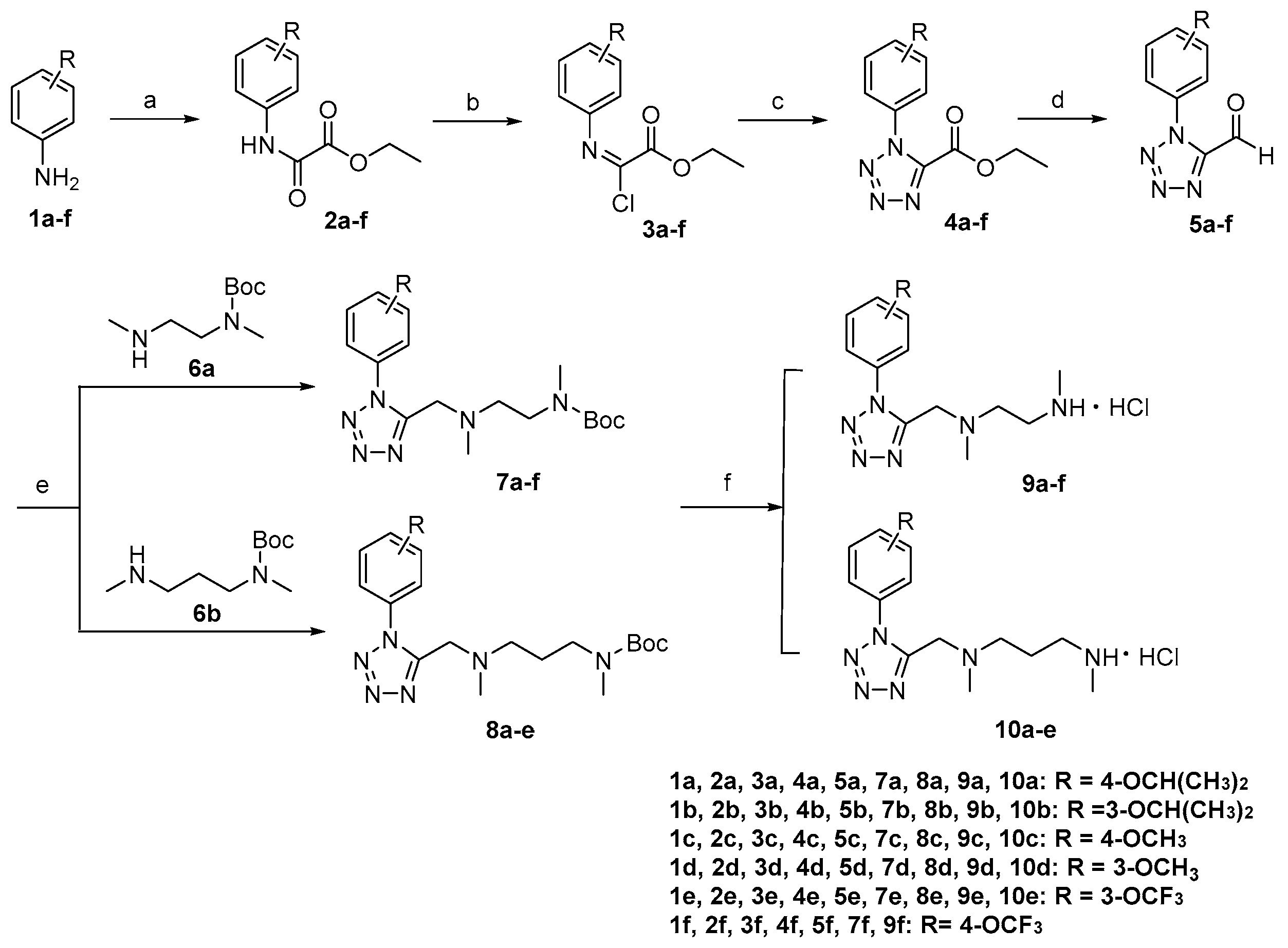
3.2.1. Synthesis of 9a–f and 10a–e
General procedure for the preparation of compounds 2a–f.
Triethylamine (2.5 eq) was added to the substituted aniline 1a–f (1.0 eq) dissolved in anhydrous dichloromethane (1.5 mL/mmol). The reaction mixture was cooled to 0 °C and ethyl oxalyl monochloride (1.2 eq) was added dropwise to the solution. Subsequently, the reaction was warmed to room temperature and stirred for 1 h. The reaction mixture was quenched with water and extracted with dichloromethane, the organic layer was dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel using EtOAc/petroleum as eluent to give 2a–f.
Ethyl N-(4-isopropoxyphenyl)oxamate (2a)
Yellow solid. Yield: 94%. MP: 128.7–129.9 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.67 (s, 1H, -NH-), 7.69–7.55 (m, 2H, -ArH), 6.98–6.77 (m, 2H, -ArH), 4.57 (Hept, J = 6.0 Hz, 1H, -OCH(CH3)2), 4.30 (q, J = 7.1 Hz, 2H, -CH2-), 1.31 (t, J = 7.1 Hz, 3H, -CH3), 1.26 (s, 3H, -CH3), 1.24 (s, 3H, -CH3).
Ethyl N-(3-isopropoxyphenyl)oxamate (2b)
Yellow oil. Yield: 92%. 1H NMR (400 MHz, Chloroform-d) δ 8.84 (s, 1H, -NH-), 7.39 (t, J = 2.2 Hz, 1H, -ArH), 7.26 (t, J = 8.1 Hz, 1H, -ArH), 7.09 (ddd, J = 8.1, 2.1, 1.0 Hz, 1H, -ArH), 6.74 (ddd, J = 8.3, 2.5, 0.9 Hz, 1H, -ArH), 4.58 (p, J = 6.1 Hz, 1H, -OCH(CH3)2), 4.44 (q, J = 7.1 Hz, 2H, -CH2-), 1.46 (t, J = 7.2 Hz, 3H, -CH3), 1.36 (d, J = 6.1 Hz, 6H, -CH(CH3)2).
Ethyl N-(4-methoxyphenyl)oxamate (2c)
Yellow solid. Yield: 94%. MP: 105.7–106.3 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.81 (s, 1H, -NH-), 7.61–7.56 (m, 2H, -ArH), 6.95–6.90 (m, 2H, -ArH), 4.44 (q, J = 7.1 Hz, 2H, -CH2-), 3.83 (s, 3H, -OCH3), 1.45 (t, J = 7.1 Hz, 3H, -CH3).
Ethyl N-(3-methoxyphenyl)oxamate (2d)
Yellow solid. Yield: 95%. MP: 88.9–99.3 °C. 1H NMR (400 MHz, Chloroform-d) δ 8.87 (s, 1H, -NH-), 7.41 (t, J = 2.3 Hz, 1H, -ArH), 7.30 (d, J = 8.1 Hz, 1H, -ArH), 7.13 (ddd, J = 8.0, 2.0, 0.9 Hz, 1H, -ArH), 6.77 (ddd, J = 8.3, 2.5, 0.9 Hz, 1H, -ArH), 4.44 (q, J = 7.2 Hz, 2H, -CH2-), 3.84 (s, 3H, -OCH3), 1.46 (t, J = 7.2 Hz, 3H, -OCH3), 1.46 (t, J = 7.2 Hz, 3H, -CH3).
Ethyl N-(3-trifluoromethoxyphenyl)oxamate (2e)
Yellow oil. Yield: 90%. 1H NMR (400 MHz, Chloroform-d) δ 8.96 (s, 1H, -NH-), 7.70 (d, J = 2.5 Hz, 1H, -ArH), 7.54 (ddd, J = 8.3, 2.1, 0.9 Hz, 1H, -ArH), 7.42 (t, J = 8.2 Hz, 1H, -ArH), 7.08 (ddt, J = 8.3, 2.3, 1.1 Hz, 1H, -ArH), 4.46 (q, J = 7.2 Hz, 2H, -CH2-), 1.46 (t, J = 7.2 Hz, 3H, -CH3).
Ethyl N-(4-trifluoromethoxyphenyl)oxamate (2f)
Yellow solid. Yield: 92%. MP: 131.2–131.9 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H, -NH-), 7.90–7.85 (m, 2H, -ArH), 7.42–7.36 (m, 2H, -ArH), 4.32 (q, J = 7.1 Hz, 2H, -CH2-), 1.32 (t, J = 7.1 Hz, 3H, -CH3).
General procedure for the preparation of compounds 3a–f.
2a–f (1.0 eq) was dissolved in CCl4 (2.0 mL/mmol), and a solution of triphenylphosphine in CCl4 (0.8 mL/mmol) was added dropwise to the reaction flask at the room temperature. The reaction was refluxed and stirred for 6 h, and then cooled to the room temperature. The precipitate was filtered off and washed with CCl4. The filtrate was concentrated in vacuo without further purification to give 3a–f.
General procedure for the preparation of compounds 4a–f.
To a solution of 3a–f (1.0 eq) in acetonitrile (1 mL/mmol) was added sodium azide (1.5 eq), and the reaction was stirred at the room temperature for 3 h and monitored by TLC. The mixture was quenched with ice water, concentrated and extracted with ethyl acetate. The combined organic layers were washed with water, and dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel using EtOAc/petroleum as eluent to give 4a–f.
Ethyl 1-(4-isopropoxyphenyl)-1H-tetrazole-5-carboxylate (4a)
White solid. Yield: 65%. MP: 150.2–151.9 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.44–7.35 (m, 2H, -ArH), 7.06–6.99 (m, 2H, -ArH), 4.64 (p, J = 6.1 Hz, 1H, -OCH(CH3)2), 4.46 (q, J = 7.1 Hz, 2H, -CH2-), 1.39 (dd, J = 7.9, 6.6 Hz, 9H, -CH3).
