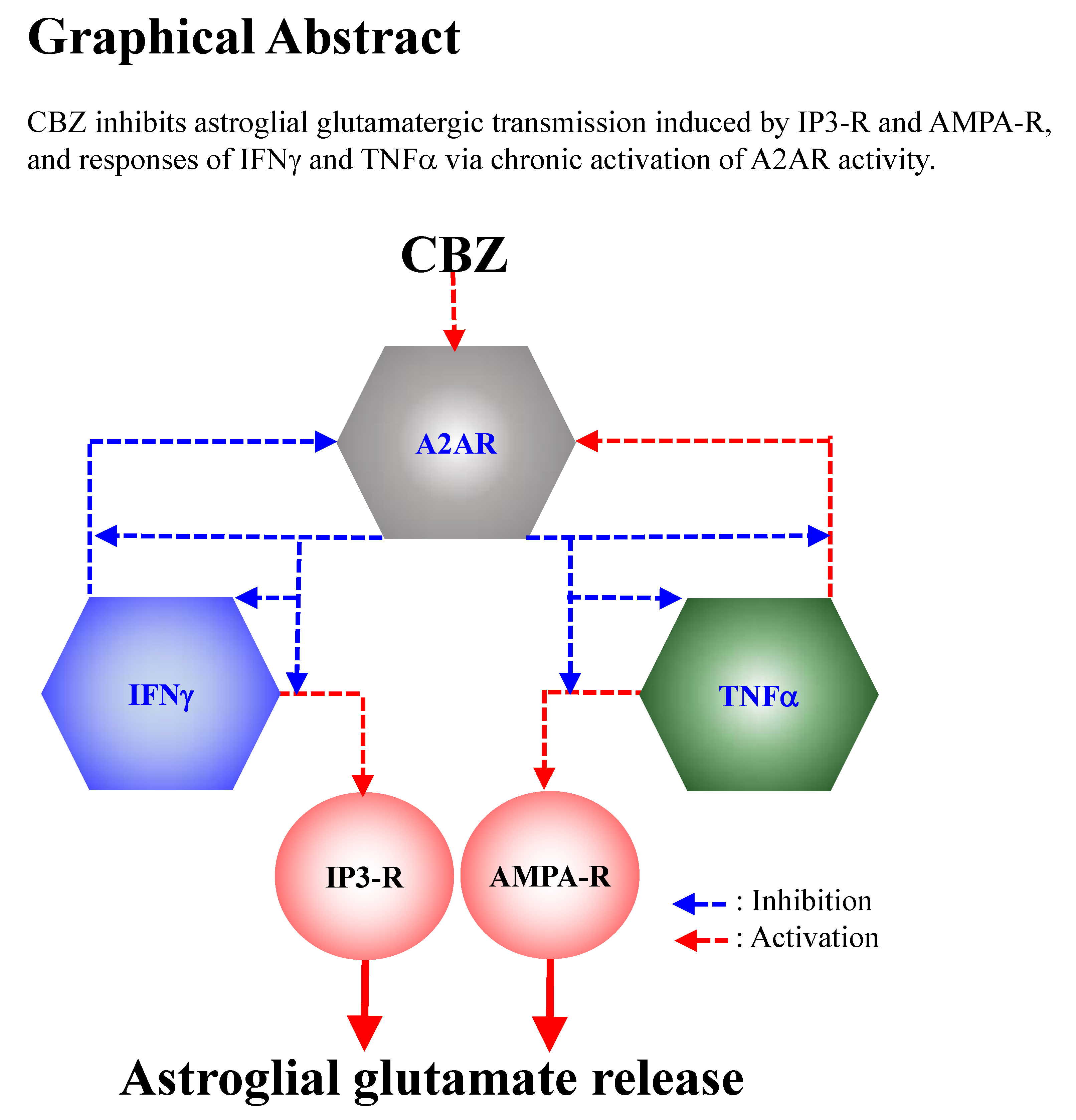
Carbamazepine Attenuates Astroglial L-Glutamate Release Induced by Pro-Inflammatory Cytokines via Chronically Activation of Adenosine A2A Receptor


Abstract
:
1. Introduction
2. Results
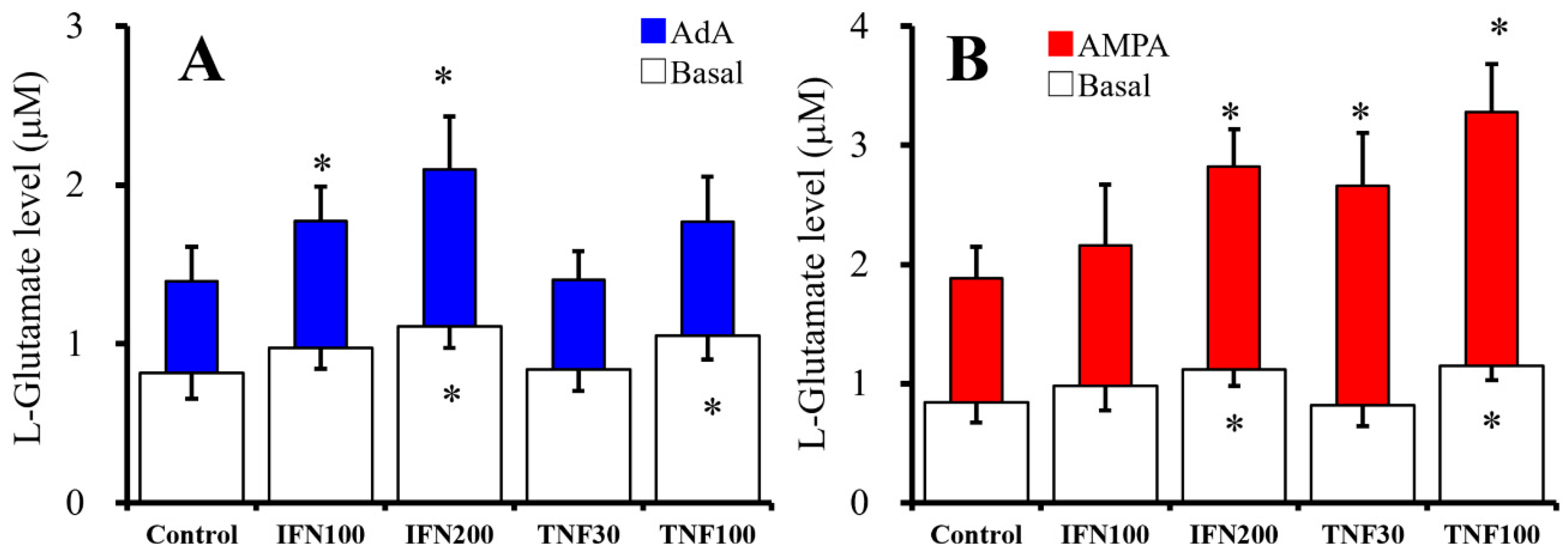
2.1. Chronic Effects of IFNγ and TNFα on Basal, AMPA-, and AdA-Evoked Astroglial L-Glutamate Releases (Study 1)
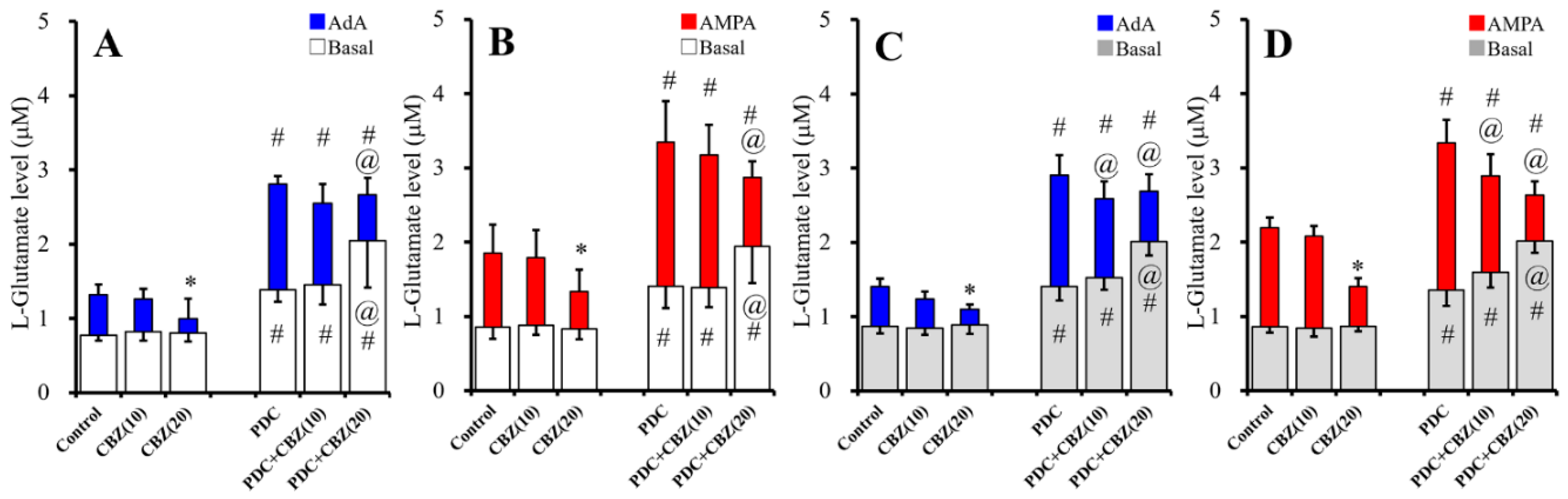
2.2. Acute and Chronic Effects of Therapeutic-Relevant Concentration of CBZ on Astroglial Releases of L-Glutamate (Study 2)
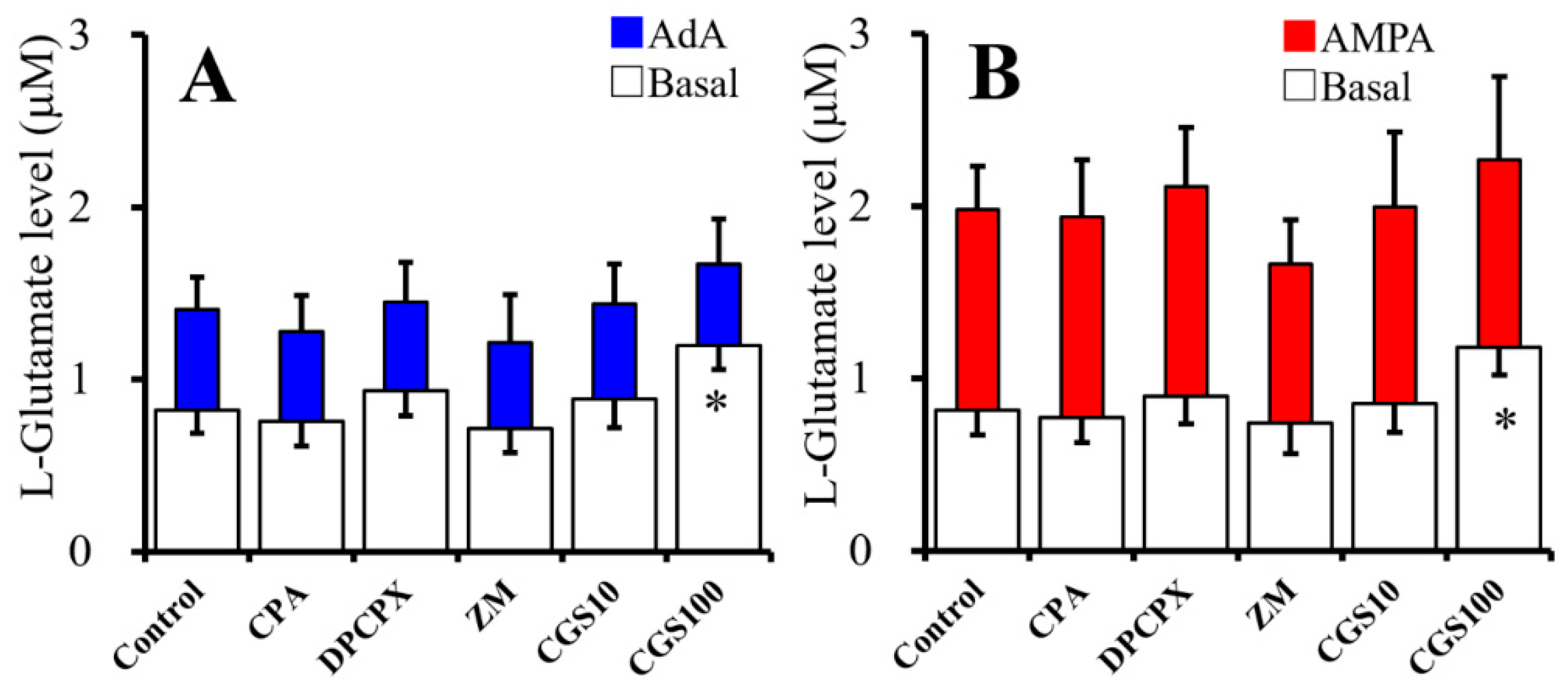
2.3. Acute Effects of Adenosine Receptor Agents on Astroglial L-Glutamate Release (Study 3)
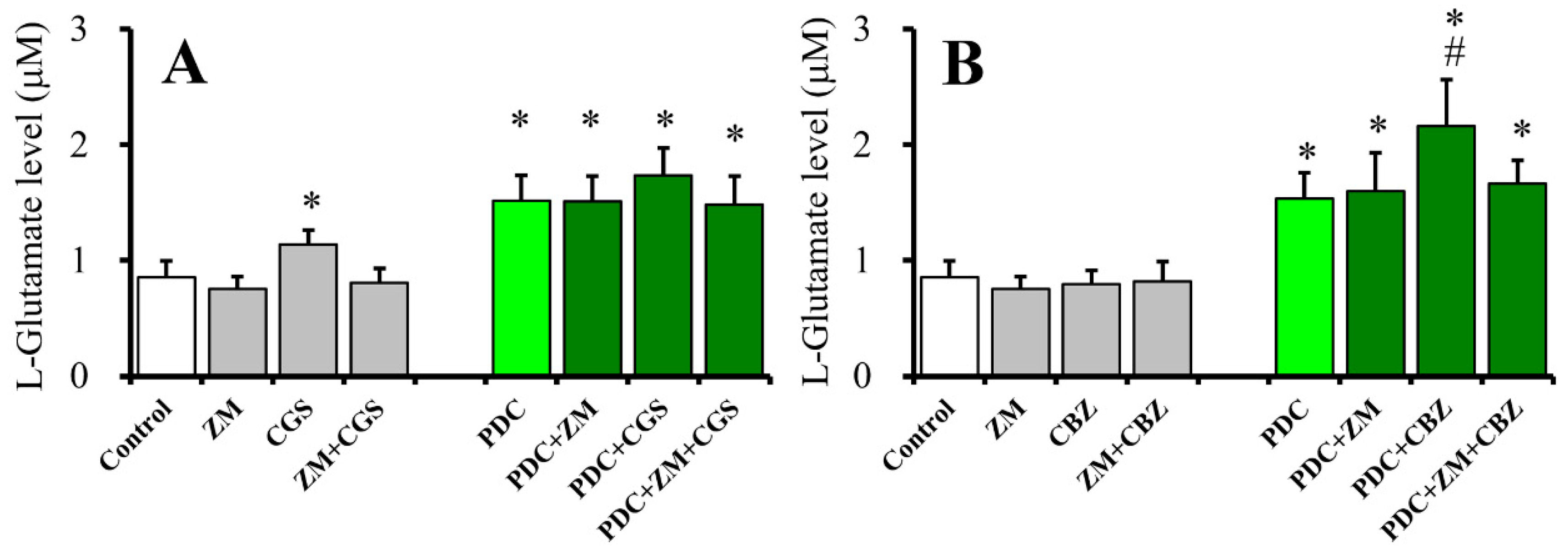
2.4. Effects of ZM241385 on CGS21680- and CBZ-evoked astroglial L-glutamate releases (Study 4)
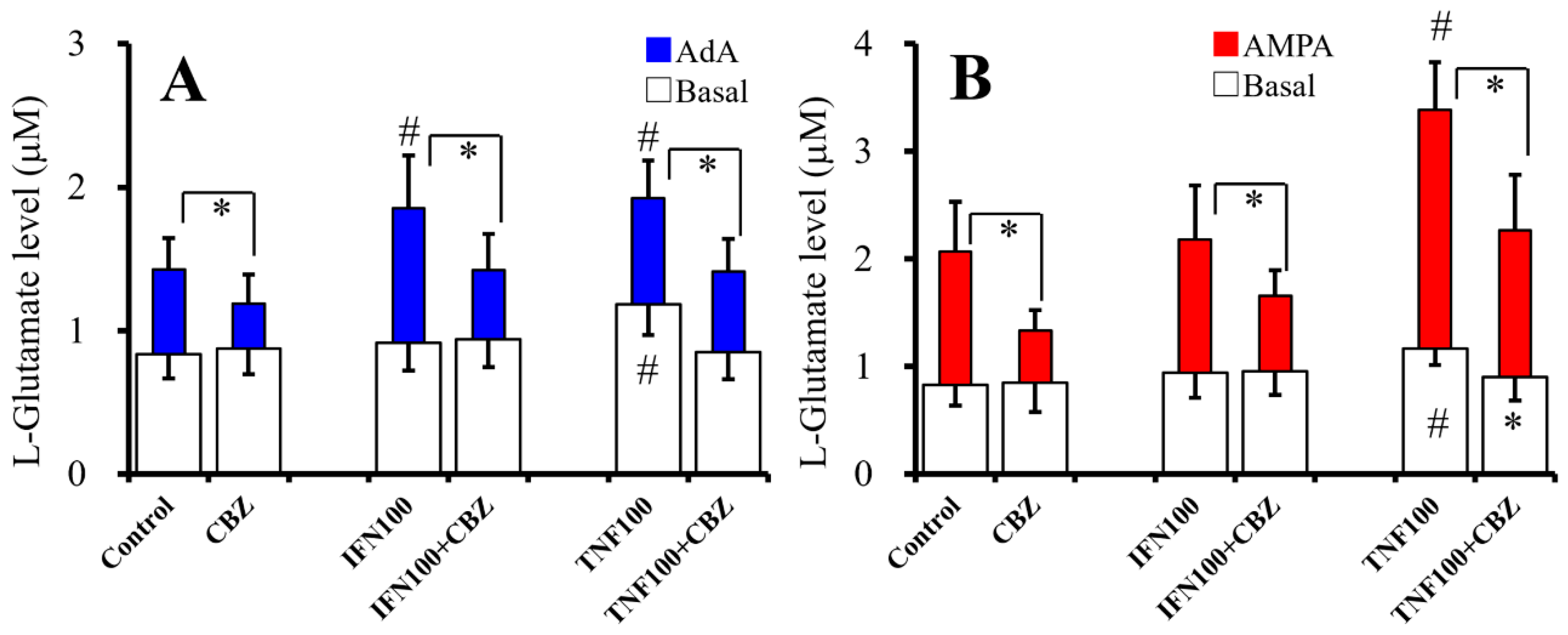
2.5. Interaction Between Chronic Cytokines Administration and Acute CBZ Administration on Basal, AdA-, and AMPA-Evoked Releases of L-Glutamate (Study 5)
2.6. Interaction Between Chronic Administration of Cytokines and CBZ on Basal AdA-, and AMPA-Evoked L-Glutamate Releases (Study 6)
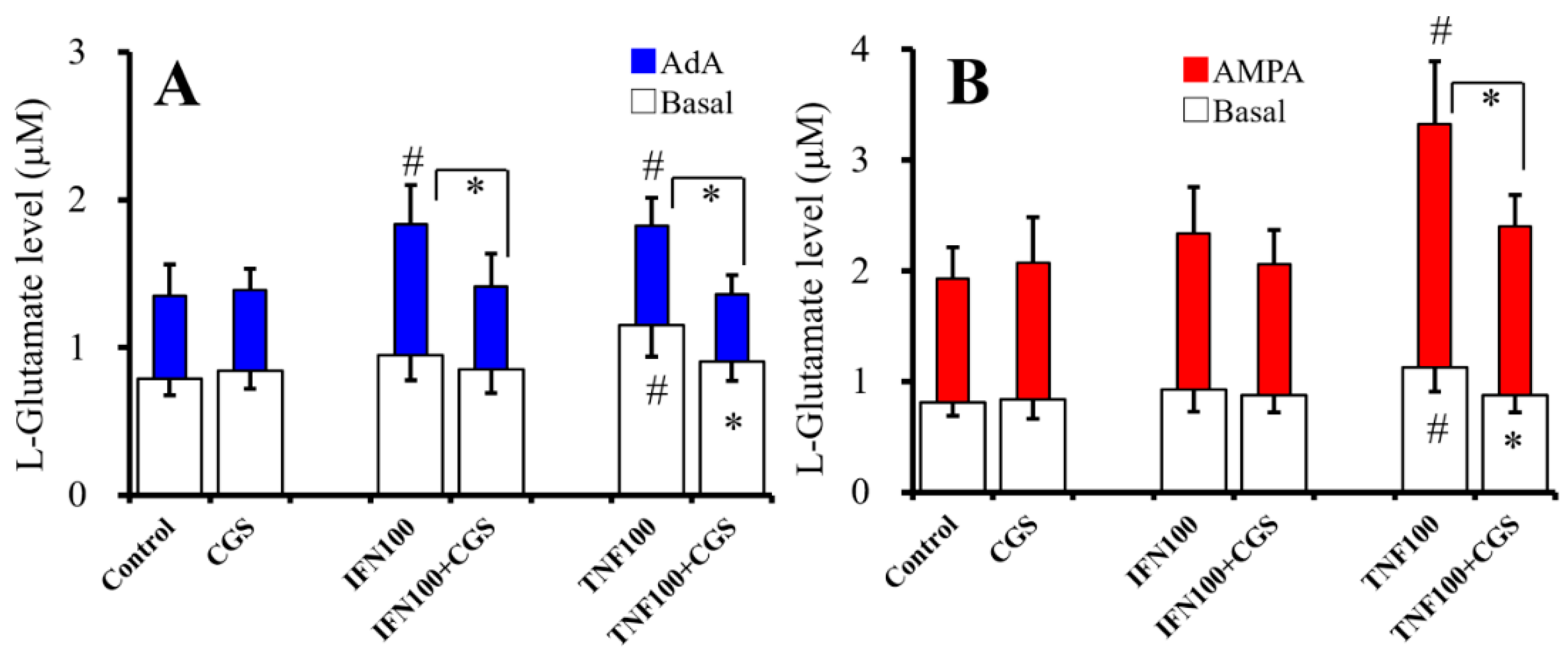
2.7. Interaction Between Chronic Administration of Cytokines and CGS21680 on Basal, AdA-, and AMPA-Evoked Releases of L-Glutamate (Study 7)
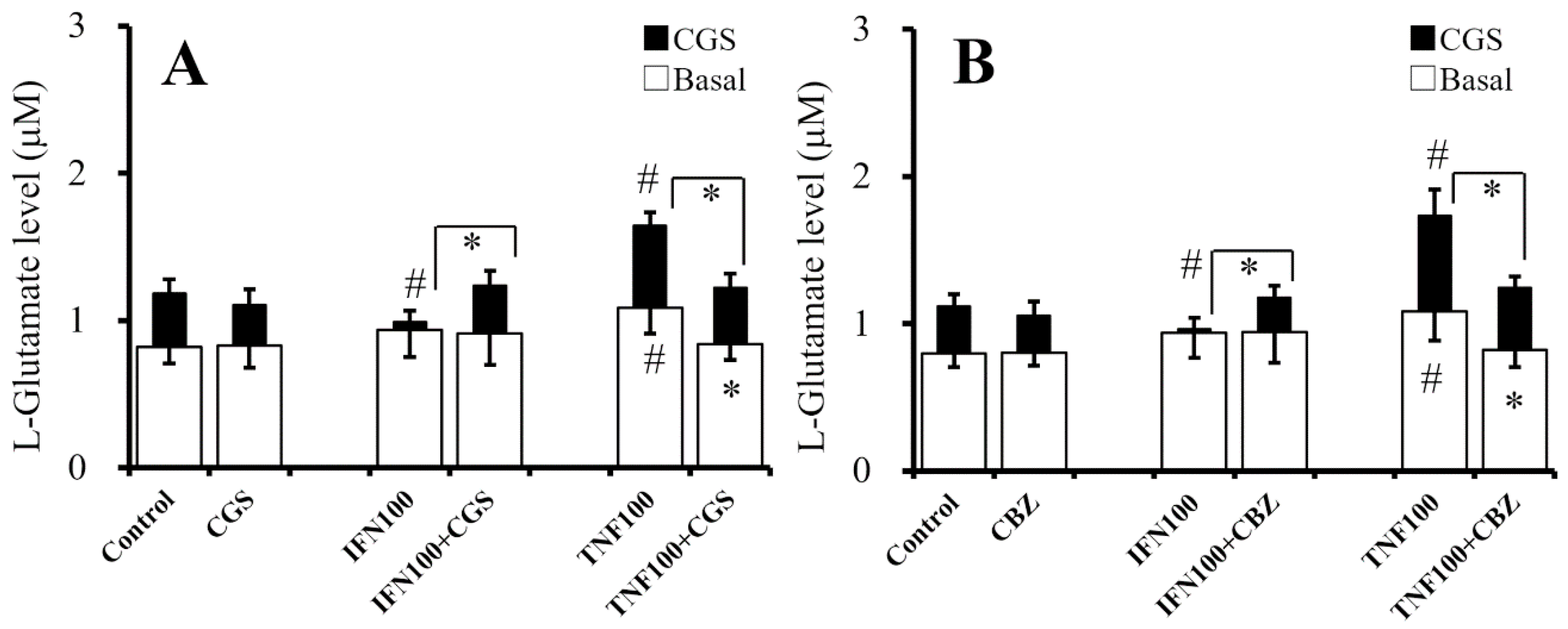
2.8. Interaction Between Chronic Administration of Cytokines, CBZ, and CGS21680 on CGS21680-Evoked L-Glutamate Release (Study 8)
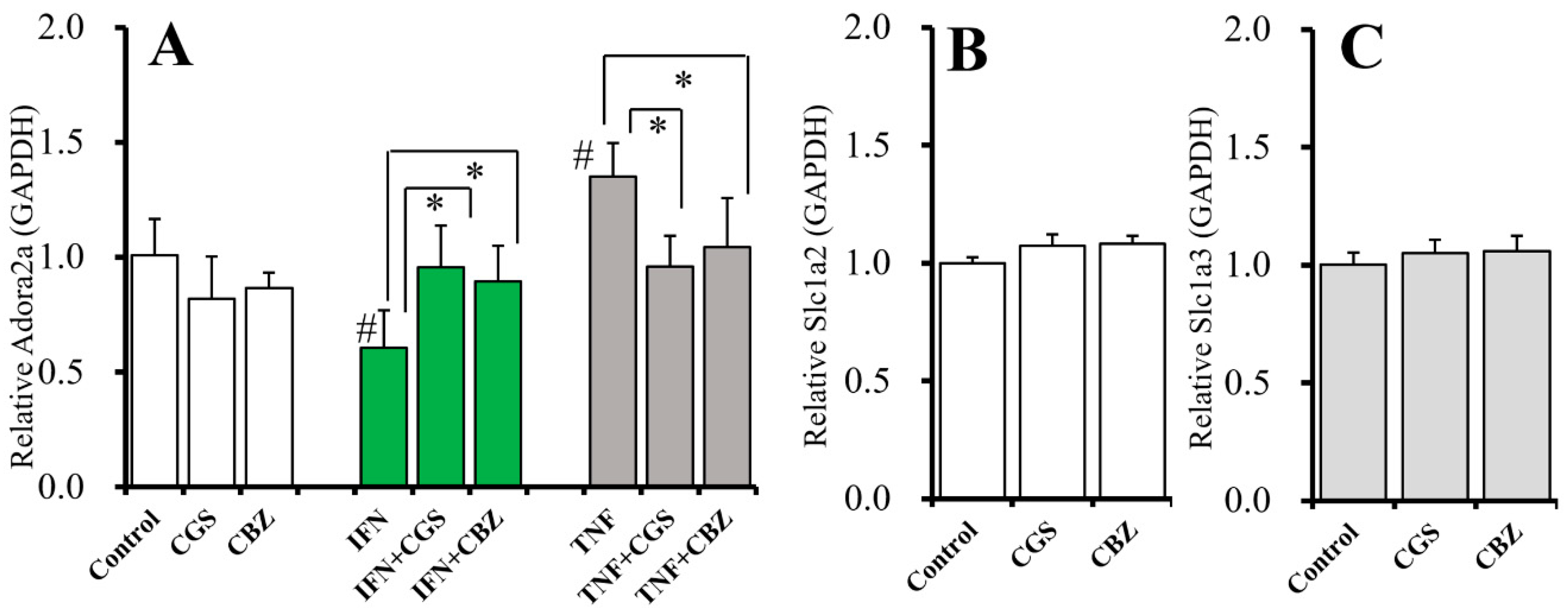
2.9. Interaction Between Chronic Administration of Cytokines, CBZ, and CGS21680 on mRNA Expression of A2AR and Glutamate Transporters
3. Discussion
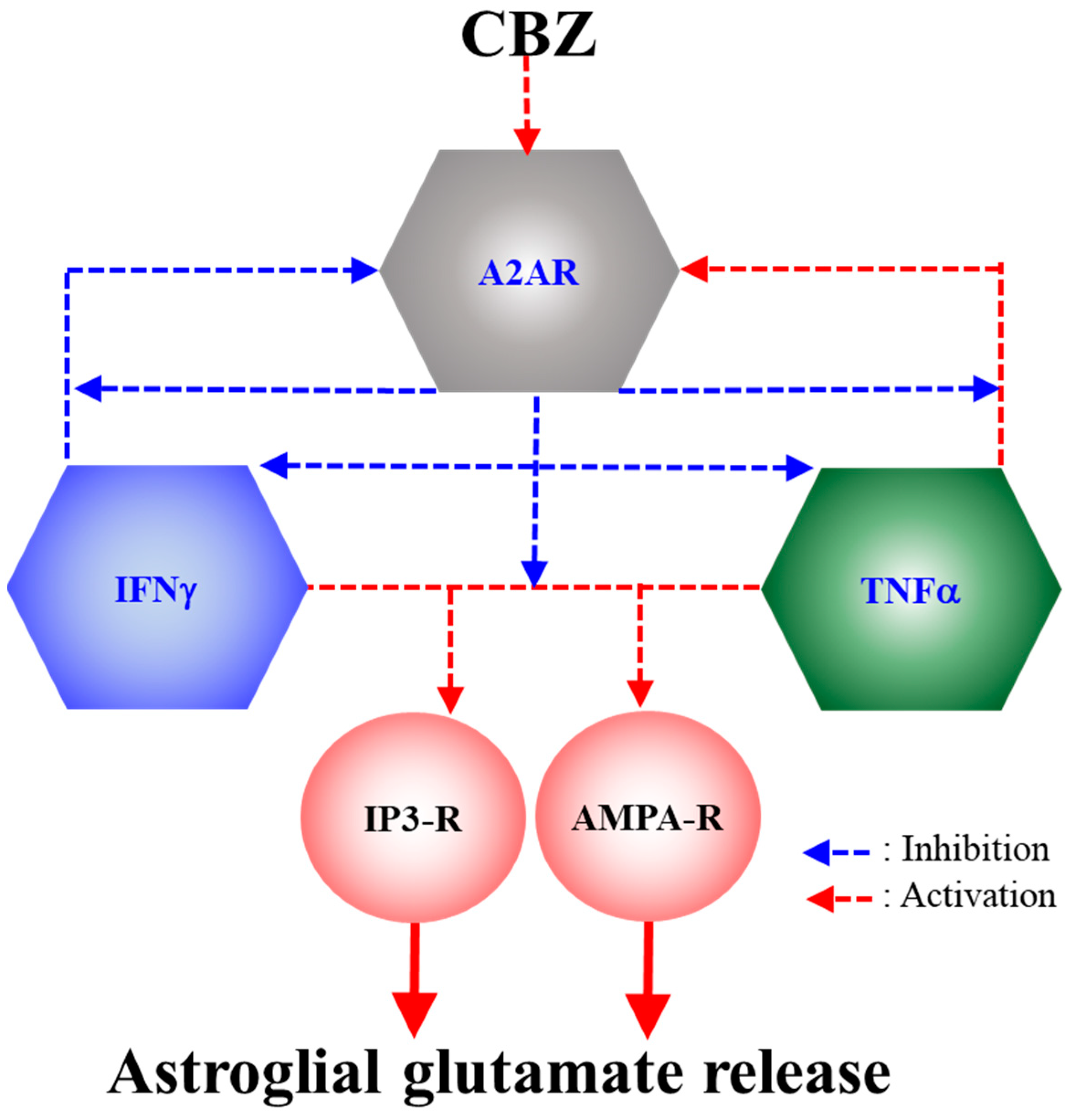
3.1. Astroglial L-Glutamate Release Mechanism Associated with A2AR
3.2. Effects of CBZ on Astroglial Transmission Induced by Chronic Exposure to Cytokines
4. Materials and Methods
4.1. Chemical Agents
4.2. Primary Astrocyte Culture
4.3. Treatment of Astrocytes and Study Design
4.4. Determination of Levels of L-Glutamate
4.5. Quantitative Real-Time PCR
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| A1R | adenosine A1 receptor |
| A2AR | adenosine A2A receptor |
| ACSF | artificial cerebrospinal fluid |
| AdA | adenophostin A |
| AMPA | amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid |
| ANOVA | analysis of variance |
| CBZ | carbamazepine |
| CPA | N6-Cyclopentyladenosine |
| fDMEM | Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum |
| DPCPX | 8-cyclopentyl-1,3-dipropylxanthine |
| IFNγ | interferon γ |
| IP3-R | inositol trisphosphate receptor |
| UHPLC | ultra-high-performance liquid chromatography. |
| TNFα | tumor necrosis factor α |
References
