Acute Functional Adaptations in Isolated Presynaptic Terminals Unveil Synaptosomal Learning and Memory


Abstract
:1. Introduction
2. Synaptosomes to Study Transmitter Release
- (i)
- (ii)
- (iii)
- (iv)
3. Synaptosomes…
4. …And Their Up–Down Superfusion to Monitor Transmitter Release
5. Presynaptic Release-Regulating Receptors
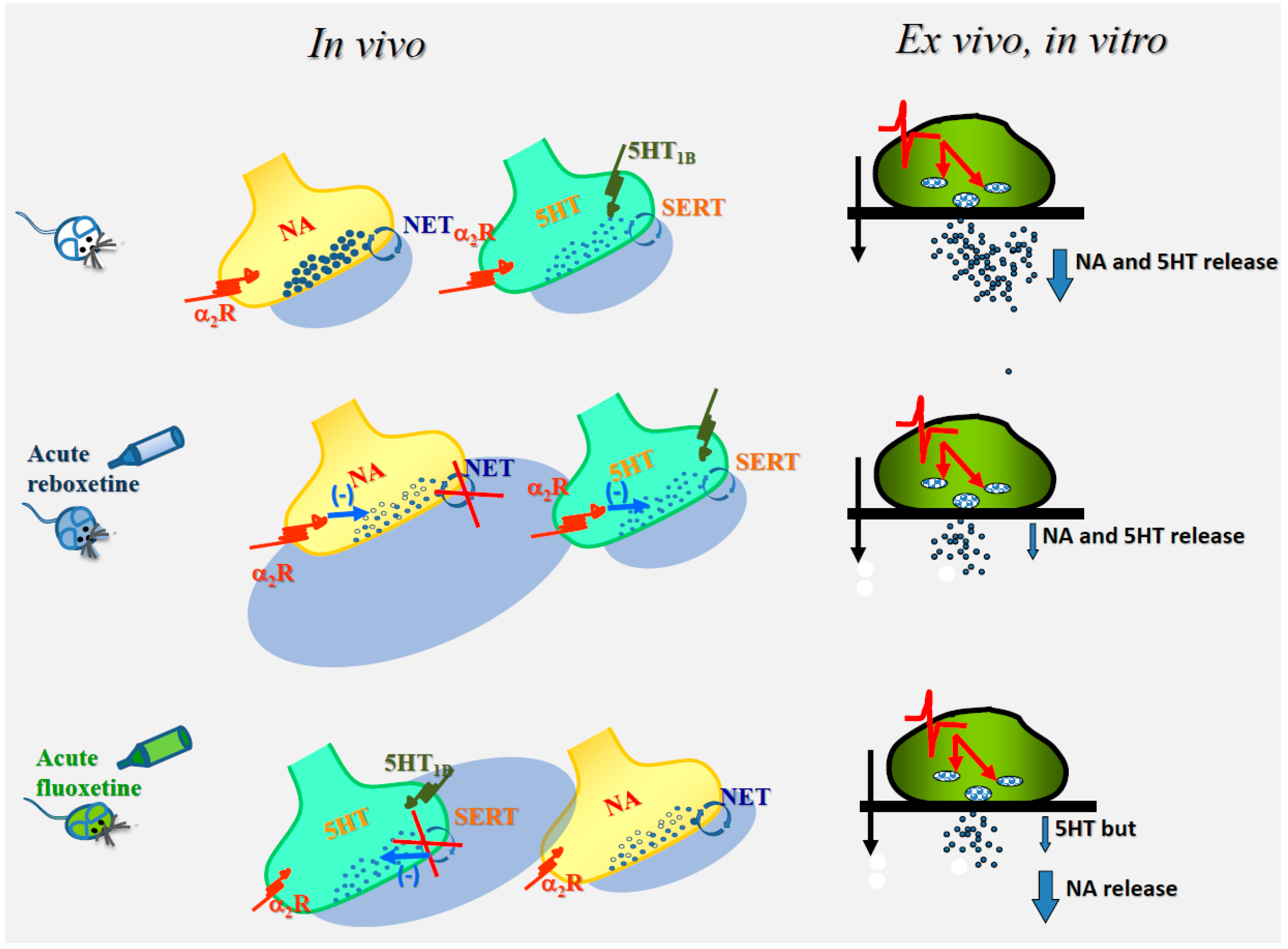
6. In Vivo Chronic Drug Administration and “Ex Vivo, In Vitro” Persistent Synaptosomal Adaptations
7. “In Vitro” Persistent “Presynaptic Adaptation” in Synaptosomes
- (i)
- synaptosomes are dynamic structures that can develop functionally relevant adaptations when exposed acutely “in vitro” to external chemical stimuli;
- (ii)
- the acute chemical-induced synaptosomal adaptations persist well beyond the physical occupancy of the selected receptor binding site(s), and possibly rely on a drug-induced cascade of events that primes persistently the functions of the isolated nerve endings.
