Flexible and Expedited Regulatory Review Processes for Innovative Medicines and Regenerative Medical Products in the US, the EU, and Japan


Abstract
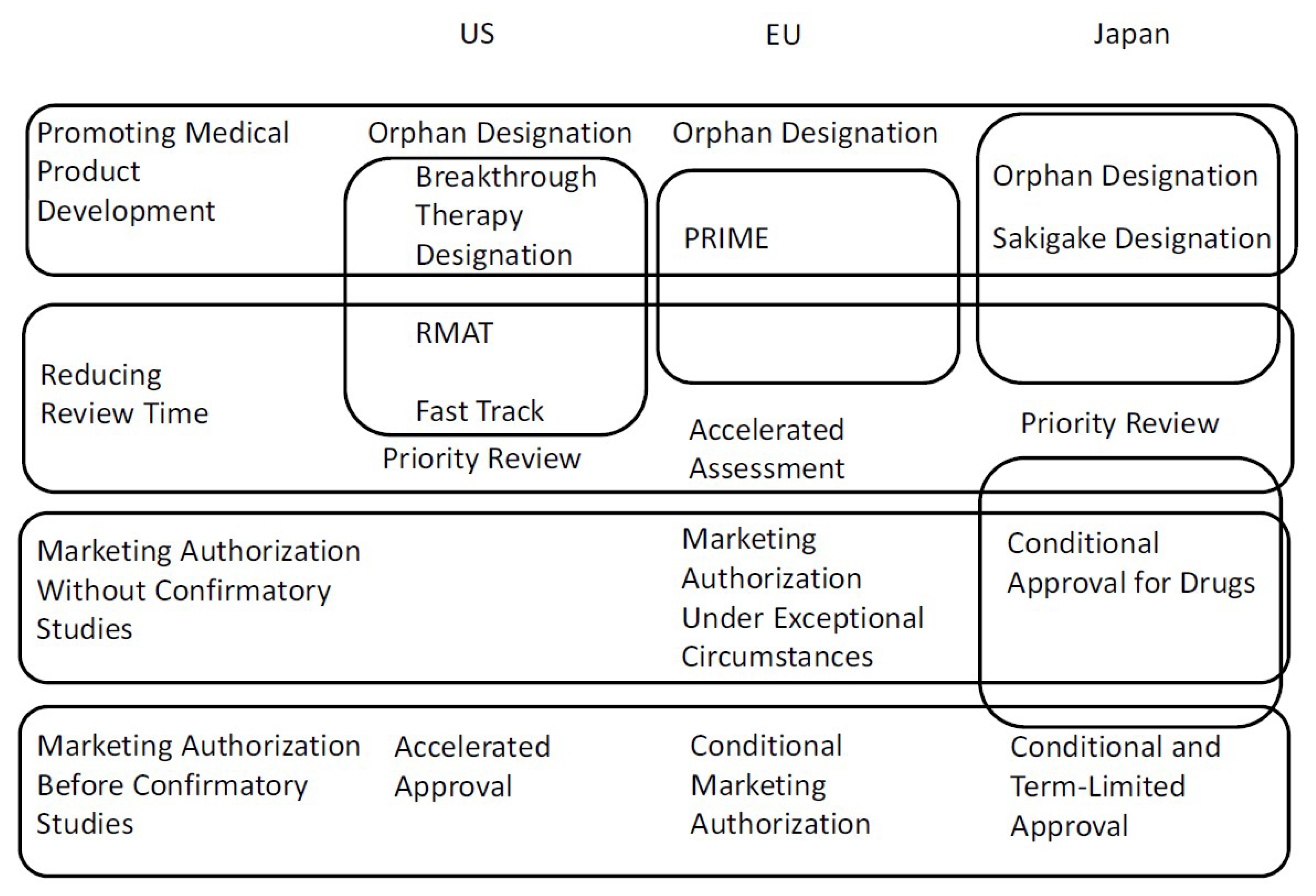
:1. Introduction
2. Standard and Priority Reviews
3. Orphan Designation
4. Accelerated or Conditional Approval
5. Breakthrough Therapy Designation
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| IVD | in vitro diagnostics |
| FDA | Food and Drug Administration |
| NDA | new drug applications |
| BLA | biologics license applications |
| EMA | European Medicines Agency |
| ATMP | advanced-therapy medicinal products |
| CHMP | Committee for Medicinal Products for Human Use |
| CAT | Committee for Advanced Therapies |
| PMDA | Pharmaceuticals and Medical Devices Agency |
| MHLW | Ministry of Health, Labour and Welfare |
| PDUFA | Prescription Drug User Fee Act |
| RMAT | regenerative medicine advanced therapy |
| PRIME | priority medicines |
References
- FDA Website. Drugs@FDA. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/ (accessed on 31 May 2019).