Ethyl 1-(3-isopropoxyphenyl)-1H-tetrazole-5-carboxylate (4b)
Yellow oil. Yield: 63%. 1H NMR (400 MHz, DMSO-d6) δ 7.48 (t, J = 8.1 Hz, 1H, -ArH), 7.35 (t, J = 2.2 Hz, 1H, -ArH), 7.19 (dddd, J = 11.7, 8.4, 2.2, 0.9 Hz, 2H, -ArH), 4.65 (h, J = 6.0 Hz, 1H, -OCH(CH3)2), 4.33 (t, J = 7.1 Hz, 2H, -CH2-), 1.30 (s, 3H, -CH3), 1.28 (s, 3H, -CH3), 1.20 (t, J = 7.1 Hz, 3H, -CH3).
Ethyl 1-(4-methoxyphenyl)-1H-tetrazole-5-carboxylate (4c)
White solid. Yield: 65%. MP: 75.9–76.3 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.46–7.41 (m, 2H, -ArH), 7.10–7.05 (m, 2H, -ArH), 4.47 (q, J = 7.1 Hz, 2H, -CH2-), 3.92 (s, 3H, -OCH3), 1.41 (t, J = 7.1 Hz, 3H, -CH3).
Ethyl 1-(3-methoxyphenyl)-1H-tetrazole-5-carboxylate (4d)
White solid. Yield: 64%. MP: 70.1–72.0 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.49 (t, J = 8.1 Hz, 1H, -ArH), 7.16 (ddd, J = 8.5, 2.5, 0.9 Hz, 1H, -ArH), 7.08 (ddd, J = 7.8, 2.0, 0.9 Hz, 1H, -ArH), 7.05 (t, J = 2.2 Hz, 1H, -ArH), 4.48 (q, J = 7.1 Hz, 2H, -CH2-), 3.89 (s, 3H, -OCH3), 1.40 (t, J = 7.1 Hz, 3H, -CH3).
Ethyl 1-(3-trifluoromethoxyphenyl)-1H-tetrazole-5-carboxylate (4e)
Yellow solid. Yield: 64%. MP: 125.2–126.0 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.66 (dd, J = 8.7, 7.7 Hz, 1H, -ArH), 7.51 (dd, J = 7.9, 2.2 Hz, 2H, -ArH), 7.46 (q, J = 1.9 Hz, 1H, -ArH), 4.49 (q, J = 7.1 Hz, 2H, -CH2-), 1.41 (t, J = 7.1 Hz, 3H, -CH3).
Ethyl 1-(4-trifluoromethoxyphenyl)-1H-tetrazole-5-carboxylate (4f)
Yellow solid. Yield: 65%. MP: 68.2–69.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.96–7.84 (m, 2H, -ArH), 7.71–7.60 (m, 2H, -ArH), 4.32 (q, J = 7.1 Hz, 2H, -CH2-), 1.19 (t, J = 7.1 Hz, 3H, -CH3).
General procedure for the preparation of compounds 5a–f.
To a solution of 4a–f (1.0 eq) in anhydrous dichloromethane (1.8 mL/mmol) was added diisobutyl aluminium hydride (1.0M in hexanes, 2.0 eq) dropwise at −78 °C and the reaction was stirred for 30 min. The mixture was quenched with methanol, concentrated and extracted with ethyl acetate. The combined organic layers were washed with water, 1 M HCl and dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel using EtOAc/petroleum as eluent to give 5a–f.
1-(4-Isopropoxyphenyl)-1H-tetrazole-5-carbaldehyde (5a)
White solid. Yield: 80%. MP: 122.5–123.1 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.05 (s, 1H, -CHO), 7.67–7.63 (m, 2H, -ArH), 7.17–7.13 (m, 2H, -ArH), 4.79–4.72 (m, 1H, -OCH(CH3)2), 1.33 (s, 3H, -CH3), 1.32 (s, 3H, -CH3).
1-(3-Isopropoxyphenyl)-1H-tetrazole-5-carbaldehyde (5b)
Colorless oil. Yield: 75%. 1H NMR (400 MHz, DMSO-d6) δ 10.07 (s, 1H, -CHO), 7.53 (t, J = 8.1 Hz, 1H, -ArH), 7.38 (t, J = 2.2 Hz, 1H, -ArH), 7.29–7.25 (m, 2H, -ArH), 4.69–4.64 (m, 1H, -OCH(CH3)2), 1.31 (d, J = 0.8 Hz, 3H, -CH3), 1.29 (d, J = 0.8 Hz, 3H, -CH3).
1-(4-Methoxyphenyl)-1H-tetrazole-5-carbaldehyde (5c)
Colorless oil. Yield: 77%. 1H NMR (400 MHz, Chloroform-d) δ 10.33 (s, 1H, -CHO), 7.51–7.47 (m, 2H, -ArH), 7.11–7.07 (m, 2H, -ArH), 3.93 (s, 3H, -OCH3).
1-(3-Methoxyphenyl)-1H-tetrazole-5-carbaldehyde (5d)
Colorless oil. Yield: 75%. 1H NMR (400 MHz, DMSO-d6) δ 10.07 (s, 1H, -CHO) 7.59–7.54 (t, 1H, -ArH), 7.41 (t, J = 2.2 Hz, 1H, -ArH), 7.31 (dddd, J = 7.9, 6.8, 2.0, 0.9 Hz, 2H, -ArH), 3.83 (s, 3H, -OCH3).
1-(3-Trifluoromethoxyphenyl)-1H-tetrazole-5-carbaldehyde (5e)
Yellow solid. Yield: 78%. MP: 97.6–98.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.11 (s, 1H, -CHO), 7.96–7.91 (m, 2H, -ArH), 7.75–7.70 (m, 1H, -ArH), 7.65 (ddt, J = 8.0, 2.3, 1.1 Hz, 1H, -ArH).
1-(4-Trifluoromethoxyphenyl)-1H-tetrazole-5-carbaldehyde (5f)
Yellow solid. Yield: 80%. MP: 118.7–119.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.10 (s, 1H, -CHO), 7.94–7.91 (m, 2H, -ArH), 7.70 (dtd, J = 8.1, 2.2, 1.2 Hz, 2H, -ArH).
General procedure for the preparation of compounds 7a–f and 8a–e.
To a solution of 5a–f (1.0 eq) in 1,2-Dichloroethane (8.5 mL/mmol) was added sodium triacetoxyborohydride (2.0 eq) and 6a or 6b (1.0 eq) and the reaction was stirred for 12 h at room temperature. The reaction mixture was diluted with water and extracted with dichloromethane. The dichloromethane layer was dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel using EtOAc/petroleum as eluent to give 7a–f or 8a–e.
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(4-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-ethylenediamine (7a)
Colorless oil. Yield: 85%. 1H NMR (400 MHz, Chloroform-d) δ 7.59 (d, J = 8.5 Hz, 2H, -ArH), 7.05 (d, J = 8.9 Hz, 2H, -ArH), 4.65 (p, J = 6.0 Hz, 1H, -OCH(CH3)2), 3.82 (s, 2H, -CH2-), 3.31 (s, 2H, -CH2-), 2.78 (s, 3H, -CH3), 2.38 (s, 3H, -CH3), 1.44 (s, 9H, -C(CH3)3), 1.41 (s, 3H, -CH3), 1.40 (s, 3H, -CH3).
N-tert-Butyloxycarbonyl-N-methyl-N-(1-(3-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-ethylenediamine (7b)
Colorless oil. Yield: 82%. 1H NMR (400 MHz, DMSO-d6) δ 7.52 (t, J = 8.1 Hz, 1H, -ArH), 7.35–7.24 (m, 2H, -ArH), 7.17 (ddd, J = 8.4, 2.5, 0.9 Hz, 1H, -ArH), 4.71 (p, J = 6.0 Hz, 1H, -OCH(CH3)2), 3.11 (t, J = 6.4 Hz, 2H, -CH2-), 2.63 (d, J = 6.4 Hz, 3H, -CH3), 2.48 (t, J = 6.6 Hz, 2H, -CH2-), 2.20 (d, J = 6.4 Hz, 3H, -CH3), 1.31 (dd, J = 12.4, 8.1 Hz, 15H, -CH3, -C(CH3)3).
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(4-methoxyphenyl)-1H-tetrazol-5-ylmethyl)-N’-methyl-ethylenediamine (7c)
Colorless oil. Yield: 85%. 1H NMR (400 MHz, DMSO-d6) δ 7.66 (dd, J = 8.3, 2.8 Hz, 2H, -ArH), 7.21–7.11 (m, 2H, -ArH), 3.85 (s, 3H, -CH3), 3.83 (s, 2H, -CH2-), 3.15 (d, J = 6.3 Hz, 2H, -CH2-), 2.64 (d, J = 9.5 Hz, 3H, -CH3), 2.47 (d, J = 6.5 Hz, 2H, -CH2-), 2.18 (d, J = 6.8 Hz, 3H, -CH3), 1.33 (d, J = 12.1 Hz, 9H, -C(CH3)3).
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(3-methoxyphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-ethylenediamine (7d)
Colorless oil. Yield: 83%. 1H NMR (400 MHz, DMSO-d6) δ 7.55 (t, J = 8.1 Hz, 1H, -ArH), 7.39–7.30 (m, 2H, -ArH), 7.20 (ddd, J = 8.4, 2.6, 0.9 Hz, 1H, -ArH), 3.90 (s, 2H, -CH2-), 3.84 (s, 3H, -CH3), 3.13 (d, J = 7.2 Hz, 2H, -CH2-), 2.62 (d, J = 6.6 Hz, 3H, -CH3), 2.48 (d, J = 6.6 Hz, 2H, -CH2-), 2.21 (s, 3H, -CH3), 1.32 (d, J = 10.5 Hz, 9H, -C(CH3)3).