- (iii)
- the timing of the onset of the synaptosomal adaptation (about 20 min) tends to exclude that genetic and/or epigenetic modifications can underlie these adaptations, provided that synaptosomes possess the repertoire permitting such a kind of events.
8. Acute “In Vivo” Drug Administration Elicits Persistent Presynaptic Adaptation in Synaptosomes
9. Acute “In Vivo” Drug-Induced Adaptation at Glutamatergic Nerve Endings: Implications in Pathological Conditions
10. Concluding Remarks
Conflicts of Interest
References
- Marchi, M.; Raiteri, M. On the presence in the cerebral cortex of muscarinic receptor subtypes which differ in neuronal localization, function and pharmacological properties. J. Pharmacol. Exp. Ther. 1985, 235, 230–233. [Google Scholar] [PubMed]
- Ghersi, C.; Bonfanti, A.; Manzari, B.; Feligioni, M.; Raiteri, M.; Pittaluga, A. Pharmacological heterogeneity of release-regulating presynaptic AMPA/kainate receptors in the rat brain: Study with receptor antagonists. Neurochem. Int. 2003, 42, 283–292. [Google Scholar] [CrossRef]
- Rodrigues, R.; Alfaro, T.M.; Rebola, N.; Oliveira, C.R.; Cunha, R.A. Co-localization and functional interaction between adenosine A2A and metabotropic group 5 receptors in glutamatergic nerve terminals of the rat striatum. J. Neurochem. 2005, 92, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Pittaluga, A.; Feligioni, M.; Longordo, F.; Arvigo, M.; Raiteri, M. Somatostatin-induced activation and up-regulation of N-methyl-D-aspartate receptor function: Mediation through calmodulin-dependent protein kinase II, phospholipase C, protein kinase C, and tyrosine kinase in hippocampal noradrenergic nerve endings. J. Pharmacol. Exp. Ther. 2005, 313, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Feligioni, M.; Holman, D.; Haglerod, C.; Davanger, S.; Henley, J.M. Ultrastructural localisation and differential agonist-induced regulation of AMPA and kainate receptors present at the presynaptic active zone and postsynaptic density. J. Neurochem. 2006, 99, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raiteri, M.; Marchi, M.; Paudice, P. Adaptation of presynaptic acetylcholine autoreceptors following long-term drug treatment. Eur. J. Pharmacol. 1981, 74, 109–110. [Google Scholar] [CrossRef]
- Raiteri, M.; Pittaluga, A.; Leardi, R.; Maura, G.; Marchi, M. Effects of long-term drug treatments on the sensitivity of presynaptic receptors regulating neurotransmitter release. Ann. Dell’Istituto Super Sanita 1984, 20, 57–64. [Google Scholar]
- Pittaluga, A.; Raiteri, L.; Longordo, F.; Luccini, E.; Barbiero, V.S.; Racagni, G.; Popoli, M.; Raiteri, M. Antidepressant treatments and function of glutamate ionotropic receptors mediating amine release in hippocampus. Neuropharmacology 2007, 53, 27–36. [Google Scholar] [CrossRef]
- Langer, S.Z. Therapeutic Use of Release-Modifying Drugs. Handb. Exp. Pharmacol. 2008, 184, 561–573. [Google Scholar]
- Langer, S.Z. Presynaptic autoreceptors regulating transmitter release. Neurochem. Int. 2008, 52, 26–30. [Google Scholar] [CrossRef]