- FDA Website. Approved Cellular and Gene Therapy Products. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (accessed on 31 May 2019).
- Pignatti, F.; Gravanis, I.; Herold, R.; Vamvakas, S.; Jonsson, B.; Marty, M. The European Medicines Agency: An overview of its mission, responsibilities, and recent initiatives in cancer drug regulation. Clin. Cancer Res. 2011, 17, 5220–5225. [Google Scholar] [CrossRef] [PubMed]
- Nagai, S.; Ozawa, K. Regulatory approval pathways for anticancer drugs in Japan, the EU and the US. Int. J. Hematol. 2016, 104, 73–84. [Google Scholar] [CrossRef] [PubMed]
- EMA Website. Authrosation of Medicines. Available online: https://www.ema.europa.eu/en/about-us/what-we-do/authorisation-medicines (accessed on 31 May 2019).
- EMA Website. Committee for Advanced Therapies (CAT). Available online: https://www.ema.europa.eu/en/committees/committee-advanced-therapies-cat (accessed on 31 May 2019).
- EMA Website. Committee for Medicinal Products for Human Use (CHMP). Available online: https://www.ema.europa.eu/en/committees/committee-medicinal-products-human-use-chmp (accessed on 31 May 2019).
- EMA Website. Human Medicines Search. Available online: https://www.ema.europa.eu/en/medicines (accessed on 31 May 2019).
- Nagai, S.; Ozawa, K. New Japanese Regulatory Frameworks for Clinical Research and Marketing Authorization of Gene Therapy and Cellular Therapy Products. Curr. Gene Ther. 2017, 17, 17–28. [Google Scholar] [CrossRef] [PubMed]
- MHLW Website. PMD Act. Available online: https://www.mhlw.go.jp/english/policy/health-medical/pharmaceuticals/dl/150407-01.pdf (accessed on 31 May 2019).
- PMDA Website. Organization. Available online: https://www.pmda.go.jp/english/about-pmda/outline/0003.html (accessed on 31 May 2019).
- PMDA Website. Approved Drugs. Available online: https://www.pmda.go.jp/english/review-services/reviews/approved-information/drugs/0003.html (accessed on 31 May 2019).
- PMDA Website. Approved Regenerative Medical Products. Available online: https://www.pmda.go.jp/english/review-services/reviews/approved-information/0003.html (accessed on 31 May 2019).
- FDA Website. Guidance for Industry Expedited Programs for Serious Conditions—Drugs and Biologics. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/expedited-programs-serious-conditions-drugs-and-biologics (accessed on 31 May 2019).
- FDA Website. Priority Review. Available online: https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/priority-review (accessed on 31 May 2019).
- FDA Website. Guidance for Industry Expedited Programs for Regenerative Medicine Therapies for Serious Conditions. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/expedited-programs-regenerative-medicine-therapies-serious-conditions (accessed on 31 May 2019).
- EMA Website. Guideline on the Scientific Application and the Practical Arrangements Necessary to Implement the Procedure for Accelerated Assessment Pursuant to Article 14(9) of Regulation (EC) No 726/2004. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-scientific-application-practical-arrangements-necessary-implement-procedure-accelerated/2004_en.pdf (accessed on 31 May 2019).
- Martinalbo, J.; Bowen, D.; Camarero, J.; Chapelin, M.; Démolis, P.; Foggi, P.; Jonsson, B.; Llinares, J.; Moreau, A.; O’Connor, D.; et al. Early market access of oncologic drugs in the EU. Ann. Oncol. 2016, 27, 96–105. [Google Scholar] [CrossRef] [PubMed]
- FDA Website. Designating an Orphan Product: Drugs and Biological Products. Available online: https://www.fda.gov/industry/developing-products-rare-diseases-conditions/designating-orphan-product-drugs-and-biological-products (accessed on 31 May 2019).
- FDA Website. Search Orphan Drug Designations and Approvals. Available online: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/ (accessed on 31 May 2019).
