Heme, Heme Oxygenase, and Endoplasmic Reticulum Stress—A New Insight into the Pathophysiology of Vascular Diseases


Abstract
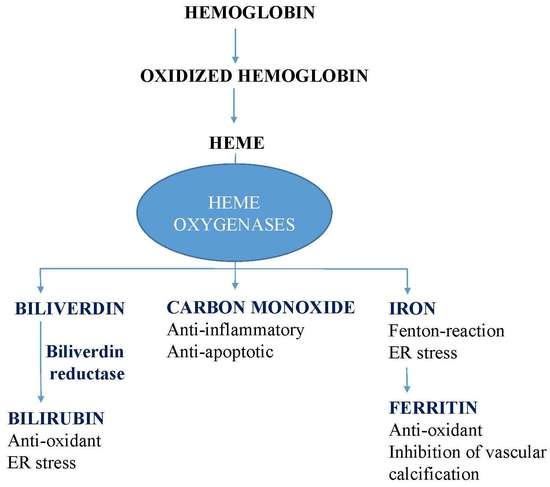
:
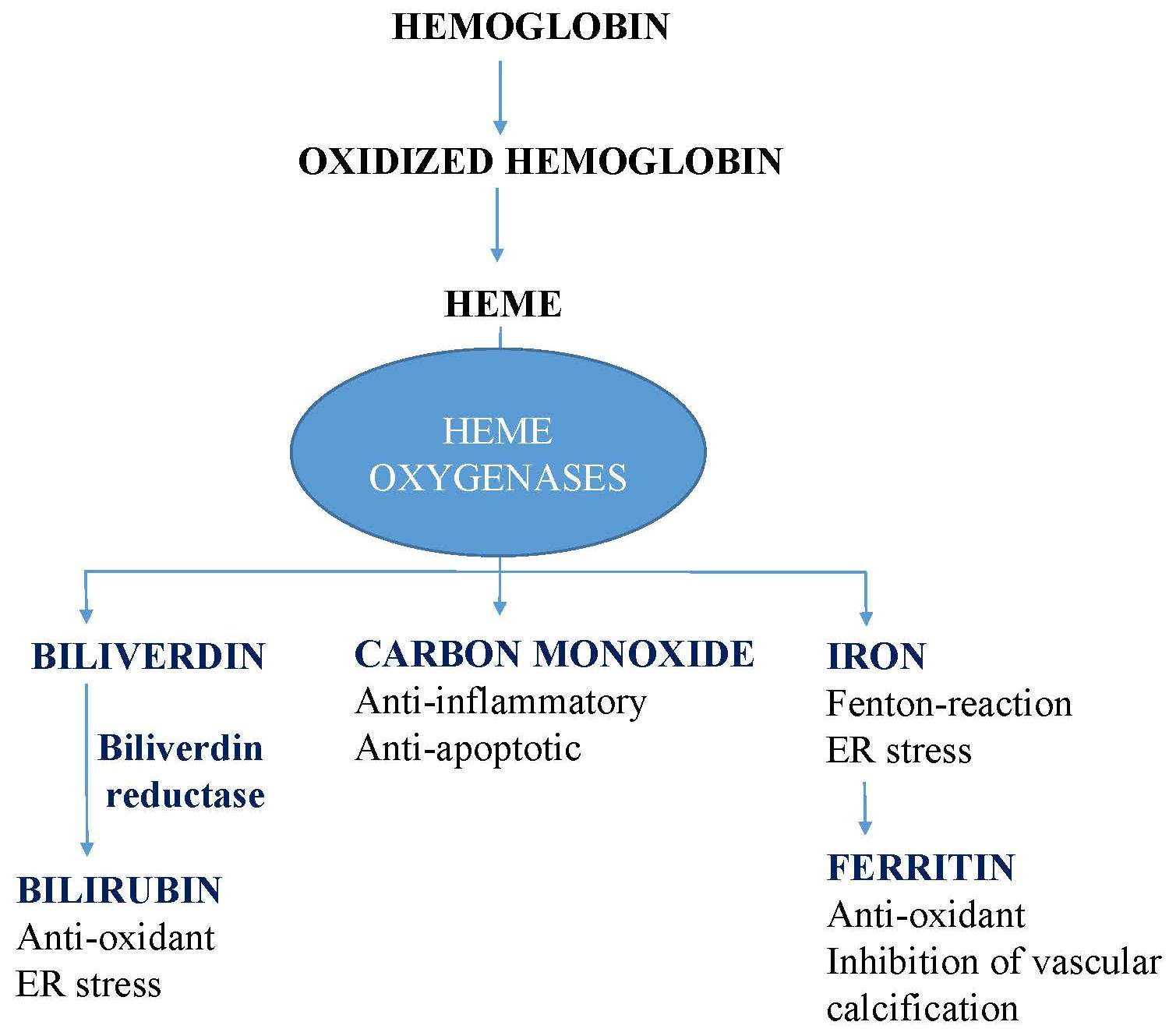{kind=link}
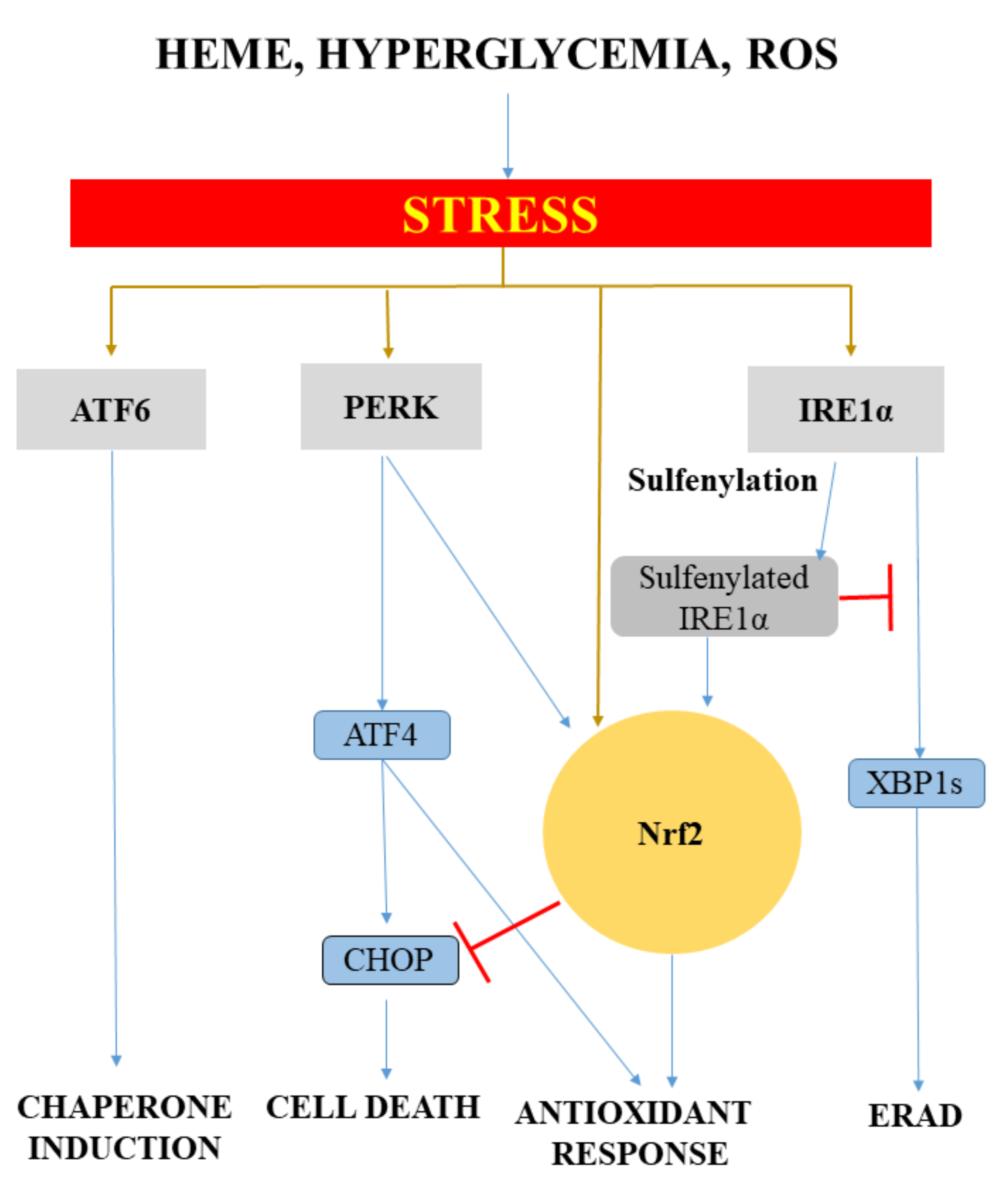{kind=link}
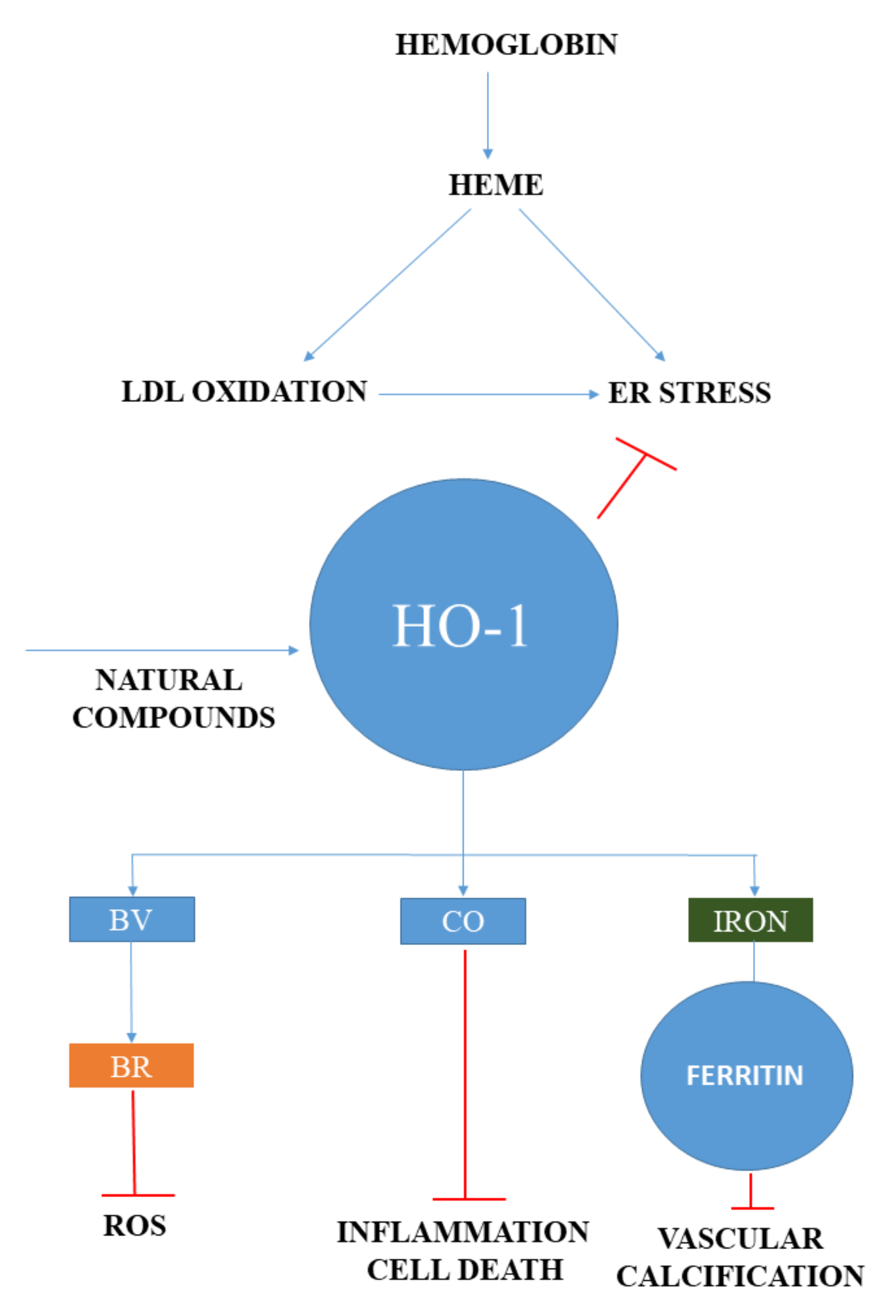{kind=link}
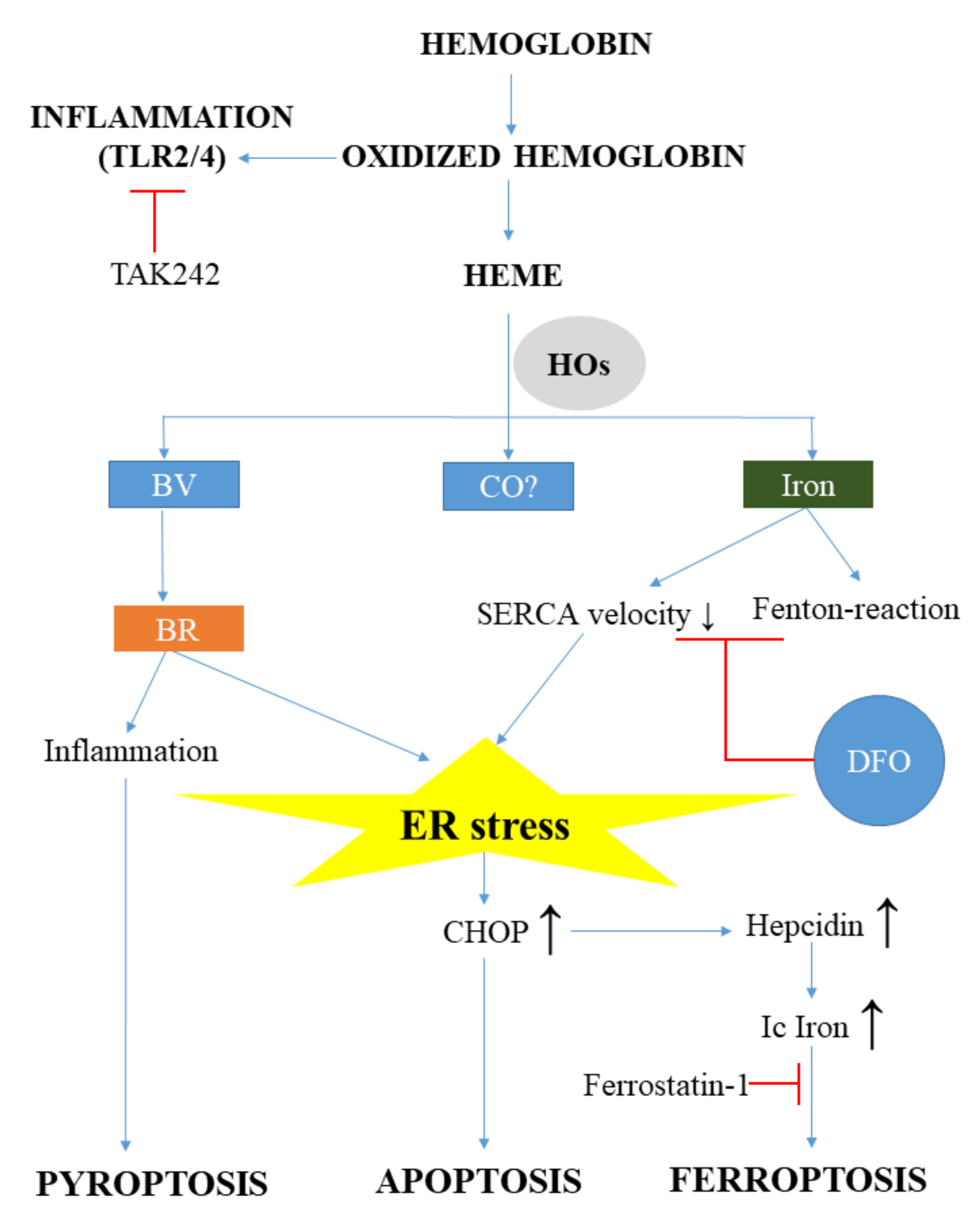{kind=link}
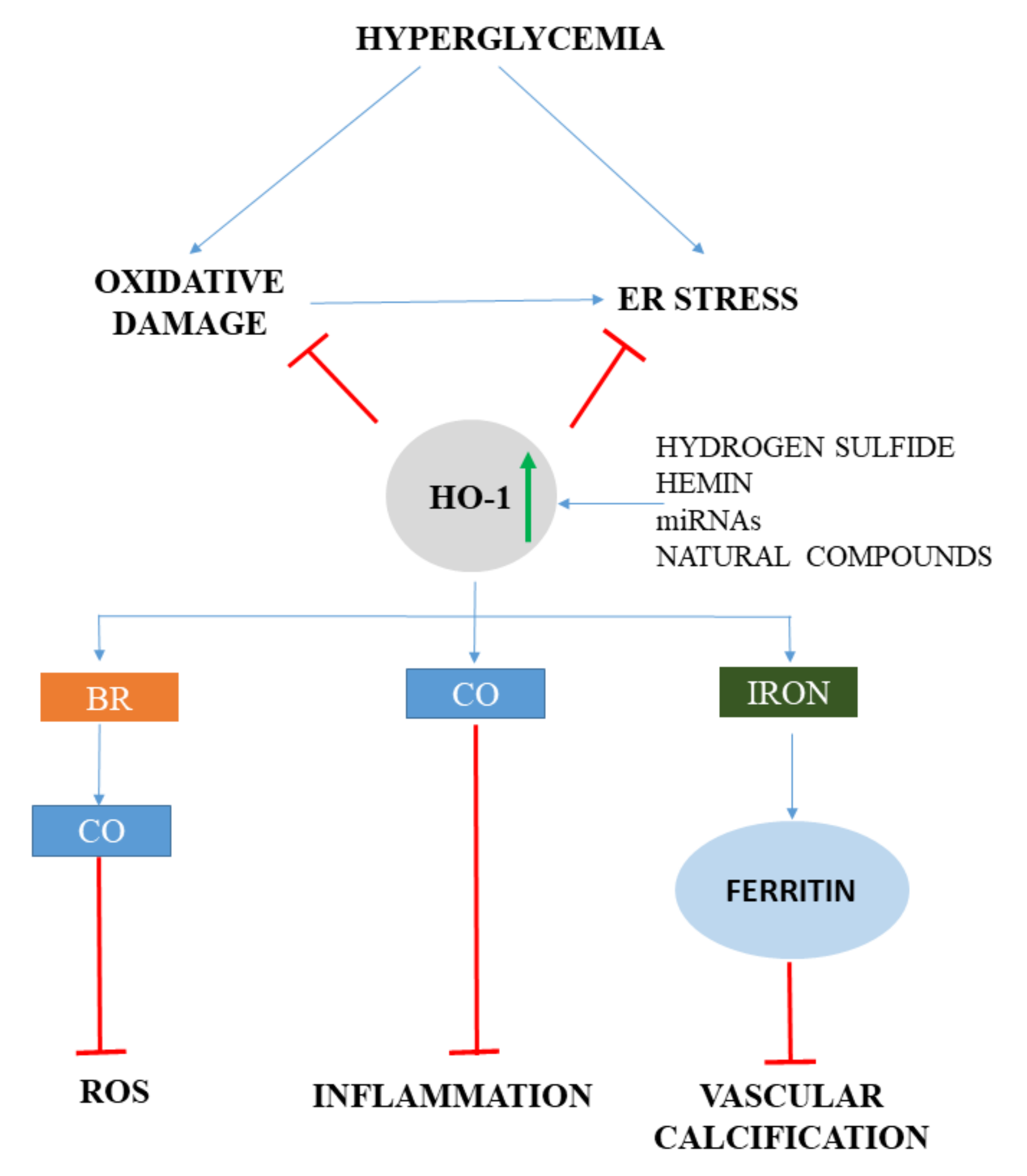{kind=link}
1. Introduction
2. Heme Stress
3. Endoplasmic Reticulum Stress
4. Atherosclerosis
4.1. Heme Stress in Atherosclerosis
4.2. HO-1 and Ferritin in Atherosclerosis
4.3. BR and CO in Atherosclerosis
4.4. ER Stress in Atherosclerosis
5. Brain
5.1. ER Stress in Brain Injury After Hemorrhage
5.2. HO-1 and HO-2 in the Brain
5.3. Iron Injury in the Brain
5.4. BR Toxicity in Brain
5.5. CO in the Brain
6. Diabetes Mellitus
6.1. Diabetic Cardiovascular Complications
6.2. HO-1 Byproducts in Diabetes
6.3. ER Stress in Diabetes
6.4. Brain and Diabetes
7. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bunn, H.F.; Jandl, J.H. Exchange of heme among hemoglobins and between hemoglobin and albumin. J. Biol. Chem. 1968, 243, 465–475. [Google Scholar] [PubMed]
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar] [PubMed]
- Stocker, R. Antioxidant activities of bile pigments. Antioxid. Redox Signal. 2004, 6, 841–849. [Google Scholar] [PubMed]
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. The heme oxygenase system: a regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef] [PubMed]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. 1968, 61. [Google Scholar] [CrossRef]
- Dunn, L.L.; Midwinter, R.G.; Ni, J.; Hamid, H.A.; Parish, C.R.; Stocker, R. New insights into intracellular locations and functions of heme oxygenase-1. Antioxid. Redox Signal. 2014, 20, 1723–1742. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M.K. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Lin, J.H.; Villalon, P.; Martasek, P.; Abraham, N.G. Regulation of heme oxygenase gene expression by cobalt in rat liver and kidney. Eur. J. Biochem. 1990, 192, 577–582. [Google Scholar] [CrossRef]
- Sardana, M.K.; Kappas, A. Dual control mechanism for heme oxygenase: tin(IV)-protoporphyrin potently inhibits enzyme activity while markedly increasing content of enzyme protein in liver. Proc. Natl. Acad. Sci. USA 1987, 84, 2464–2468. [Google Scholar] [CrossRef]
- Zhang, Y.; Furuyama, K.; Kaneko, K.; Ding, Y.; Ogawa, K.; Yoshizawa, M.; Kawamura, M.; Takeda, K.; Yoshida, T.; Shibahara, S. Hypoxia reduces the expression of heme oxygenase-2 in various types of human cell lines. A possible strategy for the maintenance of intracellular heme level. FEBS J. 2006, 273, 3136–3147. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Doré, S. Heme oxygenase 2 deficiency increases brain swelling and inflammation after intracerebral hemorrhage. Neuroscience 2008, 155, 1133–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doré, S.; Sampei, K.; Goto, S.; Alkayed, N.J.; Guastella, D.; Blackshaw, S.; Gallagher, M.; Traystman, R.J.; Hurn, P.D.; Koehler, R.C.; et al. Heme oxygenase-2 is neuroprotective in cerebral ischemia. Mol. Med. 1999, 5, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Cardozo, A.K.; Cnop, M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 2008, 29, 42–61. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Wang, Y.; Zhang, H.; Han, F. The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis 2017, 22, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Navid, F.; Colbert, R.A. Causes and consequences of endoplasmic reticulum stress in rheumatic disease. Nat. Rev. Rheumatol. 2017, 13, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J. Endoplasmic reticulum stress in lung disease. Eur. Respir. Rev. 2017, 26, 170018. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, E.A.; Orekhov, A.N. The Role of Endoplasmic Reticulum Stress and Unfolded Protein Response in Atherosclerosis. Int. J. Mol. Sci. 2016, 17, 193. [Google Scholar] [CrossRef]
- Rosen, A.D.; Frumin, N. V Focal epileptogenesis after intracortical hemoglobin injection. Exp. Neurol. 1979, 66, 277–284. [Google Scholar] [CrossRef]
- Sadrzadeh, S.M.; Anderson, D.K.; Panter, S.S.; Hallaway, P.E.; Eaton, J.W. Hemoglobin potentiates central nervous system damage. J. Clin. Invest. 1987, 79, 662–664. [Google Scholar] [CrossRef]
- Orjih, A.U.; Banyal, H.S.; Chevli, R.; Fitch, C.D. Hemin lyses malaria parasites. Science 1981, 214, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Wijayanti, N.; Katz, N.; Immenschuh, S. Biology of heme in health and disease. Curr. Med. Chem. 2004, 11, 981–986. [Google Scholar] [CrossRef] [PubMed]
- TAPPEL, A.L. Unsaturated lipide oxidation catalyzed by hematin compounds. J. Biol. Chem. 1955, 217, 721–733. [Google Scholar] [PubMed]
- Gutteridge, J.M.; Smith, A. Antioxidant protection by haemopexin of haem-stimulated lipid peroxidation. Biochem. J. 1988, 256, 861–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, T.H.; Frezzatti, W.A.; Schreier, S. Hemin-induced lipid membrane disorder and increased permeability: a molecular model for the mechanism of cell lysis. Arch. Biochem. Biophys. 1993, 307, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Aft, R.L.; Mueller, G.C. Hemin-mediated DNA strand scission. J. Biol. Chem. 1983, 258, 12069–12072. [Google Scholar] [PubMed]