- Bailey, C.P.; Trejos, J.A.; Schanne, F.A.X.; Stanton, P.K. Pairing elevation of [cyclic GMP] with inhibition of PKA produces long-term depression of glutamate release from isolated rat hippocampal presynaptic terminals. Eur. J. Neurosci. 2003, 17, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Langer, S.Z. 25 Years since the discovery of presynaptic receptors: Present knowledge and future perspectives. Trends Pharmacol. Sci. 1997, 18, 95–99. [Google Scholar] [CrossRef]
- Raiteri, L.; Raiteri, M. Synaptosomes Still Viable after 25 Years of Superfusion. Neurochem. Res. 2000, 25, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Raiteri, M. Release in vitro as a model to study neurotransmitter receptors. Pharmacol. Res. Commun. 1987, 19, 925. [Google Scholar] [CrossRef]
- Vizi, E.; Fekete, A.; Karoly, R.; Mike, A. Non-synaptic receptors and transporters involved in brain functions and targets of drug treatment. Br. J. Pharmacol. 2010, 160, 785–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittaluga, A.; Raiteri, M. N-methyl-D-aspartic acid (NMDA) and non-NMDA receptors regulating hippocampal norepinephrine release. I. Location on axon terminals and pharmacological characterization. J. Pharmacol. Exp. Ther. 1992, 260, 232–237. [Google Scholar] [PubMed]
- Pittaluga, A.; Bonfanti, A.; Raiteri, M. Differential desensitization of ionotropic non-NMDA receptors having distinct neuronal location and function. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1997, 356, 29–38. [Google Scholar] [CrossRef]
- Luccini, E.; Musante, V.; Neri, E.; Raiteri, M.; Pittaluga, A. N-methyl-D-aspartate autoreceptors respond to low and high agonist concentrations by facilitating, respectively, exocytosis and carrier-mediated release of glutamate in rat hippocampus. J. Neurosci. Res. 2007, 85, 3657–3665. [Google Scholar] [CrossRef]
- Cubelos, B.; Gime’nez, C.; Zafra, F. Localization of the GLYT1 glycine transporter at glutamatergic synapses in the rat brain. Cereb. Cortex 2005, 15, 448–459. [Google Scholar] [CrossRef]
- Musante, V.; Summa, M.; Cunha, R.A.; Raiteri, M.; Pittaluga, A. Presynaptic glycine GlyT1 transporter—NMDA receptor interaction: Relevance to NMDA autoreceptor activation in the presence of Mg2+ ions. J. Neurochem. 2011, 117, 516–527. [Google Scholar] [CrossRef]
- Ferraguti, F.; Crepaldi, L.; Nicoletti, F. Metabotropic Glutamate 1 Receptor: Current Concepts and Perspectives. Pharmacol. Rev. 2008, 60, 536–581. [Google Scholar] [CrossRef]
- Nicoletti, F.; Bockaert, J.; Collingridge, G.L.; Conn, P.J.; Ferraguti, F.; Schoepp, D.D.; Wroblewski, J.T.; Pin, J.P. Metabotropic glutamate receptors: From the workbench to the bedside. Neuropharmacology 2011, 60, 1017–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittaluga, A. Presynaptic Release-Regulating mGlu1 Receptors in Central Nervous System. Front. Pharmacol. 2016, 7, 365. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.G.; Whittaker, V.P. The isolation of nerve endings from brain: An electron-microscopic study of cell fragments derived by homogenization and centrifugation. J. Anat. 1962, 96, 79–88. [Google Scholar] [PubMed]
- Marsal, J.; Egea, G.; Solsona, C.; Rabasseda, X.; Blasi, J. Botulinum toxin type A blocks the morphological changes induced by chemical stimulation on the presynaptic membrane of Torpedo synaptosomes. Proc. Natl. Acad. Sci. USA 1989, 86, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.P.; Battenberg, E.; Wilson, M.C. SNAP-25 and synaptotagmin involvement in the final Ca(2+)-dependent triggering of neurotransmitter exocytosis. Proc. Natl. Acad. Sci. USA 1996, 93, 10471–10476. [Google Scholar] [CrossRef]