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(3-trifluoromethoxyphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-ethylenediamine (7e)
Colorless oil. Yield: 82%. 1H NMR (400 MHz, DMSO-d6) δ 7.93 (s, 1H, -ArH), 7.87–7.82 (m, 1H, -ArH), 7.79 (t, J = 8.1 Hz, 1H, -ArH), 7.68 (ddt, J = 8.0, 2.4, 1.2 Hz, 1H, -ArH), 3.94 (s, 2H, -CH2-), 3.09 (t, J = 6.3 Hz, 2H, -CH2-), 2.59 (d, J = 5.4 Hz, 3H, -CH3), 2.46 (t, J = 6.6 Hz, 2H, -CH2-), 2.19 (s, 3H, -CH3), 1.31 (d, J = 9.5 Hz, 9H, -C(CH3)3).
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(4-trifluoromethoxyphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-ethylenediamine (7f)
Colorless oil. Yield: 84%. 1H NMR (400 MHz, DMSO-d6) δ 7.94 (d, J = 8.3 Hz, 2H, -ArH), 7.69–7.64 (m, 2H, -ArH), 3.91 (s, 2H, -CH2-), 3.13 (t, J = 6.4 Hz, 2H, -CH2-), 2.61 (d, J = 6.3 Hz, 3H, -CH3), 2.48 (d, J = 6.5 Hz, 2H, -CH2-), 2.18 (d, J = 7.0 Hz, 3H, -CH3), 1.32 (d, J = 11.3 Hz, 9H, -C(CH3)3).
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(4-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-1,3-propylenediamine (8a)
Colorless oil. Yield: 80%. 1H NMR (400 MHz, DMSO-d6) δ 7.61 (dd, J = 9.1, 2.5 Hz, 2H, -ArH), 7.15–7.10 (m, 2H, -ArH), 4.76–4.69 (m, 1H, -OCH(CH3)2), 3.81 (s, 2H, -CH2-), 2.92 (s, 2H, -CH2-), 2.66 (s, 3H, -CH3), 2.29 (t, J = 7.0 Hz, 2H, -CH2-), 2.10 (s, 3H, -CH3), 1.45 (t, J = 7.2 Hz, 2H, -CH2-), 1.34 (d, J = 11.2 Hz, 9H, -C(CH3)3), 1.31 (d, J = 1.5 Hz, 3H, -CH3), 1.30 (s, 3H, -CH3).
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(3-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-1,3-propylenediamine (8b)
Colorless oil. Yield: 77%. 1H NMR (400 MHz, DMSO-d6) δ 7.51 (t, J = 8.2 Hz, 1H, -ArH), 7.33 (d, J = 9.6 Hz, 1H, -ArH), 7.28–7.22 (m, 1H, -ArH), 7.21–7.14 (m, 1H, -ArH), 4.70 (p, J = 6.0 Hz, 1H, -OCH(CH3)2), 3.86 (s, 2H, -CH2-), 2.94 (s, 2H, -CH2-), 2.67 (d, J = 4.2 Hz, 3H, -CH3), 2.29 (t, J = 7.0 Hz, 2H, -CH2-), 2.11 (s, 3H, -CH3), 1.46 (t, J = 7.1 Hz, 2H, -CH2-), 1.33 (d, J = 15.1 Hz, 9H, -C(CH3)3), 1.30 (s, 3H, -CH3), 1.28 (s, 3H, -CH3).
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(4-methoxyphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-1,3-propylenediamine (8c)
Colorless oil. Yield: 75%. 1H NMR (400 MHz, DMSO-d6) δ 7.67–7.62 (m, 2H, -ArH), 7.18–7.14 (m, 2H, -ArH), 3.85 (d, J = 4.2 Hz, 3H, -CH3), 3.81 (s, 2H, -CH2-), 2.94 (s, 2H, -CH2-), 2.67 (s, 3H, -CH3), 2.28 (t, J = 7.1 Hz, 2H, -CH2-), 2.10 (s, 3H, -CH3), 1.45 (s, 2H, -CH2-), 1.36 (s, 9H, -C(CH3)3).
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(3-methoxyphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-1,3-propylenediamine (8d)
Colorless oil. Yield: 75%. 1H NMR (400 MHz, DMSO-d6) δ 7.54 (t, J = 8.2 Hz, 1H, -ArH), 7.39 (t, J = 2.3 Hz, 1H, -ArH), 7.30 (dddd, J = 6.8, 2.8, 1.9, 0.9 Hz, 1H, -ArH), 7.23–7.17 (m, 1H, -ArH), 3.87 (s, 2H, -CH2-), 3.83 (s, 3H, -CH3), 2.93 (s, 2H, -CH2-), 2.66 (s, 3H, -CH3), 2.30 (t, J = 7.1 Hz, 2H, -CH2-), 2.12 (s, 3H, -CH3), 1.46 (t, J = 7.2 Hz, 2H, -CH2-), 1.35 (s, 9H, -C(CH3)3).
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(3-trifluoromethoxyphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-1,3-propylenediamine (8e)
Colorless oil. Yield: 75%. 1H NMR (400 MHz, DMSO-d6) δ 7.98 (s, 1H, -ArH), 7.83 (ddd, J = 8.0, 4.1, 2.0 Hz, 2H, -ArH), 7.68 (dtq, J = 7.3, 2.5, 1.2 Hz, 1H, -ArH), 3.90 (s, 2H, -CH2-), 2.87 (s, 2H, -CH2-), 2.65 (s, 3H, -CH3), 2.27 (t, J = 7.1 Hz, 2H, -CH2-), 2.08 (s, 3H, -CH3), 1.42 (d, J = 7.4 Hz, 2H, -CH2-), 1.35 (s, 9H, -C(CH3)3).
General procedure for the preparation of compounds 9a–f and 10a–e.
Compounds 7a–f or 8a–e (0.51 mmol) was added to saturated hydrochloric acid ethanol solution (10 mL) at room temperature and stirred for 30 min. The reaction solution was filtered and washed with diethylether (20 mL) to give 9a–f or 10a–e.
N,N’-Dimethyl-N’-(1-(4-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)ethylenediamine hydrochloride (9a)
White solid. Yield: 98%. MP: 187.4–189.4 °C. 1H NMR (400 MHz, Deuterium Oxide) δ 7.49–7.44 (m, 2H, -ArH), 7.19–7.13 (m, 2H, -ArH), 4.74 (p, J = 5.5, 4.9 Hz, 1H, -OCH(CH3)2), 4.35 (s, 2H, -CH2-), 3.21 (dd, J = 14.9, 6.8 Hz, 2H, -CH2-), 3.15–3.01 (m, 2H, -CH2-), 2.64 (dd, J = 4.9, 2.1 Hz, 3H, -CH3), 2.48 (d, J = 37.8 Hz, 3H, -CH3), 1.31 (s, 3H, -CH3), 1.30 (s, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 159.2, 150.1, 126.7, 124.9, 117.1, 71.9, 51.8, 49.1, 44.2, 41.3, 33.0, 20.9. HRMS (ESI) m/z [M+H]+ Calcd for C15H25N6O+: 305.20844. Found: 305.20831.
N,N’-Dimethyl-N’-(1-(3-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)ethylenediamine hydrochloride (9b)
Colorless oil. Yield: 95%. 1H NMR (400 MHz, Deuterium Oxide) δ 7.49 (ddd, J = 8.4, 7.4, 0.9 Hz, 1H, -ArH), 7.18 (ddd, J = 8.5, 2.3, 1.1 Hz, 1H, -ArH), 7.09–7.04 (m, 2H, -ArH), 4.63 (p, J = 6.1 Hz, 1H, -OCH(CH3)2), 4.37 (s, 2H, -CH2-), 3.22 (td, J = 6.2, 1.3 Hz, 2H, -CH2-), 3.13 (td, J = 6.4, 1.3 Hz, 2H, -CH2-), 2.61 (s, 3H, -CH3), 2.52 (s, 3H, -CH3), 1.24 (s, 3H, -CH3), 1.22 (s, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 158.1, 133.1, 131.3, 119.1, 117.3, 112.8, 72.1, 51.8, 49.2, 44.2, 41.3, 33.0, 20.9. HRMS (ESI) m/z [M+H]+ Calcd for C15H25N6O+: 305.20844. Found: 305.20828.
N,N’-Dimethyl-N’-(1-(4-methoxyphenyl)-1H-tetrazol-5-yl-methyl)ethylenediamine hydrochloride (9c)
White solid. Yield: 95%. MP: 163.2–163.6 °C. 1H NMR (400 MHz, Deuterium Oxide) δ 7.45–7.40 (m, 2H, -ArH), 7.14–7.10 (m, 2H, -ArH), 4.51 (s, 2H, -CH2-), 3.81 (s, 3H, -CH3), 3.31 (s, 4H, -CH2-), 2.67 (s, 3H, -CH3), 2.64 (d, J = 0.7 Hz, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 161.1, 149.8, 126.6, 124.9, 115.3, 55.7, 51.8, 49.1, 44.00, 41.4, 33.0. HRMS (ESI) m/z [M+H]+ Calcd for C13H21N6O+: 277.17714. Found: 277.17715.
N,N’-Dimethyl-N’-(1-(3-methoxyphenyl)-1H-tetrazol-5-yl-methyl)ethylenediamine hydrochloride (9d)
White solid. Yield: 98%. MP: 185.6–186.4 °C. 1H NMR (400 MHz, Deuterium Oxide) δ 7.52–7.47 (m, 1H, -ArH), 7.18 (ddd, J = 8.5, 2.5, 0.9 Hz, 1H, -ArH), 7.10–7.05 (m, 2H, -ArH), 4.47 (s, 2H, -CH2-), 3.77 (s, 3H, -CH3), 3.29–3.20 (m, 4H, -CH2-), 2.62 (s, 3H, -CH3), 2.60 (s, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 160.1, 149.7, 133.0, 131.2, 117.2, 117.1, 110.9, 55.7, 51.9, 49.2, 44.1, 41.4, 33.0. HRMS (ESI) m/z [M+H]+ Calcd for C13H21N6O+: 277.17714. Found: 277.17703.
