aguzzi, A. Prions and the lymphoreticular system. Philos. Trans. R. Soc. B Biol. Sci. 2001, 356, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.R.; Hooper, N.M. The prion protein and lipid rafts. Mol. Membr. Biol. 2006, 23, 89–99. [Google Scholar] [CrossRef]
- Wulf, M.-A.; Senatore, A.; Aguzzi, A. The biological function of the cellular prion protein: An update. BMC Biol. 2017, 15, 149. [Google Scholar] [CrossRef]
- Puig, B.; Altmeppen, H.C.; Linsenmeier, L.; Chakroun, K.; Wegwitz, F.; Piontek, U.K.; Tatzelt, J.; Bate, C.; Magnus, T.; Glatzel, M. GPI-anchor signal sequence influences PrPC sorting, shedding and signalling, and impacts on different pathomechanistic aspects of prion disease in mice. PLoS Pathog. 2019, 15, e1007520. [Google Scholar] [CrossRef]
- Zeng, L.; Zou, W.; Wang, G. Cellular prion protein (PrP(C)) and its role in stress responses. Int. J. Clin. Exp. Med. 2015, 15, 8042–8050. [Google Scholar]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; Von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.S.; Aronoff-Spencer, E.; Dunham, C.M.; Lario, P.; Avdievich, N.I.; Antholine, W.E.; Olmstead, M.M.; Vrielink, A.; Gerfen, G.J.; Peisach, J.; et al. Molecular features of the copper binding sites in the octarepeat domain of the prion protein. Biochemistry 2002, 41, 3991–4001. [Google Scholar] [CrossRef]
- Burns, C.S.; Aronoff-Spencer, E.; Legname, G.; Prusiner, S.B.; Antholine, W.E.; Gerfen, G.J.; Peisach, J.; Millhauser, G.L. Copper coordination in the full-length, recombinant prion protein. Biochemistry 2003, 42, 6794–6803. [Google Scholar] [CrossRef] [PubMed]
- Walter, E.D.; Stevens, D.J.; Spevacek, A.R.; Visconte, M.P.; Dei Rossi, A.; Millhauser, G.L. Copper binding extrinsic to the octarepeat region in the prion protein. Curr. Protein Pept. Sci. 2009, 10, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Wong, B.-S.; Hafiz, F.; Clive, C.; Haswell, S.J.; Jones, I.M. Normal prion protein has an activity like that of superoxide dismutase. Biochem. J. 1999, 344, 1–5. [Google Scholar] [CrossRef]
- Mouillet-Richard, S. Signal transduction through prion protein. Science 2000, 289, 1925–1928. [Google Scholar] [CrossRef] [PubMed]
- Castle, A.R.; Gill, A.C. Physiological functions of the cellular prion protein. Front. Mol. Biosci. 2017, 4, 19. [Google Scholar] [CrossRef]
- Machado, C.F.; Beraldo, F.H.; Santos, T.G.; Bourgeon, D.; Landemberger, M.C.; Roffé, M.; Martins, V.R. Disease-associated mutations in the prion protein impair laminin-induced process outgrowth and survival. J. Biol. Chem. 2012, 287, 43777–43788. [Google Scholar] [CrossRef]
- OMIM Entry 176640—PRION PROTEIN; PRNP. Available online: https://www.omim.org/entry/176640 (accessed on 20 May 2019).