- Gundersen, C.B. The Structure of the Synaptic Vesicle-Plasma Membrane Interface Constrains SNARE Models of Rapid, Synchronous Exocytosis at Nerve Terminals. Front. Mol. Neurosci. 2017, 10, 48. [Google Scholar] [CrossRef]
- Banerjee, A.; Larsen, R.S.; Philpot, B.P.; Paulsen, O. Roles of presynaptic NMDA receptors in neurotransmission and plasticity. Trends Neurosci. 2016, 39, 26–39. [Google Scholar] [CrossRef]
- Vergassola, M.; Olivero, G.; Cisani, F.; Usai, C.; Bossi, S.; Puliti, A.; Pittaluga, A. Presynaptic mGlu1 Receptors Control GABAB Receptors in an Antagonist-Like Manner in Mouse Cortical GABAergic and Glutamatergic Nerve Endings. Front. Mol. Neurosci. 2018, 11, 324. [Google Scholar] [CrossRef] [Green Version]
- Giribaldi, F.; Milanese, M.; Bonifacino, T.; Rossi, P.; Di Prisco, S.; Pittaluga, A.; Tacchetti, C.; Puliti, A.; Usai, C.; Bonanno, G. Group I metabotropic glutamate autoreceptors induce abnormal glutamate exocytosis in a mouse model of amyotrophic lateral sclerosis. Neuropharmacology 2012, 66, 253–263. [Google Scholar] [CrossRef]
- Raiteri, L.; Raiteri, M. Multiple functions of neuronal plasma membrane transmitter transporters. Prog. Neurobiol. 2015, 134, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Marchi, M.; Grilli, M.; Pittaluga, A.M. Nicotinic modulation of glutamate receptor function at nerve terminal level: A fine-tuning of synaptic signals. Front. Pharmacol. 2015, 6, 89. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, G.; Raiteri, M. Multiple GABAB receptors. Trends Pharmacol. Sci. 1993, 14, 259–261. [Google Scholar] [CrossRef]
- Bonanno, G.; Giambelli, R.; Raiteri, L.; Tiraboschi, E.; Zappettini, S.; Musazzi, L.; Racagni, G.; Popoli, M. Chronic Antidepressants Reduce Depolarization-Evoked Glutamate Release and Protein Interactions Favoring Formation of SNARE Complex in Hippocampus. J. Neurosci. 2005, 25, 3270–3279. [Google Scholar] [CrossRef] [PubMed]
- Raiteri, M.; Angelini, F.; Levi, G. A simple apparatus for studying the release of trasmitters from synaptosomes. Eur. J. Pharmacol. 1974, 25, 411–414. [Google Scholar] [CrossRef]
- Corti, C.; Battaglia, G.; Molinaro, G.; Riozzi, B.; Pittaluga, A.; Cors, I.M.; Mugnaini, M.; Nicoletti, F.; Bruno, V. The use of knock-out mice unravels distinct roles for mGlu2 and mGlu3 metabotropic glutamate receptors in mechanisms of neurodegeneration/neuroprotection. J. Neurosci. 2007, 27, 8297–8308. [Google Scholar] [CrossRef]
- Fredholm, B.; Cunha, R.; Svenningsson, P. Pharmacology of Adenosine A2A Receptors and Therapeutic Applications. Curr. Top. Med. Chem. 2003, 3, 413–426. [Google Scholar] [CrossRef]
- Pittaluga, A.; Segantini, D.; Feligioni, M.; Raiteri, M. Extracellular protons differentially potentiate the responses of native AMPA receptor subtypes regulating neurotransmitter release. Br. J. Pharmacol. 2005, 144, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Olivero, G.; Bonfiglio, T.; Vergassola, M.; Usai, C.; Riozzi, B.; Battaglia, G.; Nicoletti, F.; Pittaluga, A. Immuno-pharmacological characterization of group II metabotropic glutamate receptors controlling glutamate exocytosis in mouse cortex and spinal cord. Br. J. Pharmacol. 2017, 174, 4785–4796. [Google Scholar] [CrossRef] [Green Version]