- Dallerac, G.; Rouach, N. Astrocytes as new targets to improve cognitive functions. Prog. Neurobiol. 2016, 144, 48–67. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Ueda, Y. Memantine protects thalamocortical hyper-glutamatergic transmission induced by NMDA receptor antagonism via activation of system xc-. Pharmacol. Res. Perspect. 2019, 7, e00457. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Song, D.; Yan, E.; Verkhratsky, A.; Peng, L. Chronic treatment with anti-bipolar drugs suppresses glutamate release from astroglial cultures. Amino Acids 2015, 47, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Rahman, T.; Campbell, A.; O’Connell, C.R.; Nallapula, K. Carbamazepine in bipolar disorder with pain: reviewing treatment guidelines. Prim. Care Companion CNS Disord. 2014, 16. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Mane, S.; Eid, T.; Zhao, H.; Lin, A.; Guan, Z.; Kim, J.H.; Schweitzer, J.; King-Stevens, D.; Weber, P.; et al. Gene expression in temporal lobe epilepsy is consistent with increased release of glutamate by astrocytes. Mol. Med. 2007, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, L.A.; Wang, L.; Ribak, C.E. Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia 2008, 49, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somera-Molina, K.C.; Robin, B.; Somera, C.A.; Anderson, C.; Stine, C.; Koh, S.; Behanna, H.A.; Van Eldik, L.J.; Watterson, D.M.; Wainwright, M.S. Glial activation links early-life seizures and long-term neurologic dysfunction: evidence using a small molecule inhibitor of proinflammatory cytokine upregulation. Epilepsia 2007, 48, 1785–1800. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okada, M. Effects of levetiracetam on astroglial release of kynurenine-pathway metabolites. Br. J. Pharmacol. 2018, 175, 4253–4265. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Wang, N.; Xu, L.; Huang, H.; Yu, C.; Zhou, H. Effects of carbamazepine combined with vitamin B12 on levels of plasma homocysteine, hs-CRP and TNF-alpha in patients with epilepsy. Exp. Ther. Med. 2018, 15, 2327–2332. [Google Scholar]
- Gomez, C.D.; Buijs, R.M.; Sitges, M. The anti-seizure drugs vinpocetine and carbamazepine, but not valproic acid, reduce inflammatory IL-1beta and TNF-alpha expression in rat hippocampus. J. Neurochem. 2014, 130, 770–779. [Google Scholar] [CrossRef]
- Fukuyama, K.; Kato, R.; Murata, M.; Shiroyama, T.; Okada, M. Clozapine Normalizes a Glutamatergic Transmission Abnormality Induced by an Impaired NMDA Receptor in the Thalamocortical Pathway via the Activation of a Group III Metabotropic Glutamate Receptor. Biomolecules 2019, 9, 234. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Clozapine, but not haloperidol, enhances glial D-serine and L-glutamate release in rat frontal cortex and primary cultured astrocytes. Br. J. Pharmacol. 2012, 165, 1543–1555. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Hasegawa, T.; Okada, M. Cystine/Glutamate Antiporter and Aripiprazole Compensate NMDA Antagonist-Induced Dysfunction of Thalamocortical L-Glutamatergic Transmission. Int. J. Mol. Sci. 2018, 19, 3645. [Google Scholar] [CrossRef] [PubMed]