- EMA Website. Committee for Orphan Medicinal Products (COMP). Available online: https://www.ema.europa.eu/en/committees/committee-orphan-medicinal-products-comp (accessed on 31 May 2019).
- EMA Website. Orphan Designation: Overview. Available online: https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation-overview (accessed on 31 May 2019).
- MHLW Website. Overview of Orphan Drug/Medical Device Designation System. Available online: https://www.mhlw.go.jp/english/policy/health-medical/pharmaceuticals/orphan_drug.html (accessed on 31 May 2019).
- NIBIOHN Website. Available online: http://www.nibiohn.go.jp/nibio/part/promote/files/orphan%20drug%20E.pdf (accessed on 31 May 2019).
- NIBIOHN Website. Available online: http://www.nibiohn.go.jp/nibio/part/promote/files/orphan%20regenerativemedicine%20E.pdf (accessed on 31 May 2019).
- FDA Website. Accelerated Approval. Available online: https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/accelerated-approval (accessed on 31 May 2019).
- FDA Website Postmarket Requirements and Commitments. Available online: https://www.accessdata.fda.gov/scripts/cder/pmc/ (accessed on 31 May 2019).
- Nagai, S.; Ozawa, K. Drug approval based on randomized phase 3 trials for relapsed malignancy: Analysis of oncologic drugs granted accelerated approval, publications and clinical trial databases. Investig. New Drugs 2018, 36, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Beaver, J.A.; Howie, L.J.; Pelosof, L.; Kim, T.; Liu, J.; Goldberg, K.B.; Sridhara, R.; Blumenthal, G.M.; Farrell, A.T.; Keegan, P.; et al. A 25-Year Experience of US Food and Drug Administration Accelerated Approval of Malignant Hematology and Oncology Drugs and Biologics: A Review. JAMA Oncol. 2018, 4, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Hoekman, J.; Boon, W.; Bouvy, J.C.; Ebbers, H.C.; de Jong, J.P.; De Bruin, M.L. Use of the conditional marketing authorization pathway for oncology medicines in Europe. Clin. Pharmacol. Ther. 2015, 98, 534–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boon, W.P.; Moors, E.H.; Meijer, A.; Schellekens, H. Conditional approval and approval under exceptional circumstances as regulatory instruments for stimulating responsible drug innovation in Europe. Clin. Pharmacol. Ther. 2010, 88, 848–853. [Google Scholar] [CrossRef] [PubMed]
- PMDA Website. Reviews. Available online: https://www.pmda.go.jp/english/review-services/reviews/0001.html (accessed on 31 May 2019).
- Nagai, S.; Sugiyama, D. Current Trends in Clinical Development of Gene and Cellular Therapeutic Products for Cancer in Japan. Clin. Ther. 2019, 41, 174–184.e3. [Google Scholar] [CrossRef] [PubMed]
- PMDA Website. Available online: https://www.pmda.go.jp/review-services/drug-reviews/0045.html (accessed on 31 May 2019).
- FDA Website. Fast Track. Available online: https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/fast-track (accessed on 31 May 2019).
- Sherman, R.E.; Li, J.; Shapley, S.; Robb, M.; Woodcock, J. Expediting drug development—The FDA’s new “breakthrough therapy” designation. N. Engl. J. Med. 2013, 369, 1877–1880. [Google Scholar] [CrossRef] [PubMed]
- FDA Website. Breakthrough Therapy. Available online: https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/breakthrough-therapy (accessed on 31 May 2019).
- FDA. Good Review Practice: Review of Marketing Applications for Breakthrough Therapy-Designated Drugs and Biologics That Are Receiving an Expedited Review. Available online: https://www.fda.gov/media/90933/download (accessed on 31 May 2019).
- FDA Website. Frequently Asked Questions Breakthrough Therapies. Available online: https://www.fda.gov/regulatory-information/food-and-drug-administration-safety-and-innovation-act-fdasia/frequently-asked-questions-breakthrough-therapies (accessed on 31 May 2019).
- Friends of Cancer Research Website. Breakthrough Therapies. Available online: https://www.focr.org/breakthrough-therapies (accessed on 31 May 2019).
- FDA Website. Regenerative Medicine Advanced Therapy. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/regenerative-medicine-advanced-therapy-designation (accessed on 31 May 2019).
- EMA Website. PRIME Priority Medicines. Available online: https://www.ema.europa.eu/en/human-regulatory/research-development/prime-priority-medicines (accessed on 31 May 2019).