- Musante, V.; Neri, E.; Feligioni, M.; Puliti, A.; Pedrazzi, M.; Conti, V.; Usai, C.; Diaspro, A.; Ravazzolo, R.; Henley, J.M.; et al. Presynaptic mGlu1 and mGlu5 autoreceptors facilitate glutamate exocytosis from mouse cortical nerve endings. Neuropharmacology 2008, 55, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Musante, V.; Longordo, F.; Neri, E.; Pedrazzi, M.; Kalfas, F.; Severi, P.; Raiteri, M.; Pittaluga, A. RANTES Modulates the Release of Glutamate in Human Neocortex. J. Neurosci. 2008, 28, 12231–12240. [Google Scholar] [CrossRef] [PubMed]
- Grilli, M.; Neri, E.; Zappettini, S.; Massa, F.; Bisio, A.; Romussi, G.; Marchi, M.; Pittaluga, A. Salvinorin A exerts opposite presynaptic controls on neurotransmitter exocytosis from mouse brain nerve terminals. Neuropharmacology 2009, 57, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Summa, M.; Di Prisco, S.; Grilli, M.; Usai, C.; Marchi, M.; Pittaluga, A. Presynaptic mGlu7 receptors control GABA release in mouse hippocampus. Neuropharmacology 2013, 66, 215–224. [Google Scholar] [CrossRef]
- Olivero, G.; Vergassola, M.; Cisani, F.; Usai, C.; Pittaluga, A. Immuno-Pharmacological Characterization of Presynaptic GluN3A-Containing NMDA Autoreceptors: Relevance to Anti-NMDA Receptor Autoimmune Diseases. Mol. Neurobiol. 2019, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Heimann, A.; Gomes, I.; Devi, L. Antibodies Against G-Protein Coupled Receptors: Novel Uses in Screening and Drug Development. Comb. Chem. High Throughput Screen. 2008, 11, 463–467. [Google Scholar] [CrossRef] [Green Version]
- Hervás, C.; Pérez-Sen, R.; Miras-Portugal, M.T. Presence of diverse functional P2X receptors in rat cerebellar synaptic terminals. Biochem. Pharmacol. 2005, 70, 770–785. [Google Scholar] [CrossRef]
- Pereira, D.B.; Rebola, N.; Rodrigues, R.J.; Cunha, R.A.; Carvalho, A.P.; Duarte, C.B. Trkb receptors modulation of glutamate release is limited to a subset of nerve terminals in the adult rat hippocampus. J. Neurosci. Res. 2006, 83, 832–844. [Google Scholar] [CrossRef] [Green Version]
- Bitencourt, R.M.; Alpár, A.; Cinquina, V.; Ferreira, S.G.; Pinheiro, B.S.; Lemos, C.; Ledent, C.; Takahashi, R.N.; Sialana, F.J.; Lubec, G.; et al. Lack of presynaptic interaction between glucocorticoid and CB 1 cannabinoid receptors in GABA- and glutamatergic terminals in the frontal cortex of laboratory rodents. Neurochem. Int. 2015, 90, 72–84. [Google Scholar] [CrossRef]
- Rodrigues, R.J.; Almeida, T.; Díaz-Hernández, M.; Marques, J.M.; Franco, R.; Solsona, C.; Miras-Portugal, M.T.; Ciruela, F.; Cunha, R.A. Presynaptic P2X1-3 and α3-containing nicotinic receptors assemble into functionally interacting ion channels in the rat hippocampus. Neuropharmacology 2016, 105, 241–257. [Google Scholar] [CrossRef]
- Marrocco, J.; Reynaert, M.-L.; Gatta, E.; Gabriel, C.; Mocaër, E.; Di Prisco, S.; Merega, E.; Pittaluga, A.; Nicoletti, F.; Maccari, S.; et al. The Effects of Antidepressant Treatment in Prenatally Stressed Rats Support the Glutamatergic Hypothesis of Stress-Related Disorders. J. Neurosci. 2014, 34, 2015–2024. [Google Scholar] [CrossRef]