- Suliman, H.B.; Carraway, M.S.; Velsor, L.W.; Day, B.J.; Ghio, A.J.; Piantadosi, C.A. Rapid mtDNA deletion by oxidants in rat liver mitochondria after hemin exposure. Free Radic. Biol. Med. 2002, 32, 246–256. [Google Scholar] [CrossRef]
- Aft, R.L.; Mueller, G.C. Hemin-mediated oxidative degradation of proteins. J. Biol. Chem. 1984, 259, 301–305. [Google Scholar]
- Gáll, T.; Pethő, D.; Nagy, A.; Hendrik, Z.; Méhes, G.; Potor, L.; Gram, M.; Åkerström, B.; Smith, A.; Nagy, P.; et al. Heme Induces Endoplasmic Reticulum Stress (HIER Stress) in Human Aortic Smooth Muscle Cells. Front. Physiol. 2018, 9, 1595. [Google Scholar] [CrossRef]
- Silva, G.; Jeney, V.; Chora, A.; Larsen, R.; Balla, J.; Soares, M.P. Oxidized hemoglobin is an endogenous proinflammatory agonist that targets vascular endothelial cells. J. Biol. Chem. 2009, 284, 29582–29595. [Google Scholar] [CrossRef]
- Wagener, F.A.; Eggert, A.; Boerman, O.C.; Oyen, W.J.; Verhofstad, A.; Abraham, N.G.; Adema, G.; van Kooyk, Y.; de Witte, T.; Figdor, C.G. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood 2001, 98, 1802–1811. [Google Scholar] [CrossRef] [PubMed]
- Balla, G.; Vercellotti, G.M.; Muller-Eberhard, U.; Eaton, J.; Jacob, H.S. Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and toxic oxygen species. Lab. Invest. 1991, 64, 648–655. [Google Scholar] [PubMed]
- Fortes, G.B.; Alves, L.S.; de Oliveira, R.; Dutra, F.F.; Rodrigues, D.; Fernandez, P.L.; Souto-Padron, T.; De Rosa, M.J.; Kelliher, M.; Golenbock, D.; et al. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 2012, 119, 2368–2375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinecke, J.W. Mechanisms of oxidative damage of low density lipoprotein in human atherosclerosis. Curr. Opin. Lipidol. 1997, 8, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef]
- Kobayashi, A.; Ohta, T.; Yamamoto, M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol. 2004, 378, 273–286. [Google Scholar]
- Beck, K.; Wu, B.J.; Ni, J.; Santiago, F.S.; Malabanan, K.P.; Li, C.; Wang, Y.; Khachigian, L.M.; Stocker, R. Interplay between heme oxygenase-1 and the multifunctional transcription factor yin yang 1 in the inhibition of intimal hyperplasia. Circ. Res. 2010, 107, 1490–1497. [Google Scholar] [CrossRef]
- Camhi, S.L.; Alam, J.; Otterbein, L.; Sylvester, S.L.; Choi, A.M. Induction of heme oxygenase-1 gene expression by lipopolysaccharide is mediated by AP-1 activation. Am. J. Respir. Cell Mol. Biol. 1995, 13, 387–398. [Google Scholar] [CrossRef]
- Alam, J.; Igarashi, K.; Immenschuh, S.; Shibahara, S.; Tyrrell, R.M. Regulation of Heme Oxygenase-1 Gene Transcription: Recent Advances and Highlights from the International Conference (Uppsala, 2003) on Heme Oxygenase. Antioxid. Redox Signal. 2004, 6, 924–933. [Google Scholar]
- Lee, P.J.; Jiang, B.H.; Chin, B.Y.; Iyer, N.V.; Alam, J.; Semenza, G.L.; Choi, A.M. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 1997, 272, 5375–5381. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science (80-. ). 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamanaka, R.B.; Bennett, B.S.; Cullinan, S.B.; Diehl, J.A. PERK and GCN2 Contribute to eIF2 Phosphorylation and Cell Cycle Arrest after Activation of the Unfolded Protein Response Pathway. Mol. Biol. Cell 2005, 16, 5493–5501. [Google Scholar] [CrossRef] [PubMed]
- Ameri, K.; Harris, A.L. Activating transcription factor 4. Int. J. Biochem. Cell Biol. 2008, 40, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Mori, M.; Akira, S.; Gotoh, T. C/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammation. J. Immunol. 2006, 176, 6245–6253. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- He, Y.; Sun, S.; Sha, H.; Liu, Z.; Yang, L.; Xue, Z.; Chen, H.; Qi, L. Emerging roles for XBP1, a sUPeR transcription factor. Gene Expr. 2010, 15, 13–25. [Google Scholar] [CrossRef]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef] [Green Version]
- Todd, D.J.; Lee, A.-H.; Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008, 8, 663–674. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Pae, H.-O.; Zheng, M.; Park, R.; Kim, Y.-M.; Chung, H.-T. Carbon Monoxide Induces Heme Oxygenase-1 via Activation of Protein Kinase R–Like Endoplasmic Reticulum Kinase and Inhibits Endothelial Cell Apoptosis Triggered by Endoplasmic Reticulum Stress. Circ. Res. 2007, 101, 919–927. [Google Scholar] [CrossRef] [PubMed]
- He, C.H.; Gong, P.; Hu, B.; Stewart, D.; Choi, M.E.; Choi, A.M.K.; Alam, J. Identification of Activating Transcription Factor 4 (ATF4) as an Nrf2-interacting Protein. J. Biol. Chem. 2001, 276, 20858–20865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, S.; Sayers, C.M.; Verginadis, I.I.; Lehman, S.L.; Cheng, Y.; Cerniglia, G.J.; Tuttle, S.W.; Feldman, M.D.; Zhang, P.J.L.; Fuchs, S.Y.; et al. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Invest. 2015, 125, 2592–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, Z.-H.; Du, Z.-X.; Li, N.; Li, C.; Zhang, Q.; Liu, B.-Q.; Guan, Y.; Wang, H.-Q. Implication of Nrf2 and ATF4 in differential induction of CHOP by proteasome inhibition in thyroid cancer cells. Biochim. Biophys. Acta - Mol. Cell Res. 2012, 1823, 1395–1404. [Google Scholar] [CrossRef] [Green Version]