- Elahi, F.M.; Miller, B.L. A clinicopathological approach to the diagnosis of dementia. Nat. Rev. Neurol. 2017, 13, 457–476. [Google Scholar] [CrossRef] [Green Version]
- Bagyinszky, E.; Van Giau, V.; Youn, Y.C.; An, S.S.A.; Kim, S. Characterization of mutations in PRNP (prion) gene and their possible roles in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2018, 14, 2067–2085. [Google Scholar] [CrossRef]
- Minikel, E.V.; Vallabh, S.M.; Lek, M.; Estrada, K.; Samocha, K.; Sathirapongsasuti, J.F. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med. 2016, 8, 322ra9. [Google Scholar] [CrossRef]
- Nitrini, R.; Da Silva, L.S.T.; Rosemberg, S.; Caramelli, P.; Carrilho, P.E.M.; Iughetti, P.; Passos-Bueno, M.R.; Zatz, M.; Albrecht, S.; Leblanc, A. Prion disease resembling frontotemporal dementia and parkinsonism linked to chromosome 17. Arq. Neuro-Psiquiatr. 2001, 59, 161–164. [Google Scholar] [CrossRef] [Green Version]
- Hall, D.A.; Leehey, M.A.; Filley, C.M.; Steinbart, E.; Montine, T.; Schellenberg, G.D.; Bosque, P.; Nixon, R.; Bird, T. PRNP H187R mutation associated with neuropsychiatric disorders in childhood and dementia. Neurology 2005, 64, 1304–1306. [Google Scholar] [CrossRef] [PubMed]
- Woulfe, J.; Kertesz, A.; Frohn, I.; Bauer, S.; George-Hyslop, P.S.; Bergeron, C. Gerstmann-Sträussler-Scheinker disease with the Q217R mutation mimicking frontotemporal dementia. Acta Neuropathol. 2005, 110, 317–319. [Google Scholar] [CrossRef]
- Clerici, F.; Elia, A.; Girotti, F.; Contri, P.; Mariani, C.; Tagliavini, F.; Di Fede, G. Atypical presentation of Creutzfeldt–Jakob disease: The first Italian case associated with E196K mutation in the PRNP gene. J. Neurol. Sci. 2008, 275, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Giovagnoli, A.R.; Di Fede, G.; Aresi, A.; Reati, F.; Rossi, G.; Tagliavini, F. Atypical frontotemporal dementia as a new clinical phenotype of Gerstmann-Straussler-Scheinker disease with the PrP-P102L mutation. Description of a previously unreported Italian family. Neurol. Sci. 2008, 29, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Boeve, B.F.; Boot, B.P.; Orr, C.F.; Duffy, J.; Woodruff, B.K.; Nair, A.K.; Ellison, J.; Kuntz, K.; Kantarci, K.; et al. Clinical characterization of a kindred with a novel 12-octapeptide repeat insertion in the prion protein gene. Arch. Neurol. 2011, 68, 1165–1170. [Google Scholar] [CrossRef]
- Samaia, H.B.; Mari, J.D.J.; Vallada, H.P.; Moura, R.P.; Simpson, A.J.G.; Brentania, R.R.; Mari, J.J.; Brentani, R.R. A prion-linked psychiatric disorder. Nature 1997, 390, 241. [Google Scholar] [CrossRef]
- Finckh, U.; Müller-Thomsen, T.; Mann, U.; Eggers, C.; Marksteiner, J.; Meins, W.; Binetti, G.; Alberici, A.; Hock, C.; Nitsch, R.M.; et al. High Prevalence of Pathogenic Mutations in Patients with Early-Onset Dementia Detected by Sequence Analyses of Four Different Genes. Am. J. Hum. Genet. 2000, 66, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Collinge, J.; Brown, J.; Hardy, J.; Mullan, M.; Rossor, M.N.; Baker, H.; Crow, T.J.; Lofthouse, R.; Poulter, M.; Ridley, R.; et al. Inherited prion disease with 144 base pair gene insertion. Clinical and pathological features. Brain 1992, 115 Pt 3, 687–710. [Google Scholar] [CrossRef]
- Hinnell, C.; Coulthart, M.B.; Jansen, G.H.; Cashman, N.R.; Lauzon, J.; Clark, A.; Costello, F.; White, C.; Midha, R.; Wiebe, S.; et al. Gerstmann-Straussler-Scheinker disease due to a novel prion protein gene mutation. Neurology 2011, 76, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Mercer, R.C.C.; Daude, N.; Dorosh, L.; Fu, Z.-L.; Mays, C.E.; Gapeshina, H.; Wohlgemuth, S.L.; Acevedo-Morantes, C.Y.; Yang, J.; Cashman, N.R.; et al. A novel Gerstmann-Sträussler-Scheinker disease mutation defines a precursor for amyloidogenic 8 kDa PrP fragments and reveals N-terminal structural changes shared by other GSS alleles. PLoS Pathog. 2018, 14, e1006826. [Google Scholar] [CrossRef] [PubMed]