N,N’-Dimethyl-N’-(1-(3-trifluoromethoxyphenyl)-1H-tetrazol-5-yl-methyl)ethylenediamine hydrochloride (9e)
White solid. Yield: 97%. MP: 185.6–186.4 °C. 1H NMR (400 MHz, Deuterium Oxide) δ 7.66 (td, J = 8.1, 1.5 Hz, 1H, -ArH), 7.58–7.52 (m, 2H, -ArH), 7.52–7.48 (m, 1H, -ArH), 4.49 (s, 2H, -CH2-), 3.28 (d, J = 4.2 Hz, 2H, -CH2-), 3.26 (d, J = 3.9 Hz, 2H, -CH2-), 2.63 (s, 3H, -CH3), 2.62 (s, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 150.7, 149.4, 133.2, 131.8, 123.9, 123.5, 118.1, 51.9, 49.4, 44.5, 41.3, 32.9. HRMS (ESI) m/z [M+H]+ Calcd for C13H18F3N6O+: 331.14887. Found: 331.14871.
N,N’-Dimethyl-N’-(1-(4-trifluoromethoxyphenyl)-1H-tetrazol-5-yl-methyl)ethylenediamine hydrochloride (9f)
White solid. Yield: 98%. MP: 194.3–195.2 °C. 1H NMR (400 MHz, Deuterium Oxide) δ 7.61–7.56 (m, 2H, -ArH), 7.51–7.46 (m, 2H, -ArH), 4.25 (s, 2H, -CH2-), 3.15 (t, J = 6.4 Hz, 2H, -CH2-), 3.02 (t, J = 6.4 Hz, 2H, -CH2-), 2.58 (s, 3H, -CH3), 2.42 (s, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 150.6, 150.3, 130.6, 126.9, 122.5, 51.9, 49.3, 44.3, 41.3, 33.0. HRMS (ESI) m/z [M+H]+ Calcd for C13H18F3N6O+: 331.14887. Found: 331.14871.
N,N’-Dimethyl-N’-(1-(4-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-1,3-propylenediamine hydrochloride (10a)
Colorless oil. Yield: 94%. 1H NMR (400 MHz, Deuterium Oxide) δ 7.43–7.39 (m, 2H, -ArH), 7.14–7.09 (m, 2H, -ArH), 4.76 (d, J = 1.2 Hz, 2H, -CH2-), 4.67 (d, J = 6.1 Hz, 1H, -OCH(CH3)2), 3.36–3.27 (m, 2H, -CH2-), 3.05–2.97 (m, 2H, -CH2-), 2.94–2.89 (m, 3H, -CH3), 2.61 (s, 3H, -CH3), 2.14–2.03 (m, 2H, -CH2-), 1.26 (s, 3H, -CH3), 1.25 (s, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 159.4, 148.0, 126.7, 124.5, 117.2, 71.9, 53.5, 48.1, 45.4, 41.1, 32.71, 20.9. HRMS (ESI) m/z [M+H]+ Calcd for C16H27N6O+: 319.22409. Found: 319.22406.
N,N’-Dimethyl-N’-(1-(3-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-1,3-propylenediamine hydrochloride (10b)
Colorless oil. Yield: 92%. 1H NMR (400 MHz, Deuterium Oxide) δ 7.50 (t, J = 8.1 Hz, 1H, -ArH), 7.20 (ddd, J = 8.5, 2.4, 0.9 Hz, 1H, -ArH), 7.09–7.03 (m, 2H, -ArH), 4.79 (s, 2H, -CH2-), 4.68–4.60 (m, 1H, -OCH(CH3)2), 3.36–3.28 (m, 2H, -CH2-), 3.04–2.96 (m, 2H, -CH2-), 2.91 (s, 3H, -CH3), 2.61 (s, 3H, -CH3), 2.15–2.04 (m, 2H, -CH2-), 1.24 (s, 3H, -CH3), 1.23 (s, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 158.2, 147.7, 132.7, 131.4, 119.3, 117.3, 112.8, 72.1, 53.6, 48.2, 45.4, 41.1, 32.7, 20.9, 20.8. HRMS (ESI) m/z [M+H]+ Calcd for C16H27N6O+: 319.22409. Found: 319.22405.
N,N’-Dimethyl-N’-(1-(4-methoxyphenyl)-1H-tetrazol-5-yl-methyl)-1,3-propylenediamine hydrochloride (10c)
White solid. Yield: 94%. MP: 188.2–190.1 °C. 1H NMR (400 MHz, Deuterium Oxide) δ 7.48–7.42 (m, 2H, -ArH), 7.19–7.12 (m, 2H, -ArH), 4.73 (s, 2H, -CH2-), 3.84 (s, 3H, -CH3), 3.32–3.24 (m, 2H, -CH2-), 3.05–2.98 (m, 2H, -CH2-), 2.88 (s, 3H, -CH3), 2.64 (s, 3H, -CH3), 2.14–2.04 (m, 2H, -CH2-). 13C NMR (101 MHz, Deuterium Oxide) δ 148.1, 126.7, 124.5, 115.4, 55.7, 53.5, 48.1, 45.4, 41.1, 32.7, 20.9. HRMS (ESI) m/z [M+H]+ Calcd for C14H23N6O+: 291.19279. Found: 291.19271.
N,N’-Dimethyl-N’-(1-(3-methoxyphenyl)-1H-tetrazol-5-yl-methyl)-1,3-propylenediamine hydrochloride (10d)
Colorless oil. Yield: 92%. 1H NMR (400 MHz, Deuterium Oxide) δ 7.51 (dd, J = 8.5, 7.8 Hz, 1H, -ArH), 7.20 (ddd, J = 8.6, 2.5, 0.9 Hz, 1H, -ArH), 7.09 (t, J = 2.2 Hz, 1H, -ArH), 7.06 (ddd, J = 7.8, 2.1, 0.9 Hz, 1H, -ArH), 4.80 (d, J = 1.3 Hz, 2H, -CH2-), 3.78 (s, 3H, -CH3), 3.36–3.29 (m, 2H, -CH2-), 3.05–2.97 (m, 2H, -CH2-), 2.91 (d, J = 1.3 Hz, 3H, -CH3), 2.61 (s, 3H, -CH3), 2.17–2.04 (m, 2H, -CH2-). 13C NMR (101 MHz, Deuterium Oxide) δ 160.1, 147.8, 132.7, 131.4, 117.4, 117.2, 110.9, 55.8, 53.6, 48.2, 45.4, 41.1, 32.7, 20.8. HRMS (ESI) m/z [M+H]+ Calcd for C14H23N6O+: 291.19279. Found: 291.19267.
N,N’-Dimethyl-N’-(1-(3-trifluoromethoxyphenyl)-1H-tetrazol-5-yl-methyl)-1,3-propylenediamine hydrochloride (10e)
Colorless oil. Yield: 92%. 1H NMR (400 MHz, Deuterium Oxide) δ 7.70–7.64 (m, 1H, -ArH), 7.58–7.52 (m, 2H, -ArH), 7.48 (ddd, J = 7.9, 2.0, 1.0 Hz, 1H, -ArH), 4.71 (s, 2H, -CH2-), 3.26–3.20 (m, 2H, -CH2-), 3.01–2.95 (m, 2H, -CH2-), 2.83 (s, 3H, -CH3), 2.60 (s, 3H, -CH3), 2.10–2.01 (m, 2H, -CH2-). 13C NMR (101 MHz, Deuterium Oxide) δ 148.2, 132.8, 132.0, 124.2, 123.5, 118.1, 53.7, 48.5, 45.5, 41.1, 32.7, 20.9. HRMS (ESI) m/z [M+H]+ Calcd for C14H20F3N6O+: 345.16452. Found: 345.16431.