- Glauser, T.; Ben-Menachem, E.; Bourgeois, B.; Cnaan, A.; Chadwick, D.; Guerreiro, C.; Kalviainen, R.; Mattson, R.; Perucca, E.; Tomson, T. ILAE treatment guidelines: evidence-based analysis of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia 2006, 47, 1094–1120. [Google Scholar] [CrossRef] [PubMed]
- Johannessen Landmark, C. Antiepileptic drugs in non-epilepsy disorders: relations between mechanisms of action and clinical efficacy. CNS Drugs 2008, 22, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R. Carbamazepine, Mechanisms of action. In Antiepileptic Drugs (fifth ed.); Mattson, R., Meldrum, B., Perucca, E., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2002; pp. 27–235. [Google Scholar]
- Yoshida, S.; Okada, M.; Zhu, G.; Kaneko, S. Carbamazepine prevents breakdown of neurotransmitter release induced by hyperactivation of ryanodine receptor. Neuropharmacology 2007, 52, 1538–1546. [Google Scholar] [CrossRef]
- Okada, M.; Kawata, Y.; Mizuno, K.; Wada, K.; Kondo, T.; Kaneko, S. Interaction between Ca2+, K+, carbamazepine and zonisamide on hippocampal extracellular glutamate monitored with a microdialysis electrode. Br. J. Pharmacol. 1998, 124, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Hamaguchi, T.; Ohoyama, K.; Sugiura, Y.; Suzuki, D.; Kanehara, S.; Nakagawa, M.; Motomura, E.; Matsumoto, T.; Tanii, H.; et al. Topiramate and zonisamide prevent paradoxical intoxication induced by carbamazepine and phenytoin. Epilepsy Res. 2009, 84, 172–186. [Google Scholar] [CrossRef]
- Kawata, Y.; Okada, M.; Murakami, T.; Kamata, A.; Zhu, G.; Kaneko, S. Pharmacological discrimination between effects of carbamazepine on hippocampal basal, Ca(2+)- and K(+)-evoked serotonin release. Br. J. Pharmacol. 2001, 133, 557–567. [Google Scholar] [CrossRef]
- Murakami, T.; Okada, M.; Kawata, Y.; Zhu, G.; Kamata, A.; Kaneko, S. Determination of effects of antiepileptic drugs on SNAREs-mediated hippocampal monoamine release using in vivo microdialysis. Br. J. Pharmacol. 2001, 134, 507–520. [Google Scholar] [CrossRef]
- Okada, M.; Zhu, G.; Yoshida, S.; Kanai, K.; Hirose, S.; Kaneko, S. Exocytosis mechanism as a new targeting site for mechanisms of action of antiepileptic drugs. Life Sci 2002, 72, 465–473. [Google Scholar] [CrossRef]
- Zhu, G.; Okada, M.; Murakami, T.; Kawata, Y.; Kamata, A.; Kaneko, S. Interaction between carbamazepine, zonisamide and voltage-sensitive Ca2+ channel on acetylcholine release in rat frontal cortex. Epilepsy Res. 2002, 49, 49–60. [Google Scholar] [CrossRef]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Effect of lamotrigine and carbamazepine on corticotropin-releasing factor-associated serotonergic transmission in rat dorsal raphe nucleus. Psychopharmacology (Berl) 2012, 220, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Okada, M.; Murakami, T.; Kamata, A.; Zhu, G.; Kawata, Y.; Wada, K.; Kaneko, S. Effects of carbamazepine on acetylcholine release and metabolism. Epilepsy Res. 2000, 40, 187–195. [Google Scholar] [CrossRef]