- Mairesse, J.; Gatta, E.; Reynaert, M.-L.; Marrocco, J.; Morley-Fletcher, S.; Soichot, M.; DeRuyter, L.; Van Camp, G.; Bouwalerh, H.; Fagioli, F.; et al. Activation of presynaptic oxytocin receptors enhances glutamate release in the ventral hippocampus of prenatally restraint stressed rats. Psychoneuroendocrinology 2015, 62, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Morley-Fletcher, S.; Zuena, A.; Mairesse, J.; Gatta, E.; Van Camp, G.; Bouwalerh, H.; Riozzi, B.; Battaglia, G.; Pittaluga, A.; Olivero, G.; et al. The reduction in glutamate release is predictive of cognitive and emotional alterations that are corrected by the positive modulator of AMPA receptors S 47445 in perinatal stressed rats. Neuropharmacology 2018, 135, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Stanton, P.K.; Heinemann, U.; Muller, W. FM1-43 Imaging Reveals cGMP-Dependent Long-Term Depression of Presynaptic Transmitter Release. J. Neurosci. 2001, 21, RC167. [Google Scholar] [CrossRef] [PubMed]
- Grilli, M.; Summa, M.; Salamone, A.; Olivero, G.; Zappettini, S.; Di Prisco, S.; Feligioni, M.; Usai, C.; Pittaluga, A.; Marchi, M. In vitro exposure to nicotine induces endocytosis of presynaptic AMPA receptors modulating dopamine release in rat nucleus accumbens nerve terminals. Neuropharmacology 2012, 63, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Salamone, A.; Zappettini, S.; Grilli, M.; Olivero, G.; Agostinho, P.; Tomé, A.R.; Chen, J.; Pittaluga, A.; Cunha, R.A.; Marchi, M. Prolonged nicotine exposure down-regulates presynaptic NMDA receptors in dopaminergic terminals of the rat nucleus accumbens. Neuropharmacology 2014, 79, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Desce, J.M.; Godeheu, G.; Galli, T.; Artaud, F.; Chéramy, A.; Glowinski, J. Presynaptic facilitation of dopamine release through D,L-alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptors on synaptosomes from the rat striatum. J. Pharmacol. Exp. Ther. 1991, 259, 692–698. [Google Scholar]
- Pittaluga, A.; Pattarini, R.; Feligioni, M.; Raiteri, M. NMDA receptors mediating hippocampal noradrenaline and striatal dopamine release display differential sensitivity to quinolinic acid, the HIV-1 envelope protein gp120, external pH and PKC inhibition. J. Neurochem. 2001, 76, 139–148. [Google Scholar] [CrossRef]
- Risso, F.; Grilli, M.; Parodi, M.; Bado, M.; Raiteri, M.; Marchi, M. Nicotine exerts a permissive role on NMDA receptor function in hippocampal noradrenergic terminals. Neuropharmacology 2004, 47, 65–71. [Google Scholar] [CrossRef]
- Guyon, A.; Massa, F.; Rovère, C.; Nahon, J.-L. How cytokines can influence the brain: A role for chemokines? J. Neuroimmunol. 2008, 198, 46–55. [Google Scholar] [CrossRef]
- Di Prisco, S.; Olivero, G.; Merega, E.; Bonfiglio, T.; Marchi, M.; Pittaluga, A. CXCR4 and NMDA Receptors Are Functionally Coupled in Rat Hippocampal Noradrenergic and Glutamatergic Nerve Endings. J. Neuroimmune Pharmacol. 2016, 11, 645–656. [Google Scholar] [CrossRef]
- Olivero, G.; Cisani, F.; Vergassola, M.; Pittaluga, A. Prolonged activation of CXCR4 hampers the release-regulating activity of presynaptic NMDA receptors in rat hippocampal synaptosomes. Neurochem. Int. 2019, 126, 59–63. [Google Scholar] [CrossRef]
- Langer, S.Z. Presynaptic regulation of the release of catecholamines. Pharmacol. Rev. 1981, 32, 337–362. [Google Scholar] [CrossRef]