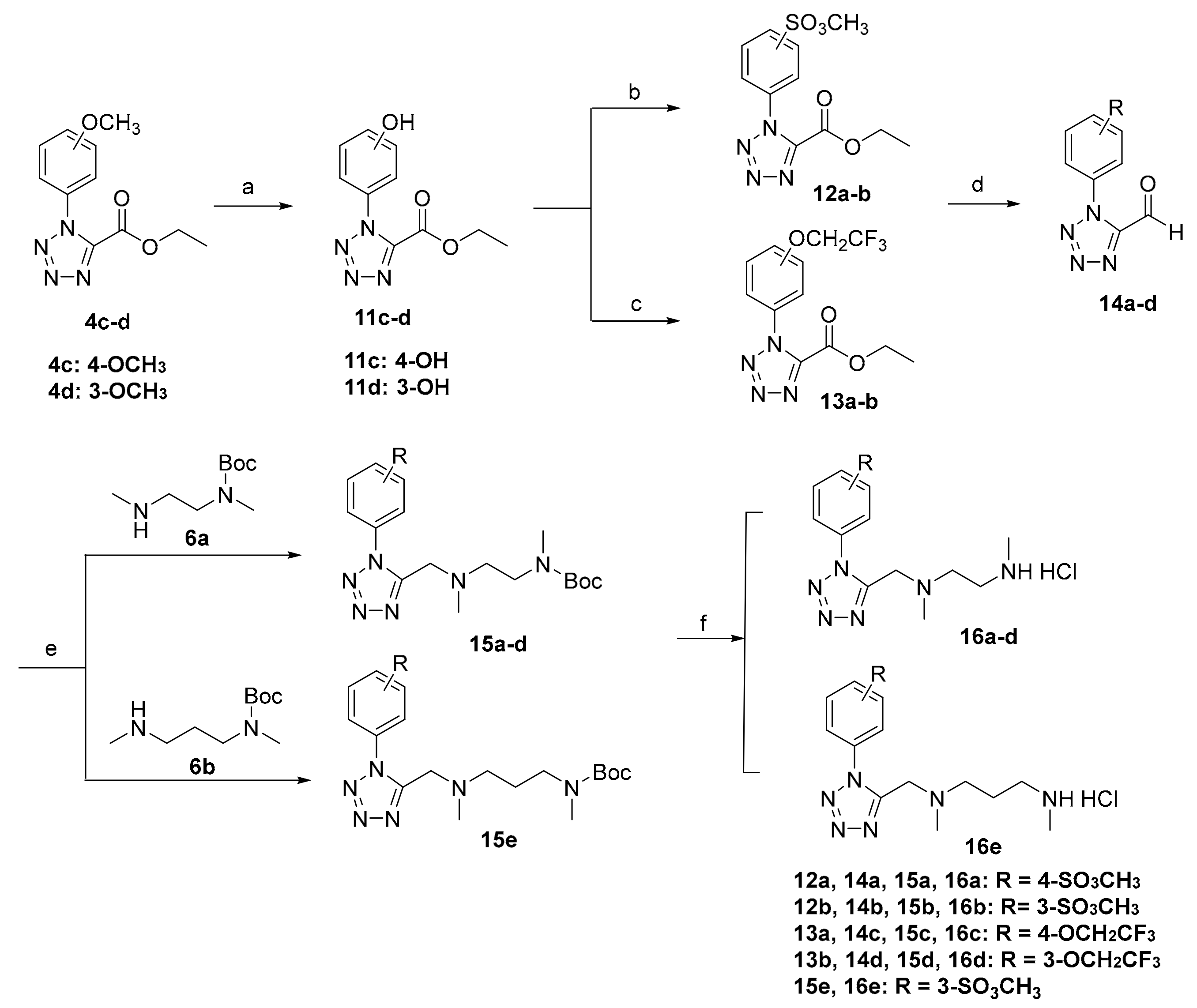
3.2.2. Synthesis of 16a–e
General procedure for the preparation of compounds 11c–d.
To a solution of compound 4c–d (2.00 g, 8.06 mmol) in dry dichloromethane was added BBr3 (16.12 mL, 1 M solution in CH2Cl2, 16.12 mmol) dropwise under argon at −78 °C and then the reaction mixture was warmed to room temperature and stirred for 12h. The mixture was washed three times with water, the organic layer was dried over Na2SO4, and the solvent removed under reduced pressure. The solid residue was purified by column chromatography on silica gel using EtOAc/petroleum as eluent to give 11c–d.
Ethyl 1-(4-hydroxylphenyl)-1H-tetrazole-5-carboxylate (11c)
White solid. Yield: 67%. MP: 136.4–137.0 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.43–7.37 (m, 2H, -ArH), 7.06–7.00 (m, 2H, -ArH), 4.47 (q, J = 7.1 Hz, 2H, -CH2-), 1.41 (t, J = 7.2 Hz, 3H, -CH3).
Ethyl 1-(3-hydroxylphenyl)-1H-tetrazole-5-carboxylate (11d)
White solid. Yield: 67%. MP: 160.4–161.3 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 1H, -OH), 7.39 (t, J = 8.0 Hz, 1H, -ArH), 7.09 (ddd, J = 7.9, 2.1, 1.0 Hz, 1H, -ArH), 7.06 (t, J = 2.1 Hz, 1H, -ArH), 7.02 (ddd, J = 8.2, 2.4, 1.0 Hz, 1H, -ArH), 4.33 (q, J = 7.1 Hz, 2H, -CH2-), 1.21 (t, J = 7.1 Hz, 3H, -CH3).
General procedure for the preparation of compounds 12a–b.
To a solution of 11c–d (0.60 g, 2.56 mmol) in ethyl acetate was added triethylamine (0.71 mL, 5.12 mmol), methanesulfonyl chloride (0.26 mL, 3.33mmol) at 0 °C and stirred for 10 min. The reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over Na2SO4, and the solvent removed under reduced pressure without further purification to give 12a–b.
Ethyl 1-(4-methylsulfonylphenyl)-1H-tetrazole-5-carboxylate (12a)
White solid, Yield: 95%. MP: 135.7–137.8 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.65–7.61 (m, 2H, -ArH), 7.57–7.51 (m, 2H, -ArH), 4.49 (q, J = 7.2 Hz, 2H, -CH2-), 3.28 (s, 3H, -SO3CH3), 1.42 (t, J = 7.1 Hz, 3H, -CH3).
Ethyl 1-(3-methylsulfonylphenyl)-1H-tetrazole-5-carboxylate (12b)
Yellow oil, Yield: 95%. 1H NMR (400 MHz, DMSO-d6) δ 7.91–7.87 (m, 1H, -ArH), 7.79–7.72 (m, 2H, -ArH), 7.66 (dt, J = 6.9, 2.3 Hz, 1H, -ArH), 4.32 (q, J = 7.1 Hz, 2H, -CH2-), 3.47 (s, 3H, -SO3CH3), 1.20 (t, J = 7.1 Hz, 3H, -CH3).
General procedure for the preparation of compounds 13a–b.
To a solution of 11c–d (0.40 g, 1.71 mmol) in dry DMF was added K2CO3 (0.28 g, 2.05 mmol), 2,2,2-trifluoroethyltrifluoromethanesulfonate (0.27 mL, 1.88 mmol) at room temperature and stirred for 3h. The mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over Na2SO4, and the solvent removed under reduced pressure without further purification to give 13a–b.
Ethyl 1-(4-(2,2,2-trifluoroethoxy)phenyl)-1H-tetrazole-5-carboxylate (13a)
White solid, Yield: 94%. MP: 119.2–120.0 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.53–7.48 (m, 2H, -ArH), 7.16–7.12 (m, 2H, -ArH), 4.47 (qd, J = 7.5, 4.4 Hz, 4H, -CH2CF3, -CH2-), 1.42 (t, J = 7.2 Hz, 3H, -CH3).
Ethyl 1-(3-(2,2,2-trifluoroethoxy)phenyl)-1H-tetrazole-5-carboxylate (13b)
White solid, Yield: 94%. MP: 77.0–77.6 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.59 (t, J = 8.2 Hz, 1H, -ArH), 7.53 (t, J = 2.2 Hz, 1H, -ArH), 7.38 (dddd, J = 9.3, 8.4, 2.3, 1.0 Hz, 2H, -ArH), 4.86 (q, J = 8.8 Hz, 2H, -CH2CF3), 4.33 (q, J = 7.1 Hz, 2H, -CH2-), 1.20 (t, J = 7.1 Hz, 3H, -CH3).
General procedure for the preparation of compounds 14a–d.
To a solution of 12a–b or 13a–b (1.0 eq) in anhydrous dichloromethane (1.8 mL/mmol) was added diisobutyl aluminium hydride (1.0 M in hexanes, 2.0 eq) dropwise at −78 °C and the reaction was stirred for 30 min. The mixture was quenched with methanol, concentrated and extracted with ethyl acetate. The combined organic layers were washed with water, 1 M HCl and dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel using EtOAc/petroleum as eluent to give 14a–d.
1-(4-Methylsulfonylphenyl)-1H-tetrazole-5-carbaldehyde (14a)
Yellow solid. Yield: 78%. MP: 146.7–147.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.10 (s, 1H, -CHO), 7.94–7.88 (m, 2H, -ArH), 7.68–7.63 (m, 2H, -ArH), 3.52 (s, 3H, -SO3CH3).
1-(3-Methylsulfonylphenyl)-1H-tetrazole-5-carbaldehyde (14b)
Yellow oil. Yield: 75%. 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H, -CHO), 7.87 (t, J = 2.1 Hz, 1H, -ArH), 7.77–7.72 (m, 2H, -ArH), 7.61 (ddd, J = 8.0, 2.3, 1.2 Hz, 1H, -ArH), 3.49 (s, 3H, -SO3CH3).
1-(4-(2,2,2-Trifluoroethoxy)phenyl)-1H-tetrazole-5-carbaldehyde (14c)
Yellow oil. Yield: 76%. 1H NMR (400 MHz, DMSO-d6) δ 10.06 (s, 1H, -CHO), 7.76–7.72 (m, 2H, -ArH), 7.33–7.30 (m, 2H, -ArH), 4.92 (q, J = 8.9, 7.1 Hz, 2H, -CH2CF3).
1-(3-(2,2,2-Trifluoroethoxy)phenyl)-1H-tetrazole-5-carbaldehyde (14d)
White solid. Yield: 75%. MP: 101.5–102.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H, -CHO), 7.63 (t, J = 8.2 Hz, 1H, -ArH), 7.49 (t, J = 2.3 Hz, 1H, -ArH), 7.42 (ddd, J = 5.5, 2.0, 0.9 Hz, 1H, -ArH), 7.41–7.39 (m, 1H, -ArH), 4.87 (q, J = 8.8, 1.3 Hz, 2H, -CH2CF3).
General procedure for the preparation of compounds 15a–e.
To a solution of 14a–d (1.0 eq) in 1,2-Dichloroethane (8.5 mL/mmol) was added sodium triacetoxyborohydride (2.0 eq) and 6a or 6b (1.0 eq) and the reaction was stirred for 12 h at room temperature. The reaction mixture was diluted with water and extracted with dichloromethane. The dichloromethane layer was dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel using EtOAc/petroleum as eluent to give 15a–e.
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(4-methylsulfonylphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-ethylenediamine (15a)
Colorless oil. Yield: 77%. 1H NMR (400 MHz, DMSO-d6) δ 7.91 (d, J = 8.6 Hz, 2H, -ArH), 7.65–7.61 (m, 2H, -ArH), 3.91 (s, 2H, -CH2-), 3.50 (s, 3H, -CH3), 3.13 (t, J = 6.5 Hz, 2H, -CH2-), 2.62 (d, J = 15.3 Hz, 3H, -CH3), 2.48 (d, J = 6.5 Hz, 2H, -CH2-), 2.19 (d, J = 6.5 Hz, 3H, -CH3), 1.33 (d, J = 11.8 Hz, 9H, -C(CH3)3).