- EMA Website. Enhanced Early Dialogue to Facilitate Accelerated Assessment of Priority Medicines (PRIME). Available online: https://www.ema.europa.eu/en/do cuments/regulatory-procedural-guideline/enhanced-early-dialogue-facilitate-accelerated-assessment-priority-medicines-prime_en.pdf (accessed on 31 May 2019).
- MHLW Website. Strategy of Sakigake. Available online: https://www.mhlw.go.jp/english/policy/health-medical/pharmaceuticals/140729-01.html (accessed on 31 May 2019).
- PMDA Website. Available online: https://www.pmda.go.jp/review-services/drug-reviews/0003.html (accessed on 31 May 2019).


{kind=link}
| US (Priority Review) | EU (Accelerated Assessment) | Japan (Priority Review) | |
|---|---|---|---|
| Features | Target total review time for original new drug applications (NDA)/biologics license applications (BLA): eight months (six months plus 60 filing days) (Standard review: 12 months (10 months plus 60 filing days)) Target total review time for efficacy supplement: six months (Standard review: 10 months) | Target total review time: 150 days (excluding the time that applicants require for responses to the CHMP’s questions) (standard assessment: 210 days) | Target total review time: nine months (standard review: 12 months) |
| Requirements |
|
|
|
| US | EU | Japan | |
|---|---|---|---|
| Features |
|
|
|
| Requirements |
|
|
|
| US (Accelerated Approval) | EU (Conditional Marketing Authorization) | Japan (Conditional and Term-Limited Approval) | |
|---|---|---|---|
| Features |
|
|
|
| Requirements |
|
|
|
| US (None) | EU (Marketing Authorization Under Exceptional Circumstances) | Japan (Conditional Approval) | |
|---|---|---|---|
| Features | - |
|
|
| Requirements | - |
|
|
| Brand Name (Company) | Non-proprietary Name | Indication | Approval Date | Number of Enrolled Patients (Japanese) | Primary Endpoint | Post-Marketing Requirement |
|---|---|---|---|---|---|---|
| JACE (Japan Tissue Engineering) | Human Autologous Epidermis-derived Cell Sheet | Serious and Extensive Burns | 29 October 2007(Regular Approval) Priority Review | 2 (2) Single-arm | Epidermal Replacement Rate at Four Weeks | Epidermal replacement rate at four weeks in a single-arm study with 30 patients (Completed) |
| JACC (Japan Tissue Engineering) | Human Autologous Cartilage Cells | Traumatic Cartilage Defects | 27 July 2012 (Regular Approval) | 33 (33) Single-arm | Improvement in Function and Cartilage Repair at 12 Months | None (Post-marketing Surveillance Only) |
| TEMCELL HS (JCR Pharma) | Human Allogeneic Bone Marrow-derived Mesenchymal Stem Cells | Acute Graft Versus Host Disease | 18 September 2015 (Regular Approval) Orphan Designation | 25 (25) Single-arm | Complete Response Rate with ≥28 Days Duration | None (Post-marketing Surveillance Only) |
| Heart Sheet (Terumo) | Human Autologous Skeletal Myoblast -derived Cell Sheet | Serious Heart Failure by Ischemic Heart Disease | 18 September 2015 (Conditional and Term-Limited Approval: five years -> eight years) | 7 (7) Single-arm | Left Ventricular Ejection Fraction at 26 Weeks | Compare time to cardiac event-related death prospectively between 60 patients treated with Heart Sheet and 120 patients with severe heart failure who are potentially eligible for Heart Sheet but are receiving other treatment as the external control group (Ongoing) |
| JACE (Japan Tissue Engineering) | Human Autologous Epidermis-derived Cell Sheet | Giant Congenital Melanocytic Nevus | 29 September 2016 (Regular Approval) Orphan Designation | 11 (11) Single-arm | ≥95% Epithelialization of Grafted Area at 12 Weeks | None (Post-marketing Surveillance Only) |
| JACE (Japan Tissue Engineering) | Human Autologous Epidermis-derived Cell Sheet | Dystrophic Epidermolysis Bullosa and Junctional Epidermolysis Bullosa | 28 December 2018 (Regular Approval) Orphan Designation | 4(4) Single-arm 3 (3) Single-arm | Epidermal Replacement Rate at four Weeks | None (Post-marketing Surveillance Only) |