- Marangos, P.J.; Weiss, S.R.; Montgomery, P.; Patel, J.; Narang, P.K.; Cappabianca, A.M.; Post, R.M. Chronic carbamazepine treatment increases brain adenosine receptors. Epilepsia 1985, 26, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Elphick, M.; Taghavi, Z.; Powell, T.; Godfrey, P.P. Chronic carbamazepine down-regulates adenosine A2 receptors: studies with the putative selective adenosine antagonists PD115,199 and PD116,948. Psychopharmacology (Berl) 1990, 100, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Okada, M.; Hirano, T.; Kondo, T.; Otani, K.; Fukushima, Y. Carbamazepine and zonisamide increase extracellular dopamine and serotonin levels in vivo, and carbamazepine does not antagonize adenosine effect in vitro: mechanisms of blockade of seizure spread. Jpn. J. Psychiatry Neurol. 1993, 47, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Kiryu, K.; Kawata, Y.; Mizuno, K.; Wada, K.; Tasaki, H.; Kaneko, S. Determination of the effects of caffeine and carbamazepine on striatal dopamine release by in vivo microdialysis. Eur. J. Pharmacol. 1997, 321, 181–188. [Google Scholar] [CrossRef]
- Okada, M.; Kaneko, S. Pharmacological interactions between magnesium ion and adenosine on monoaminergic system in the central nervous system. Magnes. Res. 1998, 11, 289–305. [Google Scholar]
- Booker, S.A.; Pires, N.; Cobb, S.; Soares-da-Silva, P.; Vida, I. Carbamazepine and oxcarbazepine, but not eslicarbazepine, enhance excitatory synaptic transmission onto hippocampal CA1 pyramidal cells through an antagonist action at adenosine A1 receptors. Neuropharmacology 2015, 93, 103–115. [Google Scholar] [CrossRef]
- Okada, M.; Hirano, T.; Mizuno, K.; Chiba, T.; Kawata, Y.; Kiryu, K.; Wada, K.; Tasaki, H.; Kaneko, S. Biphasic effects of carbamazepine on the dopaminergic system in rat striatum and hippocampus. Epilepsy Res. 1997, 28, 143–153. [Google Scholar] [CrossRef]
- Okada, M.; Hirano, T.; Mizuno, K.; Kawata, Y.; Wada, K.; Murakami, T.; Tasaki, H.; Kaneko, S. Effects of carbamazepine on hippocampal serotonergic system. Epilepsy Res. 1998, 31, 187–198. [Google Scholar] [CrossRef]