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(3-methylsulfonylphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-ethylenediamine (15b)
Colorless oil. Yield: 75%. 1H NMR (400 MHz, DMSO-d6) δ 7.84 (d, J = 23.0 Hz, 2H, -ArH), 7.77 (t, J = 8.1 Hz, 1H, -ArH), 7.64 (ddd, J = 8.1, 2.4, 1.2 Hz, 1H, -ArH), 3.94 (s, 2H, -CH2-), 3.49 (s, 3H, -CH3), 3.12 (t, J = 6.5 Hz, 2H, -CH2-), 2.62 (d, J = 11.0 Hz, 3H, -CH3), 2.48 (t, J = 6.5 Hz, 2H, -CH2-), 2.20 (d, J = 4.4 Hz, 3H, -CH3), 1.32 (d, J = 8.5 Hz, 9H, -C(CH3)3).
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(4-(2,2,2-trifluoroethoxy)phenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-ethylenediamine (15c)
Colorless oil. Yield: 82%. 1H NMR (400 MHz, DMSO-d6) δ 7.92–7.86 (m, 1H, -ArH), 7.77–7.66 (m, 2H, -ArH), 7.37–7.33 (m, 1H, -ArH), 4.91 (q, J = 8.8 Hz, 2H, -CH2-), 3.85 (s, 2H, -CH2-), 3.14 (t, J = 6.5 Hz, 2H, -CH2-), 2.64 (d, J = 13.8 Hz, 3H, -CH3), 2.48 (t, J = 6.4 Hz, 2H, -CH2-), 2.18 (s, 3H, -CH3), 1.33 (d, J = 12.7 Hz, 9H, -C(CH3)3).
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(3-(2,2,2-trifluoroethoxy)phenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-ethylenediamine (15d)
Colorless oil. Yield: 80%. 1H NMR (400 MHz, DMSO-d6) δ 7.61 (t, J = 8.2 Hz, 1H, -ArH), 7.55–7.40 (m, 2H, -ArH), 7.34 (dd, J = 8.5, 2.5 Hz, 1H, -ArH), 4.89 (q, J = 8.8 Hz, 2H, -CH2-), 3.93 (s, 2H, -CH2-), 3.11 (t, J = 6.5 Hz, 2H, -CH2-), 2.61 (d, J = 12.7 Hz, 3H, -CH3), 2.47 (t, J = 6.5 Hz, 2H, -CH2-), 2.19 (s, 3H, -CH3), 1.32 (d, J = 9.0 Hz, 9H, -C(CH3)3).
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(3-methylsulfonylphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-1,3-propylenediamine (15e)
Colorless oil. Yield: 75%.1H NMR (400 MHz, DMSO-d6) δ 7.90 (t, J = 2.1 Hz, 1H, -ArH), 7.82–7.74 (m, 2H, -ArH), 7.66–7.62 (m, 1H, -ArH), 3.89 (s, 2H, -CH2-), 3.48 (s, 3H, -CH3), 2.93 (s, 2H, -CH2-), 2.67 (d, J = 4.1 Hz, 3H, -CH3), 2.29 (t, J = 7.1 Hz, 2H, -CH2-), 2.10 (s, 3H, -CH3), 1.46 (s, 2H, -CH2-), 1.35 (s, 9H, -C(CH3)3).
General procedure for the preparation of compounds 16a–e.
Compounds 15a–e (0.51 mmol) was added to saturated hydrochloric acid ethanol solution (10 mL) at room temperature and stirred for 30 min. The reaction solution was filtered and washed with diethylether (20 mL) to give 16a–e.
N,N’-Dimethyl-N’-(1-(4-methylsulfonylphenyl)-1H-tetrazol-5-yl-methyl)ethylenediamine hydrochloride (16a)
White solid. Yield: 95%. MP: 198.9–199.3 °C. 1H NMR (400 MHz, Deuterium Oxide) δ 7.65–7.60 (m, 2H, -ArH), 7.56–7.52 (m, 2H, -ArH), 4.24 (s, 2H, -CH2-), 3.32 (s, 3H, -SO3CH3), 3.14 (t, J = 6.4 Hz, 2H, -CH2-), 3.00 (t, J = 6.3 Hz, 2H, -CH2-), 2.58 (s, 3H, -CH3), 2.41 (s, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 150.2, 150.1, 131.3, 127.1, 124.2, 51.9, 49.4, 44.1, 41.4, 37.1, 33.0. HRMS (ESI) m/z [M+H]+ Calcd for C13H21N6O3S+: 341.13904. Found: 341.13906.
N,N’-Dimethyl-N’-(1-(3-methylsulfonylphenyl)-1H-tetrazol-5-yl-methyl)ethylenediamine hydrochloride (16b)
Colorless oil. Yield: 94%. 1H NMR (400 MHz, Deuterium Oxide) δ 7.68 (td, J = 8.1, 0.7 Hz, 1H, -ArH), 7.59 (qd, J = 2.5, 0.9 Hz, 2H, -ArH), 7.57–7.53 (m, 1H, -ArH), 4.32 (s, 2H, -CH2-), 3.30 (s, 3H, -SO3CH3), 3.20–3.15 (m, 2H, -CH2-), 3.06 (t, J = 6.3 Hz, 2H, -CH2-), 2.59 (s, 3H, -CH3), 2.47 (s, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 150.6, 148.9, 133.3, 132.0, 125.2, 124.3, 119.5, 51.9, 49.3, 44.4, 41.3, 37.1, 32.9. HRMS (ESI) m/z [M+H]+ Calcd for C13H21N6O3S+: 341.13904. Found: 341.13892.
N,N’-Dimethyl-N’-(1-(4-(2,2,2-trifluoroethoxy)phenyl)-1H-tetrazol-5-yl-methyl)ethylenediamine hydrochloride (16c)
White solid. Yield: 95%. MP: 188.7–190.0 °C. 1H NMR (400 MHz, Deuterium Oxide) δ 7.49–7.44 (m, 2H, -ArH), 7.21–7.15 (m, 2H, -ArH), 4.59 (q, J = 8.4 Hz, 2H, -CH2CF3), 4.25 (s, 2H, -CH2-), 3.16 (t, J = 6.4 Hz, 2H, -CH2-), 3.02 (t, J = 6.4 Hz, 2H, -CH2-), 2.58 (s, 3H, -CH3), 2.43 (s, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 158.9, 150.1, 126.8, 126.3, 116.3, 65.6, 65.2, 51.8, 49.1, 44.2, 41.3, 33.0. HRMS (ESI) m/z [M+H]+ Calcd for C14H20F3N6O+: 345.16452. Found: 345.16437.
N,N’-Dimethyl-N’-(1-(3-(2,2,2-trifluoroethoxy)phenyl)-1H-tetrazol-5-ylmethyl)ethylenediamine hydrochloride (16d)
Colorless oil. Yield: 92%. 1H NMR (400 MHz, Deuterium Oxide) δ 7.54 (ddd, J = 8.4, 7.6, 0.7 Hz, 1H, -ArH), 7.25 (ddd, J = 8.6, 2.4, 1.0 Hz, 1H, -ArH), 7.20–7.15 (m, 2H, -ArH), 4.57 (q, J = 8.4 Hz, 2H, -CH2CF3), 4.40 (s, 2H, -CH2-), 3.23 (td, J = 6.2, 1.4 Hz, 2H, -CH2-), 3.16 (dd, J = 7.2, 5.0 Hz, 2H, -CH2-), 2.61 (s, 3H, -CH3), 2.54 (s, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 157.9, 131.5, 118.7, 117.6, 112.1, 65.7, 65.3, 51.9, 49.3, 41.2, 32.9. HRMS (ESI) m/z [M+H]+ Calcd for C14H20F3N6O+: 345.16452. Found: 345.16437.
N,N’-Dimethyl-N’-(1-(3-methylsulfonylphenyl)-1H-tetrazol-5-yl-methyl)-1,3-propylenediamine hydrochloride (16e)
Colorless oil. Yield: 92%. 1H NMR (400 MHz, Deuterium Oxide) δ 7.74–7.67 (m, 1H, -ArH), 7.60 (dt, J = 7.7, 1.4 Hz, 2H, -ArH), 7.57–7.52 (m, 1H, -ArH), 4.80 (s, 2H, -CH2-), 3.31 (s, 5H, -CH2-, -SO3CH3), 3.02–2.96 (m, 2H, -CH2-), 2.91 (s, 3H, -CH3), 2.60 (s, 3H, -CH3), 2.13–2.04 (m, 2H, -CH2-). 13C NMR (101 MHz, Deuterium Oxide) δ 149.0, 148.0, 132.8, 132.2, 125.5, 124.3, 119.5, 53.7, 48.4, 45.4, 41.2, 37.1, 32.7, 20.8. HRMS (ESI) m/z [M+H]+ Calcd for C14H23N6O3S+: 355.15469. Found: 355.15460.