| STEMIRAC (Nipuro) | Human Autologous Bone Marrow-derived Mesenchymal Stem Cell | Neurological Symptoms and Functional Disorders Associated with Spinal Cord Injury | 28 December 2018 (Conditional and Term-Limited Approval: 7 years) Sakigake Designation | 17 (17) Single-arm | Improvement in American Spinal Injury Association Impairment Scale at 220 Days | Compare improvement in American Spinal Injury Association Impairment Scale prospectively between 198 patients treated with STEMIRAC and 414 patients with spinal cord injury who are potentially eligible for STEMIRAC but are receiving other treatment as the external control group (Ongoing or Not Started) |
| Kymriah (Novartis) | Tisagenlecleucel (CD19 CAR T-cell) | Relapsed and Refractory CD19-positive B-cell Acute Lymphoblastic Leukemia (B-ALL) Relapsed and Refractory CD19-positive Diffuse Large B-cell Lymphoma (DLBCL) | 26 March 2019 (Regular Approval) Orphan Designation | B-ALL: 92 (6) Single-arm DLBCL: 165 (9) Single-arm | B-ALL: Complete Response Rate DLBCL: Overall Response Rate | None (Post-marketing Surveillance Only) |
| Collategen (AnGes) | Beperminogene Perplasmid (Hepatocyte Growth Factor Plasmid) | Chronic Arterial Occlusion (Arteriosclerosis Obliterans and Buerger’s Disease) | 26 March 2019 (Conditional and Term-Limited Approval: five years) | Arteriosclerosis Obliterans: 44 (44) Comparative Collategen: 29, Placebo: 15 Buerger’s Disease: 10 (10) Single-arm Arteriosclerosis Obliterans 2 (2), Buerger’s Disease 4 (4) Single-arm | Arteriosclerosis Obliterans: Improvement in Pain Visual Analogue Scale or Ulcer Size at 12 Weeks Buerger’s Disease: Improvement in Ulcer Size at 12 Weeks Improvement in Pain Visual Analogue Scale or Ulcer Size at 12 Weeks | Compare complete occlusion of ulcer rate prospectively between 120 patients treated with Collategen and 80 patients with Arteriosclerosis Obliterans and Buerger’s Disease who are potentially eligible for Collategen but are receiving other treatment as the external control group (Ongoing or Not Started) |
| Drug | Indication | Approval Date | Number of Enrolled Patients (Japanese) | Primary Endpoint | Post-marketing Requirement |
|---|---|---|---|---|---|
| Lorlatinib | Anaplastic Lymphoma Kinase fusion-positive Non-Small Cell Lung Cancer | 21 September 2018 | 197 (31) in one phase 2 study | Overall Response Rate | Post-marketing surveillance |
| Pembrolizumab | Microsatellite Instability-high solid cancer | 21 December 2018 | 155 (14) in two phase 2 studies | Overall Response Rate |
|
| US (Breakthrough Therapy Designation or Regenerative Medicine Advanced Therapy (RMAT) Designation) | EU Priority Medicines (PRIME) | Japan (Sakigake) | |
|---|---|---|---|
| Features |
|
|
|
| Requirements | (Breakthrough Therapy Designation)
|
|
|
| Drug | Indication | Date of Granting the Breakthrough Therapy Designation Disclosure | Approval Date |
|---|---|---|---|
| Meningococcal Group B Vaccine | Active immunization to prevent invasive meningococcal disease caused by N. meningitidis serogroup B in individuals 10 through 25 years of age | 20 March 2014 | 29 October 2014 |
| Meningococcal Group B Vaccine | Active immunization to prevent invasive meningococcal disease caused by N. meningitidis serogroup B in individuals 10 through 25 years of age | 7 April 2014 | 23 January 2015 |
| Tisagenlecleucel | For the treatment of pediatric and young adult patients (age 3-25 years) with B-cell precursor acute lymphoblastic leukemia that is refractory or in second or later relapse. | 7 July 2014 | 30 August 2017 |
| Axicabtagene Ciloleucel | Treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, primary mediastinal large B-cell lymphoma, high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. Axicabtagene ciloleucel is not indicated for the treatment of patients with primary central nervous system lymphoma. | 7 December 2015 | 18 October 2017 |