- Starke, K. Presynaptic receptors. Annu. Rev. Pharmacol. Toxicol. 1981, 21, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Luccini, E.; Musante, V.; Neri, E.; Bas, M.B.; Severi, P.; Raiteri, M.; Pittaluga, A. Functional interactions between presynaptic NMDA receptors and metabotropic glutamate receptors co-expressed on rat and human noradrenergic terminals. Br. J. Pharmacol. 2007, 151, 1087–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longordo, F.; Feligioni, M.; Chiaramonte, G.; Sbaffi, P.F.; Raiteri, M.; Pittaluga, A. The human immunodeficiency virus-1 protein transactivator of transcription up-regulates N-methyl-D-aspartate receptor function by acting at metabotropic glutamate receptor 1 receptors coexisting on human and rat brain noradrenergic neurons. J. Pharmacol. Exp. Ther. 2006, 317, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Cerrito, F.; Raiteri, M. Serotonin release is modulated by presynaptic autoreceptors. Eur. J. Pharmacol. 1979, 57, 427–430. [Google Scholar] [CrossRef]
- Weinheimer, G. Extracellular 5-hydroxytryptamine inhibits 5-hydroxytryptamine release from rat brain cortex slices. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1979, 310, 93–96. [Google Scholar]
- Göthert, M.; Huth, H. α-Adrenoceptor-mediated modulation of 5-HT release from rat brain cortex slices. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1980, 313, 21–26. [Google Scholar] [CrossRef]
- Mongeau, R.; Blier, P.; De Montigny, C. The serotonergic and noradrenergic systems of the hippocampus: Their interactions and the effects of antidepressant treatments. Brain Res. Rev. 1997, 23, 145–195. [Google Scholar] [CrossRef]
- Majeed, Z.R.; Ritter, K.; Robinson, J.; Blümich, S.L.; Brailoiu, E.; Cooper, R.L. New insights into the acute actions from a high dosage of fluoxetine on neuronal and cardiac function: Drosophila, crayfish and rodent models. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2015, 176, 52–61. [Google Scholar] [CrossRef]
- Di Prisco, S.; Merega, E.; Lanfranco, M.; Casazza, S.; Uccelli, A.; Pittaluga, A. Acute desipramine restores presynaptic cortical defects in murine experimental autoimmune encephalomyelitis by suppressing central CCL5 overproduction. Br. J. Pharmacol. 2014, 171, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Prisco, S.; Merega, E.; Bonfiglio, T.; Olivero, G.; Cervetto, C.; Grilli, M.; Usai, C.; Marchi, M.; Pittaluga, A. Presynaptic, release-regulating mGlu2-preferring and mGlu3-preferring autoreceptors in CNS: Pharmacological profiles and functional roles in demyelinating disease. Br. J. Pharmacol. 2016, 173, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Vilcaes, A.A.; Furlan, G.; Roth, G.A. Inhibition of Ca2+-dependent glutamate release from cerebral cortex synaptosomes of rats with experimental autoimmune encephalomyelitis. J. Neurochem. 2009, 108, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Cid, M.; Vilcaes, A.; Rupil, L.; Salvatierra, N.; Roth, G. Participation of the GABAergic system on the glutamate release of frontal cortex synaptosomes from Wistar rats with experimental autoimmune encephalomyelitis. Neuroscience 2011, 189, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Di Prisco, S.; Merega, E.; Milanese, M.; Summa, M.; Casazza, S.; Raffaghello, L.; Pistoia, V.; Uccelli, A.; Pittaluga, A. CCL5-Glutamate interaction in central nervous system: Early and acute presynaptic defects in EAE mice. Neuropharmacology 2013, 16, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Mandolesi, G.; Gentile, A.; Musella, A.; Fresegna, D.; De Vito, F.; Bullitta, S.; Sepman, H.; Marfia, G.A.; Centonze, D. Synaptopathy connects inflammation and neurodegeneration in multiple sclerosis. Nat. Rev. Neurol. 