- Parchi, P.; Chen, S.G.; Brown, P.; Zou, W.; Capellari, S.; Budka, H.; Hainfellner, J.; Reyes, P.F.; Golden, G.T.; Hauw, J.J.; et al. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Straussler-Scheinker disease. Proc. Natl. Acad. Sci. USA 1998, 95, 8322–8327. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, C.J.; Zhang, K.; Munn, A.L.; Wiegmans, A.; Wei, M.Q. Prion protein scrapie and the normal cellular prion protein. Prion 2016, 10, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Asante, E.A.; Smidak, M.; Grimshaw, A.; Houghton, R.; Tomlinson, A.; Jeelani, A.; Jakubcova, T.; Hamdan, S.; Richard-Londt, A.; Linehan, J.M.; et al. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 2015, 522, 478–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, S.; Higuchi, J.; Tateishi, J.; Kitamoto, T.; Shin, R.-W.; Shin, R. Codon 219 lys allele of PRNP is not found in sporadic Creutzfeldt-Jakob disease. Ann. Neurol. 1998, 43, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Perrier, V.; Kaneko, K.; Safar, J.; Vergara, J.; Tremblay, P.; DeArmond, S.J.; Cohen, F.E.; Prusiner, S.B.; Wallace, A.C. Dominant-negative inhibition of prion replication in transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 13079–13084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, K.; Zulianello, L.; Scott, M.; Cooper, C.M.; Wallace, A.C.; James, T.L.; Cohen, F.E.; Prusiner, S.B. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc. Natl. Acad. Sci. USA 1997, 94, 10069–10074. [Google Scholar] [CrossRef] [Green Version]
- Gossert, A.D.; Bonjour, S.; Lysek, D.A.; Fiorito, F.; Wüthrich, K. Prion protein NMR structures of elk and of mouse/elk hybrids. Proc. Natl. Acad. Sci. USA 2005, 102, 646–650. [Google Scholar] [CrossRef] [Green Version]
- Scialò, C.; De Cecco, E.; Manganotti, P.; Legname, G. Prion and Prion-Like Protein Strains: Deciphering the Molecular Basis of Heterogeneity in Neurodegeneration. Viruses 2019, 11, 261. [Google Scholar] [CrossRef]
- Zhang, J. The Structural Stability of Wild-type Horse Prion Protein. J. Biomol. Struct. Dyn. 2011, 29, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhang, Y. Molecular dynamics studies on the NMR and X-ray structures of rabbit prion proteins. J. Theor. Biol. 2014, 342, 70–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liu, D.D. Molecular dynamics studies on the structural stability of wild-type dog prion protein. J. Biomol. Struct. Dyn. 2011, 28, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, F.; Chatterjee, S. Molecular dynamics studies on the buffalo prion protein. J. Biomol. Struct. Dyn. 2016, 34, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, G.; Giachin, G.; Legname, G.; Carloni, P. Structural facets of disease-linked human prion protein mutants: A molecular dynamic study. Proteins Struct. Funct. Bioinform. 2010, 78, 3270–3280. [Google Scholar] [CrossRef] [PubMed]
- Benetti, F.; Legname, G. New insights into structural determinants of prion protein folding and stability. Prion 2015, 9, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Borges, N.; Parra, B.; Vidal, E.; Eraña, H.; Sánchez-Martín, M.A.; De Castro, J.; Elezgarai, S.R.; Pumarola, M.; Mayoral, T.; Castilla, J. Unraveling the key to the resistance of canids to prion diseases. PLoS Pathog. 2017, 13, e1006716. [Google Scholar] [CrossRef]
- Bett, C.; Fernández-Borges, N.; Kurt, T.D.; Lucero, M.; Nilsson, K.P.R.; Castilla, J.; Sigurdson, C.J.; Nilsson, K.P.R. Structure of the β2-α2 loop and interspecies prion transmission. FASEB J. 2012, 26, 2868–2876. [Google Scholar] [CrossRef]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional anthology of intrinsic disorder. 3 Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J. Proteome Res. 2007, 6, 1917–1932. [Google Scholar] [CrossRef]