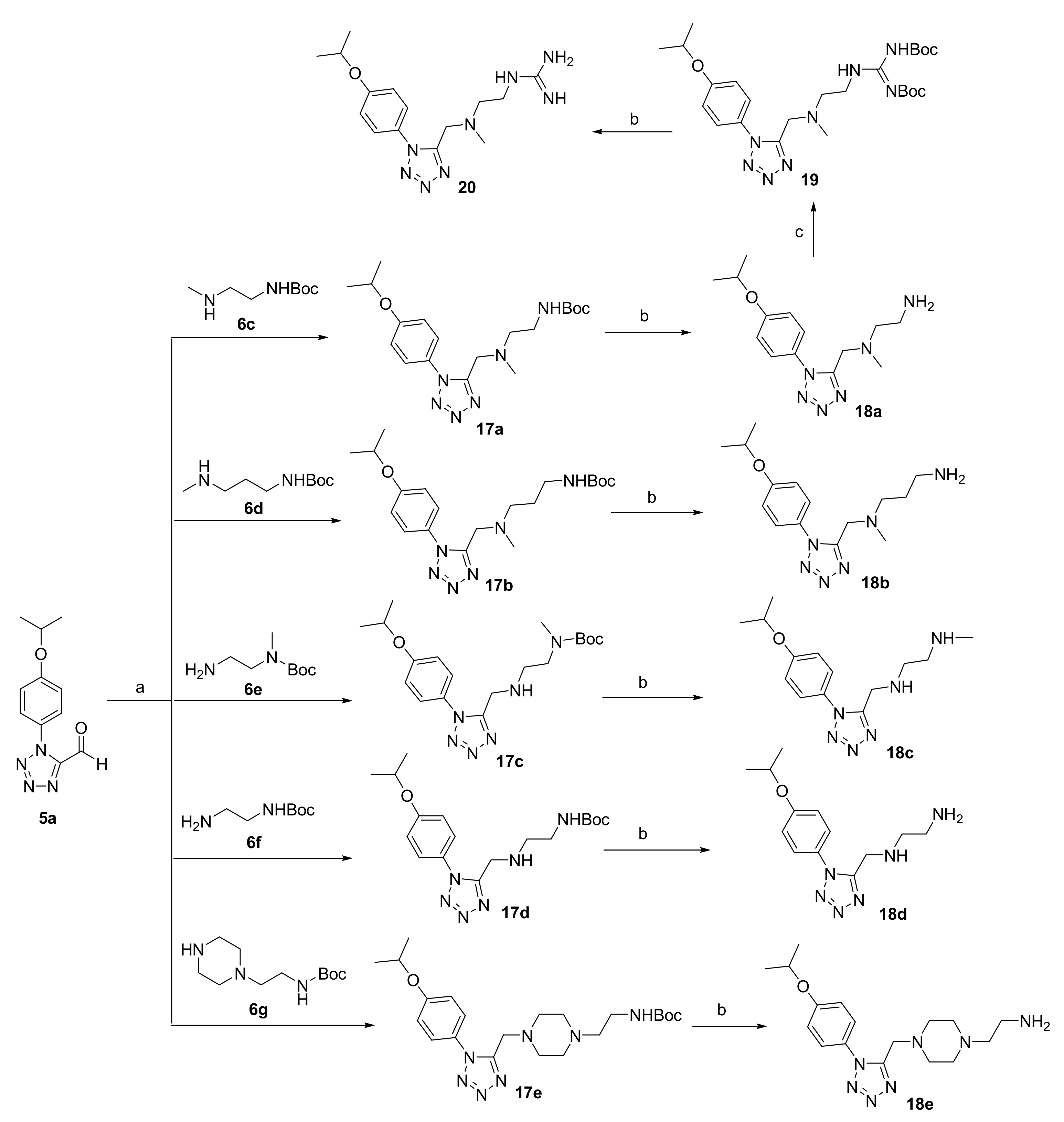
3.2.3. Synthesis of 18a–e and 20
General procedure for the preparation of compounds 17a–e.
To a solution of 5a (1.0 eq) in 1,2-Dichloroethane (8.5 mL/mmol) was added sodium triacetoxyborohydride (2.0 eq) and 6c–g (1.0 eq) and the reaction was stirred for 12 h at room temperature. The reaction mixture was diluted with water and extracted with dichloromethane. The dichloromethane layer was dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel using EtOAc/petroleum as eluent to give 17a–e.
N-tert-Butyloxycarbonyl-N’-(1-(4-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-ethylenediamine (17a)
Colorless oil. Yield: 85%. 1H NMR (400 MHz, Chloroform-d) δ 7.57–7.43 (m, 2H, -ArH), 7.12–7.03 (m, 2H, -ArH), 4.70–4.61 (m, 1H, -OCH(CH3)2), 3.31 (s, 2H, -CH2-), 2.52 (s, 2H, -CH2-), 1.61 (s, 2H, -CH2-), 1.45 (s, 9H, -C(CH3)3), 1.42 (d, J = 0.6 Hz, 3H, -CH3), 1.42–1.39 (m, 3H, -CH3), 1.27 (s, 3H, -CH3).
N-tert-Butyloxycarbonyl-N’-(1-(4-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-1,3-propylenediamine (17b)
Colorless oil. Yield: 80%. 1H NMR (400 MHz, DMSO-d6) δ 7.65–7.59 (m, 2H, -ArH), 7.15–7.09 (m, 2H, -ArH), 4.77–4.69 (m, 1H, -OCH(CH3)2), 3.78 (s, 2H, -CH2-), 2.77 (q, J = 6.6 Hz, 2H, -CH2-), 2.32 (t, J = 7.1 Hz, 2H, -CH2-), 2.09 (s, 3H, -CH3), 1.40 (t, J = 6.9 Hz, 2H, -CH2-), 1.36 (s, 9H, -C(CH3)3), 1.32 (s, 3H, -CH3), 1.31 (d, J = 2.3 Hz, 3H, -CH3).
N-tert-Butyloxycarbonyl-N-methyl-N’-(1-(4-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-ethylenediamine (17c)
Colorless oil. Yield: 85%. 1H NMR (400 MHz, Chloroform-d) δ 7.55–7.49 (m, 2H, -ArH), 7.07–7.01 (m, 2H, -ArH), 4.64 (p, J = 6.1 Hz, 1H, -OCH(CH3)2), 4.09 (s, 2H, -NH2), 3.39 (d, J = 16.0 Hz, 2H, -CH2-), 2.91 (d, J = 17.3 Hz, 2H, -CH2-), 2.86 (s, 3H, -CH3), 2.14 (s, 2H, -CH2-), 1.44 (s, 9H, -C(CH3)3), 1.41 (s, 3H, -CH3), 1.40 (s, 3H, -CH3).
N-tert-Butyloxycarbonyl-N’-(1-(4-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-ethylenediamine (17d)
Colorless oil. Yield: 85%. 1H NMR (400 MHz, Chloroform-d) δ 7.48 (d, J = 8.5 Hz, 2H, -ArH), 7.11–7.01 (m, 2H, -ArH), 4.64 (h, J = 6.0 Hz, 1H, -OCH(CH3)2), 4.19 (s, 2H, -CH2-), 3.33 (d, J = 5.9 Hz, 2H, -CH2-), 2.98 (s, 2H, -CH2-), 1.45 (s, 9H, -C(CH3)3), 1.42 (d, J = 1.3 Hz, 3H, -CH3), 1.40 (d, J = 1.4 Hz, 3H, -CH3).
N-tert-Butyloxycarbonylaminoethyl-N’-(1-(4-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-piperazine (17e)
Colorless oil. Yield: 75%. 1H NMR (400 MHz, DMSO-d6) δ 7.65–7.60 (m, 2H, -ArH), 7.17–7.10 (m, 2H, -ArH), 6.63 (t, J = 5.7 Hz, 1H, -NH-), 4.74 (hept, J = 6.0 Hz, 1H, -OCH(CH3)2), 3.77 (s, 2H, -CH2-), 2.98 (q, J = 6.4 Hz, 2H, -CH2-), 2.43–2.15 (m, 10H, -CH2-), 1.37 (s, 9H, -C(CH3)3), 1.32 (s, 3H, -CH3), 1.31 (s, 3H, -CH3).
General procedure for the preparation of compounds 18a–e.
Compounds 17a–e (0.51 mmol) was added to saturated hydrochloric acid ethanol solution (10 mL) at room temperature and stirred for 30 min. The reaction solution was filtered and washed with ethanol (20 mL) to give 18a–e.
N-(1-(4-Isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-N-methyl-ethylenediamine hydrochloride (18a)
White solid. Yield: 95%. MP: 186.5–187.7 °C. 1H NMR (400 MHz, Deuterium Oxide) δ 7.46–7.38 (m, 2H, -ArH), 7.13–7.07 (m, 2H, -ArH), 4.69–4.65 (m, 1H, -OCH(CH3)2), 4.41 (s, 2H, -CH2-), 3.14 (td, J = 6.3, 1.3 Hz, 2H, -CH2-), 3.06 (td, J = 6.3, 1.3 Hz, 2H, -CH2-), 2.49 (s, 3H, -CH3), 1.26 (s, 3H, -CH3), 1.25 (s,3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 150.5, 126.7, 125.0, 117.1, 71.9, 52.9, 49.0, 41.2, 35.2, 20.9. HRMS (ESI) m/z [M+H]+ Calcd for C14H23N6O+: 291.19279. Found: 291.19278.
N-(1-(4-Isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-N-methyl-1,3-propylenediamine hydrochloride (18b)
Colorless oil. Yield: 92%. 1H NMR (400 MHz, Deuterium Oxide) δ 7.42–7.36 (m, 2H, -ArH), 7.12–7.07 (m, 2H, -ArH), 4.69–4.63 (m, 1H, -OCH(CH3)2), 4.72 (s, 2H, -CH2-), 3.32–3.24 (m, 2H, -CH2-), 2.99–2.90 (m, 2H, -CH2-), 2.86 (s, 3H, -CH3), 2.04 (tt, J = 9.8, 6.6 Hz, 2H, -CH2-), 1.25 (s, 3H, -CH3), 1.23 (s, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 159.4, 147.9, 126.7, 124.5, 117.2, 71.9, 53.7, 48.1, 41.1, 36.2, 22.0, 20.9. HRMS (ESI) m/z [M+H]+ Calcd for C15H25N6O+: 305.20844. Found: 305.20840.