- Hourihan, J.M.; Moronetti Mazzeo, L.E.; Fernández-Cárdenas, L.P.; Blackwell, T.K. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol. Cell 2016, 63, 553–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef]
- Li, W.; Östblom, M.; Xu, L.-H.; Hellsten, A.; Leanderson, P.; Liedberg, B.; Brunk, U.T.; Eaton, J.W.; Yuan, X.-M. Cytocidal effects of atheromatous plaque components: the death zone revisited. FASEB J. 2006, 20, 2281–2290. [Google Scholar] [CrossRef] [Green Version]
- Nagy, E.; Eaton, J.W.; Jeney, V.; Soares, M.P.; Varga, Z.; Galajda, Z.; Szentmiklosi, J.; Mehes, G.; Csonka, T.; Smith, A.; et al. Red Cells, Hemoglobin, Heme, Iron, and Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1347–1353. [Google Scholar] [CrossRef]
- Balla, J.; Jacob, H.S.; Balla, G.; Nath, K.; Eaton, J.W.; Vercellotti, G.M. Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. USA 1993, 90, 9285–9289. [Google Scholar] [CrossRef] [PubMed]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercellotti, G.M. Hemin: A possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb. J. Vasc. Biol. 1991, 11, 1700–1711. [Google Scholar] [CrossRef]
- Potor, L.; Bányai, E.; Becs, G.; Soares, M.P.; Balla, G.; Balla, J.; Jeney, V. Atherogenesis may involve the prooxidant and proinflammatory effects of ferryl hemoglobin. Oxid. Med. Cell. Longev. 2013, 2013, 676425. [Google Scholar] [CrossRef] [PubMed]
- Buttari, B.; Profumo, E.; Cuccu, B.; Straface, E.; Gambardella, L.; Malorni, W.; Genuini, I.; Capoano, R.; Salvati, B.; Riganò, R. Erythrocytes from patients with carotid atherosclerosis fail to control dendritic cell maturation. Int. J. Cardiol. 2012, 155, 484–486. [Google Scholar] [CrossRef] [PubMed]
- Profumo, E.; Buttari, B.; Petrone, L.; Straface, E.; Gambardella, L.; Pietraforte, D.; Genuini, I.; Capoano, R.; Salvati, B.; Malorni, W.; et al. Redox imbalance of red blood cells impacts T lymphocyte homeostasis: implication in carotid atherosclerosis. Thromb. Haemost. 2011, 106, 1117–1126. [Google Scholar]
- Hennig, B.; Chow, C.K. Lipid peroxidation and endothelial cell injury: implications in atherosclerosis. Free Radic. Biol. Med. 1988, 4, 99–106. [Google Scholar] [CrossRef]
- Steinberg, D.; Parthasarathy, S.; Carew, T.E.; Khoo, J.C.; Witztum, J.L.; Witztum, J.L. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N. Engl. J. Med. 1989, 320, 915–924. [Google Scholar]
- Witztum, J.L.; Steinberg, D. Role of oxidized low density lipoprotein in atherogenesis. J. Clin. Invest. 1991, 88, 1785–1792. [Google Scholar] [CrossRef]
- Thomas, J.P.; Geiger, P.G.; Girotti, A.W. Lethal damage to endothelial cells by oxidized low density lipoprotein: role of selenoperoxidases in cytoprotection against lipid hydroperoxide- and iron-mediated reactions. J. Lipid Res. 1993, 34, 479–490. [Google Scholar]
- Guyton, J.R.; Lenz, M.L.; Mathews, B.; Hughes, H.; Karsan, D.; Selinger, E.; Smith, C. V Toxicity of oxidized low density lipoproteins for vascular smooth muscle cells and partial protection by antioxidants. Atherosclerosis 1995, 118, 237–249. [Google Scholar] [CrossRef]
- Marchant, C.E.; Law, N.S.; van der Veen, C.; Hardwick, S.J.; Carpenter, K.L.; Mitchinson, M.J. Oxidized low-density lipoprotein is cytotoxic to human monocyte-macrophages: protection with lipophilic antioxidants. FEBS Lett. 1995, 358, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Balla, J.; Balla, G.; Croatt, A.J.; Vercellotti, G.M.; Nath, K.A. Renal tubular epithelial cells mimic endothelial cells upon exposure to oxidized LDL. Am. J. Physiol. 1996, 271, F814–F823. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Lee, T.S.; Lee, F.Y.; Pai, R.C.; Chau, L.Y. Expression of heme oxygenase-1 in atherosclerotic lesions. Am. J. Pathol. 1998, 152, 711–720. [Google Scholar] [PubMed]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Invest. 1999, 103, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Nagy, E.; Jeney, V.; Yachie, A.; Szabó, R.P.; Wagner, O.; Vercellotti, G.M.; Eaton, J.W.; Balla, G.; Balla, J. Oxidation of hemoglobin by lipid hydroperoxide associated with low-density lipoprotein (LDL) and increased cytotoxic effect by LDL oxidation in heme oxygenase-1 (HO-1) deficiency. Cell. Mol. Biol. (Noisy-le-grand) 2005, 51, 377–385. [Google Scholar]
- Cheng, C.; Noordeloos, A.M.; Jeney, V.; Soares, M.P.; Moll, F.; Pasterkamp, G.; Serruys, P.W.; Duckers, H.J. Heme oxygenase 1 determines atherosclerotic lesion progression into a vulnerable plaque. Circulation 2009, 119, 3017–3027. [Google Scholar] [CrossRef]
- Kishimoto, Y.; Sasaki, K.; Saita, E.; Niki, H.; Ohmori, R.; Kondo, K.; Momiyama, Y. Plasma Heme Oxygenase-1 Levels and Carotid Atherosclerosis. Stroke 2018, 49, 2230–2232. [Google Scholar] [CrossRef]