- Orr, A.G.; Hsiao, E.C.; Wang, M.M.; Ho, K.; Kim, D.H.; Wang, X.; Guo, W.; Kang, J.; Yu, G.Q.; Adame, A.; et al. Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory. Nat. Neurosci. 2015, 18, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naganuma, M.; Wiznerowicz, E.B.; Lappas, C.M.; Linden, J.; Worthington, M.T.; Ernst, P.B. Cutting edge: Critical role for A2A adenosine receptors in the T cell-mediated regulation of colitis. J. Immunol. 2006, 177, 2765–2769. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Kurtz, C.C.; Wilson, J.M.; Burnette, B.R.; Wiznerowicz, E.B.; Ross, W.G.; Rieger, J.M.; Figler, R.A.; Linden, J.; Crowe, S.E.; et al. A2A adenosine receptor (AR) activation inhibits pro-inflammatory cytokine production by human CD4+ helper T cells and regulates Helicobacter-induced gastritis and bacterial persistence. Mucosal. Immunol. 2009, 2, 232–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, D.R.; Kubota, T.; Sanders, V.J.; McTiernan, C.F.; Feldman, A.M. Differential regulation of cardiac expression of IL-6 and TNF-alpha by A2- and A3-adenosine receptors. Am. J. Physiol. 1999, 276, H2141–H2147. [Google Scholar]
- Barnholt, K.E.; Kota, R.S.; Aung, H.H.; Rutledge, J.C. Adenosine blocks IFN-gamma-induced phosphorylation of STAT1 on serine 727 to reduce macrophage activation. J. Immunol. 2009, 183, 6767–6777. [Google Scholar] [CrossRef]
- Masuda, Y.; Utsui, Y.; Shiraishi, Y.; Karasawa, T.; Yoshida, K.; Shimizu, M. Relationships between plasma concentrations of diphenylhydantoin, phenobarbital, carbamazepine, and 3-sulfamoylmethyl-1,2-benzisoxazole (AD-810), a new anticonvulsant agent, and their anticonvulsant or neurotoxic effects in experimental animals. Epilepsia 1979, 20, 623–633. [Google Scholar] [CrossRef]
- Yamamura, S.; Hoshikawa, M.; Dai, K.; Saito, H.; Suzuki, N.; Niwa, O.; Okada, M. ONO-2506 inhibits spike-wave discharges in a genetic animal model without affecting traditional convulsive tests via gliotransmission regulation. Br. J. Pharmacol. 2013, 168, 1088–1100. [Google Scholar] [CrossRef]
- Nakano, T.; Hasegawa, T.; Suzuki, D.; Motomura, E.; Okada, M. Amantadine Combines Astroglial System Xc(-) Activation with Glutamate/NMDA Receptor Inhibition. Biomolecules 2019, 9, 191. [Google Scholar] [CrossRef]
- Liang, C.; Du, T.; Zhou, J.; Verkhratsky, A.; Peng, L. Ammonium increases Ca(2+) signalling and up-regulates expression of TRPC1 gene in astrocytes in primary cultures and in the in vivo brain. Neurochem. Res. 2014, 39, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Latini, S.; Pedata, F. Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J. Neurochem. 2001, 79, 463–484. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Nutt, D.J.; Murakami, T.; Zhu, G.; Kamata, A.; Kawata, Y.; Kaneko, S. Adenosine receptor subtypes modulate two major functional pathways for hippocampal serotonin release. J. Neurosci. 2001, 21, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Kawata, Y.; Murakami, T.; Wada, K.; Mizuno, K.; Kondo, T.; Kaneko, S. Differential effects of adenosine receptor subtypes on release and reuptake of hippocampal serotonin. Eur J Neurosci 1999, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, A.F.; Malva, J.O.; Carvalho, A.P.; Carvalho, C.M. Inhibition of N-,P/Q- and other types of Ca2+ channels in rat hippocampal nerve terminals by the adenosine A1 receptor. Eur. J. Pharmacol. 1997, 340, 301–310. [Google Scholar] [CrossRef]