- Walmsley, A.R.; Hooper, N.M.; Zeng, F. The N-terminal Region of the Prion Protein Ectodomain Contains a Lipid Raft Targeting Determinant. J. Biol. Chem. 2003, 278, 37241–37248. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, J.A.; Westergard, L.; Unterberger, U.; Biasini, E.; Harris, D.A. The N-Terminal, Polybasic Region Is Critical for Prion Protein Neuroprotective Activity. PLoS ONE 2011, 6, e25675. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Boil. 2011, 43, 1090–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eigenbrod, S.; Frick, P.; Bertsch, U.; Mitteregger-Kretzschmar, G.; Mielke, J.; Maringer, M.; Piening, N.; Hepp, A.; Daude, N.; Windl, O.; et al. Substitutions of PrP N-terminal histidine residues modulate scrapie disease pathogenesis and incubation time in transgenic mice. PLoS ONE 2017, 12, e0188989. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.-F.; Harischandra, D.S.; Kanthasamy, A.; Sivasankar, S. Copper-induced structural conversion templates prion protein oligomerization and neurotoxicity. Sci. Adv. 2016, 2, e1600014. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.L.; Gomes, M.P.B.; Vieira, T.C.R.G.; Cordeiro, Y. PrP interactions with nucleic acids and glycosaminoglycans in function and disease. Front. Biosci. 2010, 15, 132–150. [Google Scholar] [CrossRef]
- Vieira, T.C.R.G.; Reynaldo, D.P.; Gomes, M.P.B.; Almeida, M.S.; Cordeiro, Y.; Silva, J.L. Heparin Binding by Murine Recombinant Prion Protein Leads to Transient Aggregation and Formation of RNA-Resistant Species. J. Am. Chem. Soc. 2011, 133, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, Y. Nucleic acid-mediated protein aggregation and assembly. Chall. Oppor. Next-Gener. Seq. Biomed. Res. 2011, 84, 1–40. [Google Scholar]
- Supattapone, S. Elucidating the role of cofactors in mammalian prion propagation. Prion 2014, 8, 100–105. [Google Scholar] [CrossRef]
- Critchley, P.; Kazlauskaite, J.; Eason, R.; Pinheiro, T.J. Binding of prion proteins to lipid membranes. Biochem. Biophys. Res. Commun. 2004, 313, 559–567. [Google Scholar] [CrossRef]
- Dong, C.-F.; Shi, S.; Wang, X.-F.; An, R.; Li, P.; Chen, J.-M.; Wang, X.; Wang, G.-R.; Shan, B.; Zhang, B.-Y.; et al. The N-terminus of PrP is responsible for interacting with tubulin and fCJD related PrP mutants possess stronger inhibitive effect on microtubule assembly in vitro. Arch. Biochem. Biophys. 2008, 470, 83–92. [Google Scholar] [CrossRef]
- Torrent, J.; Vilchez-Acosta, A.; Muñoz-Torrero, D.; Trovaslet, M.; Nachon, F.; Chatonnet, A.; Grznarova, K.; Van Ba, I.A.-T.; Le Goffic, R.; Herzog, L.; et al. Interaction of prion protein with acetylcholinesterase: potential pathobiological implications in prion diseases. Acta Neuropathol. Commun. 2015, 3, 195. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, T.; Nishizawa, K.; Sakasegawa, Y.; Oguma, A.; Teruya, K.; Kurahashi, H.; Hara, H.; Sakaguchi, S.; Doh-Ura, K. Melanin or a Melanin-Like Substance Interacts with the N-Terminal Portion of Prion Protein and Inhibits Abnormal Prion Protein Formation in Prion-Infected Cells. J. Virol. 2017, 91, e01862-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macedo, B.; Cordeiro, Y. Unraveling Prion Protein Interactions with Aptamers and Other PrP-Binding Nucleic Acids. Int. J. Mol. Sci. 2017, 18, 1023. [Google Scholar] [CrossRef] [PubMed]
- Sonati, T.; Reimann, R.R.; Falsig, J.; Baral, P.K.; O’Connor, T.; Hornemann, S.; Yaganoglu, S.; Li, B.; Herrmann, U.S.; Wieland, B.; et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 2013, 501, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, K.A.; Henderson, D.M.; Mathiason, C.K.; Hoover, E.A. Assessment of the PrPc Amino-Terminal Domain in Prion Species Barriers. J. Virol. 2016, 90, 10752–10761. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Christensen, H.M.; Stewart, L.R.; A Roth, K.; Chiesa, R.; A Harris, D. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues. EMBO J. 2007, 26, 548–558. [Google Scholar] [CrossRef]
- Wu, B.; McDonald, A.J.; Markham, K.; Rich, C.B.; McHugh, K.P.; Tatzelt, J.; Colby, D.W.; Millhauser, G.L.; Harris, D.A. The N-terminus of the prion protein is a toxic effector regulated by the C-terminus. Elife 2017, 6, e23473. [Google Scholar] [CrossRef] [PubMed]
- Watt, N.T.; Griffiths, H.H.; Hooper, N.M. Neuronal zinc regulation and the prion protein. Prion 2013, 7, 203–208. [Google Scholar] [CrossRef] [Green Version]
- Vanhoof, G.; Goossens, F.; De Meester, I.; Hendriks, D.; Scharpé, S. Proline motifs in peptides and their biological processing. FASEB J. 1995, 9, 736–744. [Google Scholar] [CrossRef]