2015, 11, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Bonfiglio, T.; Olivero, G.; Merega, E.; Di Prisco, S.; Padolecchia, C.; Grilli, M.; Milanese, M.; Di Cesare Mannelli, L.; Ghelardini, C.; Bonanno, G.; et al. Prophylactic versus Therapeutic Fingolimod: Restoration of Presynaptic Defects in Mice Suffering from Experimental Autoimmune Encephalomyelitis. PLoS ONE 2017, 12, e0170825. [Google Scholar] [CrossRef]


{kind=link}
| CNS Region | Transmitter | Drug Pre-Treatment | Depolarizing Stimulus Applied | Pretreatment Output | Reference |
|---|---|---|---|---|---|
| Rat hippocampus | Endogenous glutamate | PDE type V inhibitor PKA inhibitor | 25 mM KCl for 30 s | ⇓ | [11] |
| Rat Nucleus Accumbens | [3H]dopamine | Nicotine (30 µM) | 100 µM NMDA | ⇓ | [32,55] |
| Rat Nucleus Accumbens | [3H]dopamine | Nicotine (30 µM) | 100 µM AMPA | ⇓ | [32,54] |
| Rat hippocampus | [3H] aspartate | CXCL12 (3 nM) | 30 µM NMDA/3 nM CXCL12 | ⇓ | [60,61] |
| Rat hippocampus | [3H] noradrenaline | CXCL12 (3 nM) | 100 µM NMDA/3 nM CXCL12 | ⇓ | [60,61] |
| In Vivo Treatment | “Ex Vivo, In Vitro” Synaptosomes Adaptations | |||||
|---|---|---|---|---|---|---|
| Drug | Treatment | Synaptosomal Preparation | Transmitter | Stimulus Applied | Outcome | Ref. |
| reboxetine | 10 mg/Kg, os | rat hippocampus | [3H]noradrenaline | 12 mM KCl | ⇓ | [8] |
| reboxetine | 10 mg/Kg, os | rat hippocampus | [3H]serotonin | 12 mM KCl | ⇓ | [8] |
| desipramine | 10 mg/Kg, i.p. | mice cortex | [3H]noradrenaline | 12 mM KCl | ⇓ | [71] |
| Desipramine-yohimbine | 10 mg/Kg i.p. 0.5 mg/Kg i.p. | mice cortex | [3H]noradrenaline | 12 mM KCl | No effect | [71] |
| fluoxetine | 10 mg/Kg, i.p. | Rat hippocampus | [3H]serotonin | 12 mM KCl | ⇓ | [8] |
| fluoxetine | 10 mg/Kg, i.p. | Rat hippocampus | [3H]noradrenaline | 12 mM KCl | No effect | [8] |
| LY379268 | 1 mg/Kg, i.p. | Mouse spinal cord | [3H]D-aspartate | 15 mM KCl | ⇓ | [76] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pittaluga, A. Acute Functional Adaptations in Isolated Presynaptic Terminals Unveil Synaptosomal Learning and Memory. Int. J. Mol. Sci. 2019, 20, 3641. https://doi.org/10.3390/ijms20153641
Pittaluga A. Acute Functional Adaptations in Isolated Presynaptic Terminals Unveil Synaptosomal Learning and Memory. International Journal of Molecular Sciences. 2019; 20(15):3641. https://doi.org/10.3390/ijms20153641
Chicago/Turabian StylePittaluga, Anna. 2019. "Acute Functional Adaptations in Isolated Presynaptic Terminals Unveil Synaptosomal Learning and Memory" International Journal of Molecular Sciences 20, no. 15: 3641. https://doi.org/10.3390/ijms20153641
APA StylePittaluga, A. (2019). Acute Functional Adaptations in Isolated Presynaptic Terminals Unveil Synaptosomal Learning and Memory. International Journal of Molecular Sciences, 20(15), 3641. https://doi.org/10.3390/ijms20153641