- Kim, C.S.; Johnston, D. A1 adenosine receptor-mediated GIRK channels contribute to the resting conductance of CA1 neurons in the dorsal hippocampus. J. Neurophysiol. 2015, 113, 2511–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciruela, F.; Casado, V.; Rodrigues, R.J.; Lujan, R.; Burgueno, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.R.; Mallol, J.; et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J. Neurosci. 2006, 26, 2080–2087. [Google Scholar] [CrossRef]
- Matos, M.; Augusto, E.; Agostinho, P.; Cunha, R.A.; Chen, J.F. Antagonistic interaction between adenosine A2A receptors and Na+/K+-ATPase-alpha2 controlling glutamate uptake in astrocytes. J. Neurosci. 2013, 33, 18492–18502. [Google Scholar] [CrossRef]
- Varani, K.; Portaluppi, F.; Merighi, S.; Ongini, E.; Belardinelli, L.; Borea, P.A. Caffeine alters A2A adenosine receptors and their function in human platelets. Circulation 1999, 99, 2499–2502. [Google Scholar] [CrossRef]
- Rekik, M.; Mustafa, J.S. Modulation of A2A adenosine receptors and associated Galphas proteins by ZM 241385 treatment of porcine coronary artery. J. Cardiovasc. Pharmacol. 2003, 42, 736–744. [Google Scholar] [CrossRef]
- Pereira, M.R.; Hang, V.R.; Vardiero, E.; de Mello, F.G.; Paes-de-Carvalho, R. Modulation of A1 adenosine receptor expression by cell aggregation and long-term activation of A2a receptors in cultures of avian retinal cells: involvement of the cyclic AMP/PKA pathway. J. Neurochem. 2010, 113, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Palmer, T.M.; Gettys, T.W.; Jacobson, K.A.; Stiles, G.L. Desensitization of the canine A2a adenosine receptor: delineation of multiple processes. Mol. Pharmacol. 1994, 45, 1082–1094. [Google Scholar] [PubMed]
- Paterniti, I.; Melani, A.; Cipriani, S.; Corti, F.; Mello, T.; Mazzon, E.; Esposito, E.; Bramanti, P.; Cuzzocrea, S.; Pedata, F. Selective adenosine A2A receptor agonists and antagonists protect against spinal cord injury through peripheral and central effects. J. Neuroinflammation 2011, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, J.; Rocha, A.; Nunes, F.; Costa, M.S.; Schein, V.; Kazlauckas, V.; Kalinine, E.; Souza, D.O.; Cunha, R.A.; Porciuncula, L.O. Caffeine consumption prevents memory impairment, neuronal damage, and adenosine A2A receptors upregulation in the hippocampus of a rat model of sporadic dementia. J. Alzheimers Dis. 2013, 34, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Sahu, S.K.; Gummadi, S.N.; Manoj, N.; Aradhyam, G.K. Phospholipid scramblases: an overview. Arch. Biochem. Biophys 2007, 462, 103–114. [Google Scholar] [CrossRef]