| Voretigene Neparvovec | The treatment of patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. | 24 September 2014 | 19 December 2017 |
| Tisagenlecleucel | For the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma who are ineligible for autologous transplant. | 18 April 2017 | 1 May 2018 |
| Coagulation Factor Xa (Recombinant), Inactivated-zhzo | Indicated for patients treated with rivaroxaban and apixaban, when reversal of anticoagulation is needed due to life-threatening or uncontrolled bleeding. | 25 November 2013 | 3 May 2018 |
| Onasemnogene abeparvovec | Indication-treatment of pediatric patients with spinal muscular atrophy (SMA) Type 1 with or without disease onset. | 20 July 2016 | 24 May 2019 |
| Drug | Indication | Date of Granting the PRIME | Approval Date |
|---|---|---|---|
| Tisagenlecleucel | Treatment of pediatric patients with relapsed or refractory B cell acute lymphoblastic leukemia | 23 June 2016 | 22 August 2018 |
| Axicabtagene ciloleucel | Treatment of adult patients with diffuse large B-cell lymphoma who have not responded to their prior therapy, or have had disease progression after autologous stem cell transplant | 26 May 2016 | 23 August 2018 |
| Autologous CD34+ hematopoietic stem cells transduced with lentiviral vector encoding the human βA-T87Q-globin gene (Lentiglobin) | Treatment of transfusion-dependent beta-thalassemia | 15 September 2016 | 29 May 2019 |
| Non-Proprietary Name (Company) | Country Where the Head Quarter Office of the Company That Is Primarily Developing the Product Exists | Characteristics | Indication | Designation Date | Marketing Authorization |
|---|---|---|---|---|---|
| Sirolimus (Nobelpharma) | Japan | Mammalian Target of Rapamycin inhibitor | Skin lesions associated with tuberous sclerosis complex | 27 October 2015 | Approved on 23 March 2017 |
| NS-065/NCNP01 (Nippon Shinyaku) | Japan | Exon 53 skipping of a dystrophin mRNA | Duchenne Muscle Dystrophy | 27 October 2015 | No |
| Baloxavir marboxil (Shionogi) | Japan | Cap-dependent endonuclease inhibitor | Influenza A or B virus infections | 27 October 2015 | Approved on 23 February 2018 |
| BCX7353 (BioCyst Pharmaceuticals -> Integrated Development Associates) | Foreign | Kallikrein inhibitor | Hereditary Angioedema | 27 October 2015 | No |
| Gilteritinib fumarate (Astellas Pharma) | Japan | FLT3 inhibitor | Relapsed or refractory FLT3 mutation-positive acute myeloid leukemia | 27 October 2015 | Approved on 21 September 2018 |
| Pembrolizumab (MSD) | Foreign | Anti-PD 1 antibody | Advanced Gastric Cancer | 27 October 2015 Currently declined | No |
| Olipudase Alfa (Sanofi) | Foreign | Recombinant human acid sphingomyelinase | Acid sphingomyelinase deficiency | 21 April 2017 | No |
| Aducanumab (Biogen) | Foreign | Anti-Amyloid beta antibody | Alzheimer’s Disease | 21 April 2017 | No |
| DS-5141b (DAIICHI SANKYO) | Japan | Exon 45 skipping of a dystrophin mRNA | Duchenne muscular dystrophy | 21 April 2017 | No |
| SPM-011 (STELLA PHARMA) | Japan | Boron Neutron Capture Therapy | Recurrent malignant glioma, unresectable local recurred head and neck cancer, and advanced non squamous cell carcinoma | 21 April 2017 | No |
| Nivolumab (ONO PHARMACEUTICAL) | Japan | Anti-PD 1 antibody | Cholangiocarcinoma | 21 April 2017 | No |
| RTA402 (Kyowa Kirin) | Japan | Nrf2 activator | Diabetic Nephropathy | 27 March 2018 | No |
| JR-141 (JCR Pharma) | Japan | Anti-transferin receptor antibody-fused iduronate-2-sulfatase | Mucopolysaccharidosis type II (Hunter syndrome) | 27 March 2018 | No |
| tafamidis meglumine (Pfizer) | Foreign | Transthyretin stabilizer | Transthyretin-mediated amyloidosis | 27 March 2018 | Approved on 26 March 2019 |
| MSC2156119J (Merck Biopharma) | Foreign | c-MET inhibitor | MET exon 14 skipping-positive NSCLC | 27 March 2018 | No |