- Linden, R. The Biological Function of the Prion Protein: A Cell Surface Scaffold of Signaling Modules. Front. Mol. Neurosci. 2017, 10, 336. [Google Scholar] [CrossRef]
- Lim, K.H.; Nguyen, T.N.; Damo, S.M.; Mazur, T.; Ball, H.L.; Prusiner, S.B.; Pines, A.; Wemmer, D.E. Solid-state NMR structural studies of the fibril form of a mutant mouse prion peptide PrP89–143(P101L). Solid State Nucl. Magn. Reson. 2006, 29, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Norstrom, E.M.; Mastrianni, J.A. The AGAAAAGA Palindrome in PrP Is Required to Generate a Productive PrPSc-PrPC Complex That Leads to Prion Propagation. J. Biol. Chem. 2005, 280, 27236–27243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.H.; Koriath, C.; Jaunmuktane, Z.; Campbell, T.; Joiner, S.; Wadsworth, J.D.; Hosszu, L.L.; Brandner, S.; Parvez, A.; Truelsen, T.C.; et al. Evaluating the causality of novel sequence variants in the prion protein gene by example. Neurobiol. Aging 2018, 71, 265.e1–265.e7. [Google Scholar] [CrossRef] [PubMed]
- Di Fede, G.; Catania, M.; Atzori, C.; Moda, F.; Pasquali, C.; Indaco, A.; Grisoli, M.; Zuffi, M.; Guaita, M.C.; Testi, R.; et al. Clinical and neuropathological phenotype associated with the novel V189I mutation in the prion protein gene. Acta Neuropathol. Commun. 2019, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Cescatti, M.; Saverioni, D.; Capellari, S.; Tagliavini, F.; Kitamoto, T.; Ironside, J.; Giese, A.; Parchi, P. Analysis of conformational stability of abnormal prion protein aggregates across the spectrum of creutzfeldt-jakob disease prions. J. Virol. 2016, 90, 6244–6254. [Google Scholar] [CrossRef] [PubMed]
- Brandner, S.; Jaunmuktane, Z. Prion disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 197–222. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Mutation | Domain | Clinical Phenotype |
|---|---|---|
| Pro39Leu | N-terminal | FTLD, FTD |
| Pro102Leu | N-terminal | Classical CJD-like symptoms, GSS |
| Pro105Leu | N-terminal | GSS, spastic paraparesis and progressive dementia |
| Pro105Ser, Pro105Thr | N-terminal | GSS |
| Gly114Val | N-terminal | CJD, neuropsychiatric symptoms |
| Ala117Val | N-terminal | CJD, Progressive cortical dementia and cerebellar ataxia |
| Octapeptide insertions (from 4 to 9 OR insertions) | N-terminal | CJD |
| Gly131Val | C-terminal | GSS, tremor and apraxia |
| Gln160-nonsense; Tyr163-nonsense | C-terminal | Alzheimer’s disease-type pathology |
| Val176Gly | C-terminal | Cerebellum ataxia, personality changes and progressive dementia |
| Asp178Asn | C-terminal | CJD and FFI depends on the allele on codon 129, Met or Val |
| Val189Ile | C-terminal | Classical and atypical CJD (behavioral abnormalities, ataxia and extrapyramidal features) |
| Val180Ile, Thr183Ala, Thr188Lys, Glu196Lys, Glu196Ala, Glu200Lys, Glu200Gly, Val203Ile, Arg208His, Val210Ile, Glu211Gln, Ile215Val | C-terminal | Classical and atypical CJD |
| Gln160-nonsense, His187Arg, Phe198Ser, Asp202Asn, Glu2011Gln, Gln212Pro, Gln217Arg, Tyr226-nonsense, Gln227-nonsense | C-terminal | Classical and atypical GSS |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernardi, L.; Bruni, A.C. Mutations in Prion Protein Gene: Pathogenic Mechanisms in C-Terminal vs. N-Terminal Domain, a Review. Int. J. Mol. Sci. 2019, 20, 3606. https://doi.org/10.3390/ijms20143606
Bernardi L, Bruni AC. Mutations in Prion Protein Gene: Pathogenic Mechanisms in C-Terminal vs. N-Terminal Domain, a Review. International Journal of Molecular Sciences. 2019; 20(14):3606. https://doi.org/10.3390/ijms20143606
Chicago/Turabian StyleBernardi, Livia, and Amalia C. Bruni. 2019. "Mutations in Prion Protein Gene: Pathogenic Mechanisms in C-Terminal vs. N-Terminal Domain, a Review" International Journal of Molecular Sciences 20, no. 14: 3606. https://doi.org/10.3390/ijms20143606
APA StyleBernardi, L., & Bruni, A. C. (2019). Mutations in Prion Protein Gene: Pathogenic Mechanisms in C-Terminal vs. N-Terminal Domain, a Review. International Journal of Molecular Sciences, 20(14), 3606. https://doi.org/10.3390/ijms20143606