- Mao, H.; Tao, T.; Wang, X.; Liu, M.; Song, D.; Liu, X.; Shi, D. Zedoarondiol Attenuates Endothelial Cells Injury Induced by Oxidized Low-Density Lipoprotein via Nrf2 Activation. Cell. Physiol. Biochem. 2018, 48, 1468–1479. [Google Scholar] [CrossRef]
- Li, W.; Zhi, W.; Liu, F.; He, Z.; Wang, X.; Niu, X. Atractylenolide I restores HO-1 expression and inhibits Ox-LDL-induced VSMCs proliferation, migration and inflammatory responses in vitro. Exp. Cell Res. 2017, 353, 26–34. [Google Scholar] [CrossRef]
- Vogel, M.E.; Idelman, G.; Konaniah, E.S.; Zucker, S.D. Bilirubin Prevents Atherosclerotic Lesion Formation in Low-Density Lipoprotein Receptor-Deficient Mice by Inhibiting Endothelial VCAM-1 and ICAM-1 Signaling. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Lapenna, D.; Ciofani, G.; Pierdomenico, S.D.; Giamberardino, M.A.; Ucchino, S.; Davì, G. Association of serum bilirubin with oxidant damage of human atherosclerotic plaques and the severity of atherosclerosis. Clin. Exp. Med. 2018, 18, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Wu, J.; Wang, A.; Xu, C.; Wang, Z.; Zhao, X. Lower serum indirect bilirubin levels are inversely related to carotid intima-media thickness progression. Curr. Neurovasc. Res. 2019, 16. [Google Scholar] [CrossRef] [PubMed]
- Vítek, L.; Jirsa, M.; Brodanová, M.; Kalab, M.; Marecek, Z.; Danzig, V.; Novotný, L.; Kotal, P. Gilbert syndrome and ischemic heart disease: a protective effect of elevated bilirubin levels. Atherosclerosis 2002, 160, 449–456. [Google Scholar] [CrossRef]
- Soni, H.; Pandya, G.; Patel, P.; Acharya, A.; Jain, M.; Mehta, A.A. Beneficial effects of carbon monoxide-releasing molecule-2 (CORM-2) on acute doxorubicin cardiotoxicity in mice: role of oxidative stress and apoptosis. Toxicol. Appl. Pharmacol. 2011, 253, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Segersvärd, H.; Lakkisto, P.; Hänninen, M.; Forsten, H.; Siren, J.; Immonen, K.; Kosonen, R.; Sarparanta, M.; Laine, M.; Tikkanen, I. Carbon monoxide releasing molecule improves structural and functional cardiac recovery after myocardial injury. Eur. J. Pharmacol. 2018, 818, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-J.; Xu, D.-Y.; Sun, Y.-X.; Xue, T.; Zhang, C.-X.; Zhang, Z.-X.; Lin, W.; Li, K.-X. CO-releasing molecules-2 attenuates ox-LDL-induced injury in HUVECs by ameliorating mitochondrial function and inhibiting Wnt/β-catenin pathway. Biochem. Biophys. Res. Commun. 2017, 490, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Petrick, L.; Rosenblat, M.; Aviram, M. In vitro effects of exogenous carbon monoxide on oxidative stress and lipid metabolism in macrophages. Toxicol. Ind. Health 2016, 32, 1318–1323. [Google Scholar] [CrossRef]
- Torti, S.V.; Kwak, E.L.; Miller, S.C.; Miller, L.L.; Ringold, G.M.; Myambo, K.B.; Young, A.P.; Torti, F.M. The molecular cloning and characterization of murine ferritin heavy chain, a tumor necrosis factor-inducible gene. J. Biol. Chem. 1988, 263, 12638–12644. [Google Scholar]
- Zarjou, A.; Jeney, V.; Arosio, P.; Poli, M.; Antal-Szalmás, P.; Agarwal, A.; Balla, G.; Balla, J. Ferritin prevents calcification and osteoblastic differentiation of vascular smooth muscle cells. J. Am. Soc. Nephrol. 2009, 20, 1254–1263. [Google Scholar] [CrossRef]
- Sikura, K.É.; Potor, L.; Szerafin, T.; Zarjou, A.; Agarwal, A.; Arosio, P.; Poli, M.; Hendrik, Z.; Méhes, G.; Oros, M.; et al. Potential Role of H-Ferritin in Mitigating Valvular Mineralization. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 413–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becs, G.; Zarjou, A.; Agarwal, A.; Kovács, K.É.; Becs, Á.; Nyitrai, M.; Balogh, E.; Bányai, E.; Eaton, J.W.; Arosio, P.; et al. Pharmacological induction of ferritin prevents osteoblastic transformation of smooth muscle cells. J. Cell. Mol. Med. 2016, 20, 217–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.; Bai, Y.-P.; Gao, H.-C.; Wang, X.; Li, L.-F.; Zhang, G.-G.; Hu, C.-P. Ox-LDL induces endothelial cell apoptosis via the LOX-1-dependent endoplasmic reticulum stress pathway. Atherosclerosis 2014, 235, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Myoishi, M.; Hao, H.; Minamino, T.; Watanabe, K.; Nishihira, K.; Hatakeyama, K.; Asada, Y.; Okada, K.-I.; Ishibashi-Ueda, H.; Gabbiani, G.; et al. Increased Endoplasmic Reticulum Stress in Atherosclerotic Plaques Associated With Acute Coronary Syndrome. Circulation 2007, 116, 1226–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.K.; Yu, P.L.; Bai, Y.P.; Yan, S.T.; Zhao, S.P.; Zhang, G.Q. Role of PERK/eIF2α/CHOP Endoplasmic Reticulum Stress Pathway in Oxidized Low-density Lipoprotein Mediated Induction of Endothelial Apoptosis. Biomed. Environ. Sci. 2016, 29, 868–876. [Google Scholar]
- Yao, S.; Miao, C.; Tian, H.; Sang, H.; Yang, N.; Jiao, P.; Han, J.; Zong, C.; Qin, S. Endoplasmic reticulum stress promotes macrophage-derived foam cell formation by up-regulating cluster of differentiation 36 (CD36) expression. J. Biol. Chem. 2014, 289, 4032–4042. [Google Scholar] [CrossRef]