| Trastuzumab deruxtecan (DAIICHI SANKYO) | Japan | Anti-HER2 antibody-drug conjugate | Relapsed HER2-positive gastric cancer | 27 March 2018 | No |
| Entrectinib (Chugai Pharmaceutical) | Japan | TRK inhibitor | Adult and pediatric NTRK fusion gene-positive solid cancer | 27 March 2018 | No |
| Valemetostat (DAIICHI SANKYO) | Japan | EZH 1/2 inhibitor | Relapsed and refractory peripheral T-cell lymphoma | 8 April 2019 | No |
| Ixazomib (Takeda Pharmaceutical) | Japan | Proteasome inhibitor | AL amyloidosis | 8 April 2019 | No |
| TAK-925 (Takeda Pharmaceutical) | Japan | Orexin 2 Receptor-selective agonist | Narcolepsy | 8 April 2019 | No |
| ASP-1929 (Rakuten Medical) | Japan | Cetuximab and IRDye 700DX activated with red light using a proprietary investigational laser and fiber optics | Head and neck cancer | 8 April 2019 | No |
| E7090 (Eisai) | Japan | FGFR 1/2/3 inhibitor | FGFR2 fusion gene-positive Cholangiocarcinoma | 8 April 2019 | No |
| Product Name (Company) | Country where the Head Quarter Office of the Company That Is Primarily Developing the Product Exists | Characteristics | Indication | Designation Date |
|---|---|---|---|---|
| STEMIRAC (Nipuro) | Japan | Human Autologous Bone Marrow-derived Mesenchymal Stem Cell | Neurological Symptoms and Functional Disorders Associated with Spinal Cord Injury | 10 February 2016 |
| G47 Delta (DaiichiSankyo) | Japan | Oncolytic Herpes Simplex Virus-1 | Malignant Glioma | 10 February 2016 |
| JRM-001 (Japan Regenerative Medicine) | Japan | Human Autologous Cardiac Progenitor/Stem Cells | Pediatric Congenital Heart Disease (Single Ventricle Physiology) | 10 February 2016 |
| CLS2702C/D (CellSeed) | Japan | Human Autologous Oral Mucosal Epithelial Cell Sheet | Prevention of Esophageal Partial Narrowing after Endoscopic Submucosal Dissection | 28 February 2017 |
| None (Sumitomo Dainippon Pharma) | Japan | Human Allogeneic iPS-derived Dopamine Neural Progenitor Cells | Parkinson’s Disease | 28 February 2017 |
| HLCM051 (Healios) | Japan | Human Allogeneic Bone Marrow Progenitor/Stem Cells | Cerebral Infarction | 28 February 2017 |
| TBI-1301 (Takara Bio and Otsuka Pharmaceutical) | Japan | NY-ESO-1 siTCR Gene Therapy | Synovial Sarcoma | 27 March 2018 |
| CLBS12 (Caladrius Biosciences) | Foreign | Human Autologous CD34-positive Peripheral Blood Cells | Critical Limb Ischemia | 27 March 2018 |
| AVXS-101 (AveXis and Novartis) | Foreign | Human SMN Adeno-associated Virus 9 Gene Therapy | Spinal Muscular Atrophy | 27 March 2018 |
| OBP-301 (Oncolys BioPharma) | Japan | hTERT Promotor Oncolytic Adenovirus | Esophageal Cancer | 8 April 2019 |
| SB623 (SanBio) | Japan | Adult Allogeneic Bone Marrow-derived Mesenchymal Stem Cells | Chronic Motor Deficit Resulting from Traumatic Brain Injury | 8 April 2019 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagai, S. Flexible and Expedited Regulatory Review Processes for Innovative Medicines and Regenerative Medical Products in the US, the EU, and Japan. Int. J. Mol. Sci. 2019, 20, 3801. https://doi.org/10.3390/ijms20153801
Nagai S. Flexible and Expedited Regulatory Review Processes for Innovative Medicines and Regenerative Medical Products in the US, the EU, and Japan. International Journal of Molecular Sciences. 2019; 20(15):3801. https://doi.org/10.3390/ijms20153801
Chicago/Turabian StyleNagai, Sumimasa. 2019. "Flexible and Expedited Regulatory Review Processes for Innovative Medicines and Regenerative Medical Products in the US, the EU, and Japan" International Journal of Molecular Sciences 20, no. 15: 3801. https://doi.org/10.3390/ijms20153801
APA StyleNagai, S. (2019). Flexible and Expedited Regulatory Review Processes for Innovative Medicines and Regenerative Medical Products in the US, the EU, and Japan. International Journal of Molecular Sciences, 20(15), 3801. https://doi.org/10.3390/ijms20153801