- Vikman, K.S.; Owe-Larsson, B.; Brask, J.; Kristensson, K.S.; Hill, R.H. Interferon-gamma-induced changes in synaptic activity and AMPA receptor clustering in hippocampal cultures. Brain Res. 2001, 896, 18–29. [Google Scholar] [CrossRef]
- Stellwagen, D.; Beattie, E.C.; Seo, J.Y.; Malenka, R.C. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J. Neurosci. 2005, 25, 3219–3228. [Google Scholar] [CrossRef] [PubMed]
- Park, K.M.; Yule, D.I.; Bowers, W.J. Tumor necrosis factor-alpha-mediated regulation of the inositol 1,4,5-trisphosphate receptor promoter. J. Biol. Chem. 2009, 284, 27557–27566. [Google Scholar] [CrossRef]
- Khoa, N.D.; Montesinos, M.C.; Reiss, A.B.; Delano, D.; Awadallah, N.; Cronstein, B.N. Inflammatory cytokines regulate function and expression of adenosine A(2A) receptors in human monocytic THP-1 cells. J. Immunol. 2001, 167, 4026–4032. [Google Scholar] [CrossRef]
- Morello, S.; Ito, K.; Yamamura, S.; Lee, K.Y.; Jazrawi, E.; Desouza, P.; Barnes, P.; Cicala, C.; Adcock, I.M. IL-1 beta and TNF-alpha regulation of the adenosine receptor (A2A) expression: differential requirement for NF-kappa B binding to the proximal promoter. J. Immunol. 2006, 177, 7173–7183. [Google Scholar] [CrossRef]
- Khoa, N.D.; Postow, M.; Danielsson, J.; Cronstein, B.N. Tumor necrosis factor-alpha prevents desensitization of Galphas-coupled receptors by regulating GRK2 association with the plasma membrane. Mol. Pharmacol. 2006, 69, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Giustizieri, M.; Armogida, M.; Berretta, N.; Federici, M.; Piccirilli, S.; Mercuri, N.B.; Nistico, R. Differential effect of carbamazepine and oxcarbazepine on excitatory synaptic transmission in rat hippocampus. Synapse 2008, 62, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Huang, Y.; Washington, J.M.; Briggs, N.W.; Zuo, Z. Carbamazepine enhances the activity of glutamate transporter type 3 via phosphatidylinositol 3-kinase. Epilepsy Res. 2005, 66, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Li, L.B.; Toan, S.V.; Zelenaia, O.; Watson, D.J.; Wolfe, J.H.; Rothstein, J.D.; Robinson, M.B. Regulation of astrocytic glutamate transporter expression by Akt: evidence for a selective transcriptional effect on the GLT-1/EAAT2 subtype. J. Neurochem. 2006, 97, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Domenici, M.R.; Ferrante, A.; Martire, A.; Chiodi, V.; Pepponi, R.; Tebano, M.T.; Popoli, P. Adenosine A2A receptor as potential therapeutic target in neuropsychiatric disorders. Pharmacol. Res. 2019, 104338. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Marrion, N.V.; Peters, J.A.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; et al. The Concise Guide to Pharmacology 2017/18: G protein-coupled receptors. Br. J. Pharmacol. 2017, 174, S17–S129. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.C.; Drummond, G.B.; McLachlan, E.M.; Kilkenny, C.; Wainwright, C.L. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1573–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuyama, K.; Tanahashi, S.; Hoshikawa, M.; Shinagawa, R.; Okada, M. Zonisamide regulates basal ganglia transmission via astroglial kynurenine pathway. Neuropharmacology 2014, 76 Pt A, 137–145. [Google Scholar] [CrossRef]