- Erbay, E.; Babaev, V.R.; Mayers, J.R.; Makowski, L.; Charles, K.N.; Snitow, M.E.; Fazio, S.; Wiest, M.M.; Watkins, S.M.; Linton, M.F.; et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat. Med. 2009, 15, 1383–1391. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhang, D.; Liu, X.; Li, X.; Liu, F.; Yu, Y.; Jia, S.; Zhou, Y.; Zhao, Y. Endoplasmic Reticulum Stress Affects Lipid Metabolism in Atherosclerosis Via CHOP Activation and Over-Expression of miR-33. Cell. Physiol. Biochem. 2018, 48, 1995–2010. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Mozzini, C.; Fratta Pasini, A.; Garbin, U.; Stranieri, C.; Pasini, A.; Vallerio, P.; Cominacini, L. Increased endoplasmic reticulum stress and Nrf2 repression in peripheral blood mononuclear cells of patients with stable coronary artery disease. Free Radic. Biol. Med. 2014, 68, 178–185. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Q.; Shi, L.; Qin, L.; Zhang, Q.; Mi, M. Equol Attenuates Atherosclerosis in Apolipoprotein E-Deficient Mice by Inhibiting Endoplasmic Reticulum Stress via Activation of Nrf2 in Endothelial Cells. PLoS ONE 2016, 11, e0167020. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Sun, G.; Dong, X.; Wang, M.; Qin, M.; Yu, Y.; Sun, X. Isorhamnetin attenuates atherosclerosis by inhibiting macrophage apoptosis via PI3K/AKT activation and HO-1 induction. PLoS ONE 2015, 10, e0120259. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Tong, M.; Ou, B.; Liu, C.; Hu, C.; Yang, Y. Isorhamnetin protects against bleomycin-induced pulmonary fibrosis by inhibiting endoplasmic reticulum stress and epithelial-mesenchymal transition. Int. J. Mol. Med. 2019, 43, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-C.; Zhou, Y.; Fang, H.; Lin, S.; Wang, P.-F.; Xiong, R.-P.; Chen, J.; Xiong, X.-Y.; Lv, F.-L.; Liang, Q.-L.; et al. Toll-like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann. Neurol. 2014, 75, 876–889. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.S.; Woo, S.K.; Kurland, D.B.; Yoon, S.H.; Palmer, A.F.; Banerjee, U.; Iqbal, S.; Ivanova, S.; Gerzanich, V.; Simard, J.M. Methemoglobin is an endogenous toll-like receptor 4 ligand-relevance to subarachnoid hemorrhage. Int. J. Mol. Sci. 2015, 16, 5028–5046. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graça-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Yin, Q.; Zhong, Q.; Lv, F.-L.; Zhou, Y.; Li, J.-Q.; Wang, J.-Z.; Su, B.; Yang, Q.-W. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J. Neuroinflammation 2012, 9, 46. [Google Scholar] [CrossRef]
- Min, H.; Choi, B.; Jang, Y.H.; Cho, I.-H.; Lee, S.J. Heme molecule functions as an endogenous agonist of astrocyte TLR2 to contribute to secondary brain damage after intracerebral hemorrhage. Mol. Brain 2017, 10, 27. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-C.; Wang, P.-F.; Fang, H.; Chen, J.; Xiong, X.-Y.; Yang, Q.-W. Toll-like receptor 4 antagonist attenuates intracerebral hemorrhage-induced brain injury. Stroke 2013, 44, 2545–2552. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, Y.; He, C.; Wang, T.; Piao, Q.; Liu, Q. Silencing the gene encoding C/EBP homologous protein lessens acute brain injury following ischemia/reperfusion. Neural Regen. Res. 2012, 7, 2432–2438. [Google Scholar]
- Yan, F.; Cao, S.; Li, J.; Dixon, B.; Yu, X.; Chen, J.; Gu, C.; Lin, W.; Chen, G. Pharmacological Inhibition of PERK Attenuates Early Brain Injury After Subarachnoid Hemorrhage in Rats Through the Activation of Akt. Mol. Neurobiol. 2017, 54, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Zhang, J.; Dang, B.; Li, H.; Shen, H.; Li, X.; Wang, Z. PERK Pathway Activation Promotes Intracerebral Hemorrhage Induced Secondary Brain Injury by Inducing Neuronal Apoptosis Both in Vivo and in Vitro. Front. Neurosci. 2018, 12, 111. [Google Scholar] [CrossRef] [PubMed]
- Schallner, N.; Pandit, R.; LeBlanc, R.; Thomas, A.J.; Ogilvy, C.S.; Zuckerbraun, B.S.; Gallo, D.; Otterbein, L.E.; Hanafy, K.A. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J. Clin. Invest. 2015, 125, 2609–2625. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Doré, S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain 2007, 130, 1643–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeppen, A.H.; Dickson, A.C.; Smith, J. Heme oxygenase in experimental intracerebral hemorrhage: the benefit of tin-mesoporphyrin. J. Neuropathol. Exp. Neurol. 2004, 63, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Song, Y.; Zhang, Z.; Li, D.; Zhu, H.; Liang, R.; Gu, Y.; Pang, Y.; Qi, J.; Wu, H.; et al. Distinct role of heme oxygenase-1 in early- and late-stage intracerebral hemorrhage in 12-month-old mice. J. Cereb. Blood Flow Metab. 2017, 37, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yang, Q.; Li, G.; Wang, L.; Hu, W.; Tang, Q.; Li, D.; Sun, Z. Time course of heme oxygenase-1 and oxidative stress after experimental intracerebral hemorrhage. Acta Neurochir. (Wien) 2011, 153, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Day, J.P.; Phillips, H.; Slootsky, B.; Tolosano, E.; Doré, S. Deletion of the hemopexin or heme oxygenase-2 gene aggravates brain injury following stroma-free hemoglobin-induced intracerebral hemorrhage. J. Neuroinflammation 2016, 13, 26. [Google Scholar] [CrossRef] [Green Version]
- Chen-Roetling, J.; Cai, Y.; Regan, R.F. Neuroprotective effect of heme oxygenase-2 knockout in the blood injection model of intracerebral hemorrhage. BMC Res. Notes 2014, 7, 561. [Google Scholar] [CrossRef]
- Graf, E.; Mahoney, J.R.; Bryant, R.G.; Eaton, J.W. Iron-catalyzed hydroxyl radical formation. Stringent requirement for free iron coordination site. J. Biol. Chem. 1984, 259, 3620–3624. [Google Scholar]
- Kaplán, P.; Doval, M.; Majerová, Z.; Lehotský, J.; Racay, P. Iron-induced lipid peroxidation and protein modification in endoplasmic reticulum membranes. Protection by stobadine. Int. J. Biochem. Cell Biol. 2000, 32, 539–547. [Google Scholar] [CrossRef]
- Kaplan, P.; Babusikova, E.; Lehotsky, J.; Dobrota, D. Free radical-induced protein modification and inhibition of Ca2+-ATPase of cardiac sarcoplasmic reticulum. Mol. Cell. Biochem. 2003, 248, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Vecchi, C.; Montosi, G.; Zhang, K.; Lamberti, I.; Duncan, S.A.; Kaufman, R.J.; Pietrangelo, A. ER stress controls iron metabolism through induction of hepcidin. Science 2009, 325, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Xiang, X.; Zhang, H.; Jiang, D.; Liang, Y.; Qing, W.; Liu, L.; Zhao, Q.; He, Z. CHOP induces apoptosis by affecting brain iron metabolism in rats with subarachnoid hemorrhage. Exp. Neurol. 2018, 302, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Han, X.; Lan, X.; Gao, Y.; Wan, J.; Durham, F.; Cheng, T.; Yang, J.; Wang, Z.; Jiang, C.; et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI insight 2017, 2, e90777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selim, M. Deferoxamine Mesylate: A New Hope for Intracerebral Hemorrhage: From Bench to Clinical Trials. Stroke 2009, 40, S90–S91. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wan, J.; Lan, X.; Han, X.; Wang, Z.; Wang, J. Neuroprotection of brain-permeable iron chelator VK-28 against intracerebral hemorrhage in mice. J. Cereb. Blood Flow Metab. 2017, 37, 3110–3123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBlanc, R.H.; Chen, R.; Selim, M.H.; Hanafy, K.A. Heme oxygenase-1-mediated neuroprotection in subarachnoid hemorrhage via intracerebroventricular deferoxamine. J. Neuroinflammation 2016, 13, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef]
- Stocker, R.; Glazer, A.N.; Ames, B.N. Antioxidant activity of albumin-bound bilirubin. Proc. Natl. Acad. Sci. USA 1987, 84, 5918–5922. [Google Scholar] [CrossRef]
- Cremer, R.J.; Perryman, P.W.; Richards, D.H. Influence of light on the hyperbilirubinÆmia of infants. Lancet 1958, 271, 1094–1097. [Google Scholar] [CrossRef]
- McDonagh, A.F.; Palma, L.A.; Trull, F.R.; Lightner, D.A. Phototherapy for neonatal jaundice. Configurational isomers of bilirubin. J. Am. Chem. Soc. 1982, 104, 6865–6867. [Google Scholar] [CrossRef]
- Diamond, L.K.; Allen, F.H.; Thomas, W.O. Erythroblastosis Fetalis. N. Engl. J. Med. 1951, 244, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Newman, T.B.; Maisels, M.J. Evaluation and treatment of jaundice in the term newborn: a kinder, gentler approach. Pediatrics 1992, 89, 809–818. [Google Scholar] [PubMed]
- Valaes, T.; Petmezaki, S.; Henschke, C.; Drummond, G.S.; Kappas, A. Control of jaundice in preterm newborns by an inhibitor of bilirubin production: studies with tin-mesoporphyrin. Pediatrics 1994, 93, 1–11. [Google Scholar] [PubMed]
- Kamisako, T.; Kobayashi, Y.; Takeuchi, K.; Ishihara, T.; Higuchi, K.; Tanaka, Y.; Gabazza, E.C.; Adachi, Y. Recent advances in bilirubin metabolism research: the molecular mechanism of hepatocyte bilirubin transport and its clinical relevance. J. Gastroenterol. 2000, 35, 659–664. [Google Scholar] [CrossRef]
- Qaisiya, M.; Brischetto, C.; Jašprová, J.; Vitek, L.; Tiribelli, C.; Bellarosa, C. Bilirubin-induced ER stress contributes to the inflammatory response and apoptosis in neuronal cells. Arch. Toxicol. 2017, 91, 1847–1858. [Google Scholar] [CrossRef]
- Schiavon, E.; Smalley, J.L.; Newton, S.; Greig, N.H.; Forsythe, I.D. Neuroinflammation and ER-stress are key mechanisms of acute bilirubin toxicity and hearing loss in a mouse model. PLoS ONE 2018, 13, e0201022. [Google Scholar] [CrossRef]
- Feng, J.; Li, M.; Wei, Q.; Li, S.; Song, S.; Hua, Z. Unconjugated bilirubin induces pyroptosis in cultured rat cortical astrocytes. J. Neuroinflammation 2018, 15, 23. [Google Scholar] [CrossRef]
- Yabluchanskiy, A.; Sawle, P.; Homer-Vanniasinkam, S.; Green, C.J.; Foresti, R.; Motterlini, R. CORM-3, a carbon monoxide-releasing molecule, alters the inflammatory response and reduces brain damage in a rat model of hemorrhagic stroke*. Crit. Care Med. 2012, 40, 544–552. [Google Scholar] [CrossRef]