- Tateishi, N.; Shimoda, T.; Manako, J.; Katsumata, S.; Shinagawa, R.; Ohno, H. Relevance of astrocytic activation to reductions of astrocytic GABAA receptors. Brain Res. 2006, 1089, 79–91. [Google Scholar] [CrossRef]
- Tanahashi, S.; Ueda, Y.; Nakajima, A.; Yamamura, S.; Nagase, H.; Okada, M. Novel delta1-receptor agonist KNT-127 increases the release of dopamine and L-glutamate in the striatum, nucleus accumbens and median pre-frontal cortex. Neuropharmacology 2012, 62, 2057–2067. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glutamate Transporter Functional | Glutamate Transporter Blockade | ||||||
|---|---|---|---|---|---|---|---|
| Basal | AdA-Evoke | AMPA-Evoke | CGS-Evoke | Basal | AdA-Evoke | AMPA-Evoke | |
| CBZ (Acute) | → | ↓ | ↓ | ↑ | ↓ | ↓ | |
| CBZ (chronic) | → | ↓ | ↓ | → | ↑ | ↓ | ↓ |
| CGS (Acute) | ↑ | → | → | → | |||
| CGS (Chronic) | → | → | → | → | |||
| After Chronic IFNγ Administraion | After Chronic TNFα Administraion | |||||||
|---|---|---|---|---|---|---|---|---|
| Basal | AdA-Evoke | AMPA-Evoke | CGS-Evoke | Basal | AdA-Evoke | AMPA-Evoke | CGS-Evoke | |
| CBZ (Acute) | → | ↓ | ↓ | ↓ | ↓ | ↓ | ||
| CBZ (chronic) | → | ↓ | ↓ | ↑ | ↓ | ↓ | ↓ | ↓ |
| CGS (Chronic) | → | ↓ | → | ↑ | ↓ | ↓ | ↓ | ↓ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okada, M.; Fukuyama, K.; Shiroyama, T.; Ueda, Y. Carbamazepine Attenuates Astroglial L-Glutamate Release Induced by Pro-Inflammatory Cytokines via Chronically Activation of Adenosine A2A Receptor. Int. J. Mol. Sci. 2019, 20, 3727. https://doi.org/10.3390/ijms20153727
Okada M, Fukuyama K, Shiroyama T, Ueda Y. Carbamazepine Attenuates Astroglial L-Glutamate Release Induced by Pro-Inflammatory Cytokines via Chronically Activation of Adenosine A2A Receptor. International Journal of Molecular Sciences. 2019; 20(15):3727. https://doi.org/10.3390/ijms20153727
Chicago/Turabian StyleOkada, Motohiro, Kouji Fukuyama, Takashi Shiroyama, and Yuto Ueda. 2019. "Carbamazepine Attenuates Astroglial L-Glutamate Release Induced by Pro-Inflammatory Cytokines via Chronically Activation of Adenosine A2A Receptor" International Journal of Molecular Sciences 20, no. 15: 3727. https://doi.org/10.3390/ijms20153727
APA StyleOkada, M., Fukuyama, K., Shiroyama, T., & Ueda, Y. (2019). Carbamazepine Attenuates Astroglial L-Glutamate Release Induced by Pro-Inflammatory Cytokines via Chronically Activation of Adenosine A2A Receptor. International Journal of Molecular Sciences, 20(15), 3727. https://doi.org/10.3390/ijms20153727