- Maritim, A.C.; Sanders, R.A.; Watkins, J.B. Diabetes, oxidative stress, and antioxidants: a review. J. Biochem. Mol. Toxicol. 2003, 17, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Regidor, D.L.; Kopple, J.D.; Kovesdy, C.P.; Kilpatrick, R.D.; McAllister, C.J.; Aronovitz, J.; Greenland, S.; Kalantar-Zadeh, K. Associations between changes in hemoglobin and administered erythropoiesis-stimulating agent and survival in hemodialysis patients. J. Am. Soc. Nephrol. 2006, 17, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Quan, S.; Kaminski, P.M.; Yang, L.; Morita, T.; Inaba, M.; Ikehara, S.; Goodman, A.I.; Wolin, M.S.; Abraham, N.G. Heme oxygenase-1 prevents superoxide anion-associated endothelial cell sloughing in diabetic rats. Biochem. Biophys. Res. Commun. 2004, 315, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Turkseven, S.; Mingone, C.J.; Gupte, S.A.; Wolin, M.S.; Abraham, N.G. Heme oxygenase-1 gene expression increases vascular relaxation and decreases inducible nitric oxide synthase in diabetic rats. Cell. Mol. Biol. (Noisy-le-grand) 2005, 51, 371–376. [Google Scholar]
- Di Pascoli, M.; Rodella, L.; Sacerdoti, D.; Bolognesi, M.; Turkseven, S.; Abraham, N.G. Chronic CO levels have [corrected] a beneficial effect on vascular relaxation in diabetes. Biochem. Biophys. Res. Commun. 2006, 340, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Li, M.; Vanella, L.; Kim, D.H.; Rezzani, R.; Rodella, L.; Sodhi, K.; Canestraro, M.; Martasek, P.; Peterson, S.J.; et al. Adipocyte heme oxygenase-1 induction attenuates metabolic syndrome in both male and female obese mice. Hypertens. (Dallas, Tex. 1979) 2010, 56, 1124–1130. [Google Scholar] [CrossRef]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen Sulfide Induces Keap1 S-sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef] [Green Version]
- Gupta, I.; Goyal, A.; Singh, N.K.; Yadav, H.N.; Sharma, P.L. Hemin, a heme oxygenase-1 inducer, restores the attenuated cardioprotective effect of ischemic preconditioning in isolated diabetic rat heart. Hum. Exp. Toxicol. 2017, 36, 867–875. [Google Scholar] [CrossRef]
- Ali, M.A.M.; Heeba, G.H.; El-Sheikh, A.A.K. Modulation of heme oxygenase-1 expression and activity affects streptozotocin-induced diabetic nephropathy in rats. Fundam. Clin. Pharmacol. 2017, 31, 546–557. [Google Scholar] [CrossRef]
- Gou, L.; Zhao, L.; Song, W.; Wang, L.; Liu, J.; Zhang, H.; Huang, Y.; Lau, C.W.; Yao, X.; Tian, X.Y.; et al. Inhibition of miR-92a Suppresses Oxidative Stress and Improves Endothelial Function by Upregulating Heme Oxygenase-1 in db/db Mice. Antioxid. Redox Signal. 2018, 28, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, Q.; Li, S. MicroRNA-218 promotes high glucose-induced apoptosis in podocytes by targeting heme oxygenase-1. Biochem. Biophys. Res. Commun. 2016, 471, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.I.; Chander, P.N.; Rezzani, R.; Schwartzman, M.L.; Regan, R.F.; Rodella, L.; Turkseven, S.; Lianos, E.A.; Dennery, P.A.; Abraham, N.G. Heme oxygenase-2 deficiency contributes to diabetes-mediated increase in superoxide anion and renal dysfunction. J. Am. Soc. Nephrol. 2006, 17, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Nitti, M.; Piras, S.; Furfaro, A.L.; Traverso, N.; Pronzato, M.A.; Mann, G.E. Heme oxygenase-1-derived bilirubin protects endothelial cells against high glucose-induced damage. Free Radic. Biol. Med. 2015, 89, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Tian, X.Y.; Liu, L.; Wong, W.T.; Zhang, Y.; Han, Q.-B.; Ho, H.-M.; Wang, N.; Wong, S.L.; et al. Unconjugated Bilirubin Mediates Heme Oxygenase-1–Induced Vascular Benefits in Diabetic Mice. Diabetes 2015, 64, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Hamur, H.; Duman, H.; Demirtas, L.; Bakirci, E.M.; Durakoglugil, M.E.; Degirmenci, H.; Kalkan, K.; Yildirim, E.; Vuruskan, E. Total Bilirubin Levels Predict Subclinical Atherosclerosis in Patients With Prediabetes. Angiology 2016, 67, 909–915. [Google Scholar] [CrossRef]
- Dong, H.; Huang, H.; Yun, X.; Kim, D.; Yue, Y.; Wu, H.; Sutter, A.; Chavin, K.D.; Otterbein, L.E.; Adams, D.B.; et al. Bilirubin Increases Insulin Sensitivity in Leptin-Receptor Deficient and Diet-Induced Obese Mice Through Suppression of ER Stress and Chronic Inflammation. Endocrinology 2014, 155, 818–828. [Google Scholar] [CrossRef]
- Lee, D.W.; Shin, H.Y.; Jeong, J.H.; Han, J.; Ryu, S.; Nakahira, K.; Moon, J.-S. Carbon monoxide regulates glycolysis-dependent NLRP3 inflammasome activation in macrophages. Biochem. Biophys. Res. Commun. 2017, 493, 957–963. [Google Scholar] [CrossRef]
- Wang, Y.; Ying, L.; Chen, Y.; Shen, Y.; Guo, R.; Jin, K.; Wang, L. Induction of heme oxygenase-1 ameliorates vascular dysfunction in streptozotocin-induced type 2 diabetic rats. Vascul. Pharmacol. 2014, 61, 16–24. [Google Scholar] [CrossRef]
- Yahagi, K.; Kolodgie, F.D.; Lutter, C.; Mori, H.; Romero, M.E.; Finn, A.V.; Virmani, R. Pathology of Human Coronary and Carotid Artery Atherosclerosis and Vascular Calcification in Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 191–204. [Google Scholar] [CrossRef] [Green Version]
- Boon, A.; Cheriex, E.; Lodder, J.; Kessels, F. Cardiac valve calcification: characteristics of patients with calcification of the mitral annulus or aortic valve. Heart 1997, 78, 472–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Horany, H.E.-S.; Watany, M.M.; Hagag, R.Y.; El-Attar, S.H.; Basiouny, M.A. Expression of LRP1 and CHOP genes associated with peripheral neuropathy in type 2 diabetes mellitus: Correlations with nerve conduction studies. Gene 2019, 702, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Brosius, F.C.; Kaufman, R.J. Is the ER stressed out in diabetic kidney disease? J. Am. Soc. Nephrol. 2008, 19, 2040–2042. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Chávez, G.; Hernández-Ramírez, E.; Osorio-Paz, I.; Hernández-Espinosa, C.; Salceda, R. Potential Role of Endoplasmic Reticulum Stress in Pathogenesis of Diabetic Retinopathy. Neurochem. Res. 2016, 41, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-W.; Zhu, H.-T.; Chen, K.-L.; Dong, X.; Wei, J.; Qiu, C.; Xue, J.-H. Protein kinase RNA-like endoplasmic reticulum kinase (PERK) signaling pathway plays a major role in reactive oxygen species (ROS)-mediated endoplasmic reticulum stress-induced apoptosis in diabetic cardiomyopathy. Cardiovasc. Diabetol. 2013, 12, 158. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Yang, S.; An, N.; Wang, G.; Xu, Q.; Liu, J.; Mao, Y. Astragalus polysaccharides inhibits cardiomyocyte apoptosis during diabetic cardiomyopathy via the endoplasmic reticulum stress pathway. J. Ethnopharmacol. 2019, 238, 111857. [Google Scholar] [CrossRef] [PubMed]
- Mozzini, C.; Garbin, U.; Stranieri, C.; Pasini, A.; Solani, E.; Tinelli, I.A.; Cominacini, L.; Fratta Pasini, A.M. Endoplasmic reticulum stress and Nrf2 repression in circulating cells of type 2 diabetic patients without the recommended glycemic goals. Free Radic. Res. 2015, 49, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Maamoun, H.; Zachariah, M.; McVey, J.H.; Green, F.R.; Agouni, A. Heme oxygenase (HO)-1 induction prevents Endoplasmic Reticulum stress-mediated endothelial cell death and impaired angiogenic capacity. Biochem. Pharmacol. 2017, 127, 46–59. [Google Scholar] [CrossRef]
- Yang, X.; Yao, W.; Liu, H.; Gao, Y.; Liu, R.; Xu, L. Tangluoning, a traditional Chinese medicine, attenuates in vivo and in vitro diabetic peripheral neuropathy through modulation of PERK/Nrf2 pathway. Sci. Rep. 2017, 7, 1014. [Google Scholar] [CrossRef]
- Ding, Y.; Dai, X.; Zhang, Z.; Jiang, Y.; Ma, X.; Cai, X.; Li, Y. Proanthocyanidins protect against early diabetic peripheral neuropathy by modulating endoplasmic reticulum stress. J. Nutr. Biochem. 2014, 25, 765–772. [Google Scholar] [CrossRef]
- Kang, M.-K.; Lee, E.-J.; Kim, Y.-H.; Kim, D.Y.; Oh, H.; Kim, S.-I.; Kang, Y.-H. Chrysin Ameliorates Malfunction of Retinoid Visual Cycle through Blocking Activation of AGE-RAGE-ER Stress in Glucose-Stimulated Retinal Pigment Epithelial Cells and Diabetic Eyes. Nutrients 2018, 10, 1046. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-S.; Lii, C.-K.; Lin, A.-H.; Yeh, Y.-W.; Yao, H.-T.; Li, C.-C.; Wang, T.-S.; Chen, H.-W. Protection by chrysin, apigenin, and luteolin against oxidative stress is mediated by the Nrf2-dependent up-regulation of heme oxygenase 1 and glutamate cysteine ligase in rat primary hepatocytes. Arch. Toxicol. 2013, 87, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cao, P.; Gui, J.; Wang, X.; Han, J.; Wang, Y.; Wang, G. Arctigenin ameliorates renal impairment and inhibits endoplasmic reticulum stress in diabetic db/db mice. Life Sci. 2019, 223, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.-H.; Park, J.-S.; Kim, D.-H.; Kim, H.-S. Arctigenin Increases Hemeoxygenase-1 Gene Expression by Modulating PI3K/AKT Signaling Pathway in Rat Primary Astrocytes. Biomol. Ther. (Seoul) 2014, 22, 497–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, F.-J.; Ma, L.-L.; Guo, J.-J.; Xu, L.-H.; Li, Y.; Qu, S. Endoplasmic reticulum stress/autophagy pathway is involved in diabetes-induced neuronal apoptosis and cognitive decline in mice. Clin. Sci. (Lond.) 2018, 132, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-M.; Lin, C.-C.; Hsieh, H.-L. High-Glucose-Derived Oxidative Stress-Dependent Heme Oxygenase-1 Expression from Astrocytes Contributes to the Neuronal Apoptosis. Mol. Neurobiol. 2017, 54, 470–483. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gáll, T.; Balla, G.; Balla, J. Heme, Heme Oxygenase, and Endoplasmic Reticulum Stress—A New Insight into the Pathophysiology of Vascular Diseases. Int. J. Mol. Sci. 2019, 20, 3675. https://doi.org/10.3390/ijms20153675
Gáll T, Balla G, Balla J. Heme, Heme Oxygenase, and Endoplasmic Reticulum Stress—A New Insight into the Pathophysiology of Vascular Diseases. International Journal of Molecular Sciences. 2019; 20(15):3675. https://doi.org/10.3390/ijms20153675
Chicago/Turabian StyleGáll, Tamás, György Balla, and József Balla. 2019. "Heme, Heme Oxygenase, and Endoplasmic Reticulum Stress—A New Insight into the Pathophysiology of Vascular Diseases" International Journal of Molecular Sciences 20, no. 15: 3675. https://doi.org/10.3390/ijms20153675
APA StyleGáll, T., Balla, G., & Balla, J. (2019). Heme, Heme Oxygenase, and Endoplasmic Reticulum Stress—A New Insight into the Pathophysiology of Vascular Diseases. International Journal of Molecular Sciences, 20(15), 3675. https://doi.org/10.3390/ijms20153675