N-Methyl-N’-(1-(4-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-ethylenediamine hydrochloride (18c)
White solid. Yield: 92%. MP: 179.7–180.7 °C. 1H NMR (400 MHz, Deuterium Oxide) δ 7.43–7.36 (m, 2H, -ArH), 7.12–7.06 (m, 2H, -ArH), 4.67 (d, J = 6.1 Hz, 1H, -OCH(CH3)2), 4.49 (s, 2H, -CH2-), 3.37 (ddd, J = 7.3, 6.1, 1.5 Hz, 2H, -CH2-), 3.29 (ddd, J = 7.7, 6.1, 1.6 Hz, 2H, -CH2-), 2.65 (s, 3H, -CH3), 1.25 (s, 3H, -CH3), 1.24 (s, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 159.2, 149.3, 126.4, 124.7, 117.2, 71.9, 44.5, 43.4, 40.7, 33.1, 20.9. HRMS (ESI) m/z [M+H]+ Calcd for C14H23N6O+: 291.19279. Found: 291.19269.
N-(1-(4-Isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-ethylenediaminehydrochloride (18d)
White solid. Yield: 93%. MP: 183.1–183.9 °C. 1H NMR (400 MHz, Deuterium Oxide) δ 7.43–7.38 (m, 2H, -ArH), 7.13–7.08 (m, 2H, -ArH), 4.69–4.65 (m, 1H, -OCH(CH3)2), 4.52 (s, 2H, -CH2-), 3.37 (dd, J = 7.2, 5.9 Hz, 2H, -CH2-), 3.26 (ddd, J = 7.4, 6.3, 1.1 Hz, 2H, -CH2-), 1.26 (s, 3H, -CH3), 1.25 (s, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 159.2, 149.3, 126.4, 124.7, 117.2, 71.9, 44.6, 40.7, 35.5, 20.9. HRMS (ESI) m/z [M+H]+ Calcd for C13H21N6O+: 277.17714. Found: 291.17709.
N-aminoethyl-N’-(1-(4-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-piperazinehydrochloride (18e)
Yellow solid. Yield: 92%. MP: 183.2–183.9 °C. 1H NMR (400 MHz, Deuterium Oxide) δ 7.46–7.40 (m, 2H, -ArH), 7.11–7.05 (m, 2H, -ArH), 4.00 (s, 2H, -CH2-), 3.35–3.26 (m, 4H, -CH2-), 3.19 (s, 4H, -CH2-), 2.82 (s, 4H, -CH2-), 1.25 (s, 3H, -CH3), 1.23 (s, 3H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 158.9, 151.7, 126.7, 116.9, 71.9, 52.5, 51.8, 49.3, 48.3, 33.7, 20.9. HRMS (ESI) m/z [M+H]+ Calcd for C17H28N7O+: 346.23499. Found: 346.23494.
N-2,3-bis-(tert-Butoxycarbonyl)guanidino-N’-(1-(4-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-ethylenediamine (19)
To a solution of 18a (0.26g, 0.80mmol) in dry DMF was added to stirred for 10 min and then 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (0.23g, 0.80mmol) was added and stirred for 5 h. The reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over Na2SO4, and the solvent removed under reduced pressure. The solid residue thus obtained was purified by column chromatography on silica gel using EtOAc/petroleum (1/2, V/V) as eluent to give 19 as yellow oil (0.22g, 82%). 1H NMR (400 MHz, Chloroform-d) δ 11.45 (s, 1H, -NH-), 8.48 (s, 1H, -NH-), 7.65–7.59 (m, 2H, -ArH), 7.06–6.99 (m, 2H, -ArH), 4.63 (p, J = 6.1 Hz, 1H, -OCH(CH3)2), 3.79 (s, 2H, -CH2-), 3.52 (q, J = 5.5 Hz, 2H, -CH2-), 2.78 (t, J = 6.0 Hz, 2H, -CH2-), 2.39 (s, 3H, -CH3), 1.53 (s, 3H, -CH3), 1.52 (s, 9H, -C(CH3)3), 1.41 (s, 9H, -C(CH3)3), 1.39 (s, 3H, -CH3).
N-Guanidino-N’-(1-(4-isopropoxyphenyl)-1H-tetrazol-5-yl-methyl)-N’-methyl-ethylenediamine hydrochloride (20)
Compounds 19 (0.55 mmol) was added to saturated hydrochloric acid ethanol solution (10 mL) at room temperature and stirred for 30min. The reaction solution was filtered and washed with ethanol (20 mL) to give 20. Yellow oil. Yield: 92%. 1H NMR (400 MHz, Deuterium Oxide) δ 7.40 (dd, J = 8.7, 5.3 Hz, 2H, -ArH), 7.10 (dd, J = 8.7, 4.7 Hz, 2H, -ArH), 4.76 (s, 1H, -NH-), 4.66 (d, J = 5.0 Hz, 2H, -CH2-), 3.59 (dt, J = 25.7, 6.0 Hz, 2H, -CH2-), 3.42 (dd, J = 14.0, 7.2 Hz, 2H, -CH2-), 2.87 (d, J = 34.7 Hz, 3H, -NH-, -NH2), 1.24 (d, J = 6.0 Hz, 6H, -CH3). 13C NMR (101 MHz, Deuterium Oxide) δ 159.4, 148.1, 126.7, 124.5, 117.2, 71.9, 54.5, 48.4, 41.6, 36.3, 29.5, 20.9. HRMS (ESI) m/z [M+H]+ Calcd for C15H25N8O+: 333.21458. Found: 333.21451.

 and
and 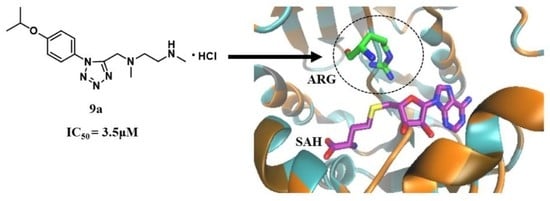











{kind=link}
{kind=link}
{kind=link}
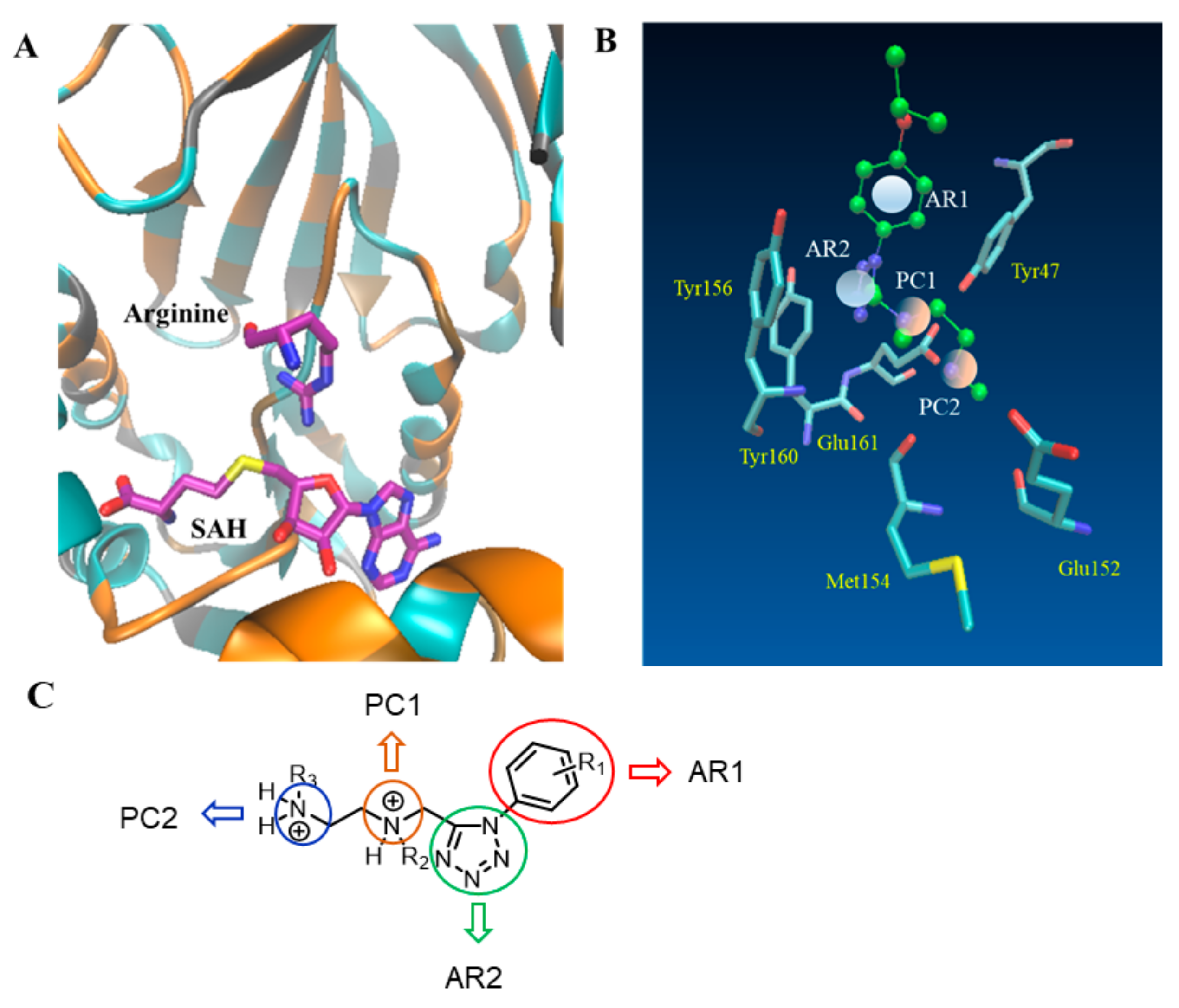{kind=link}
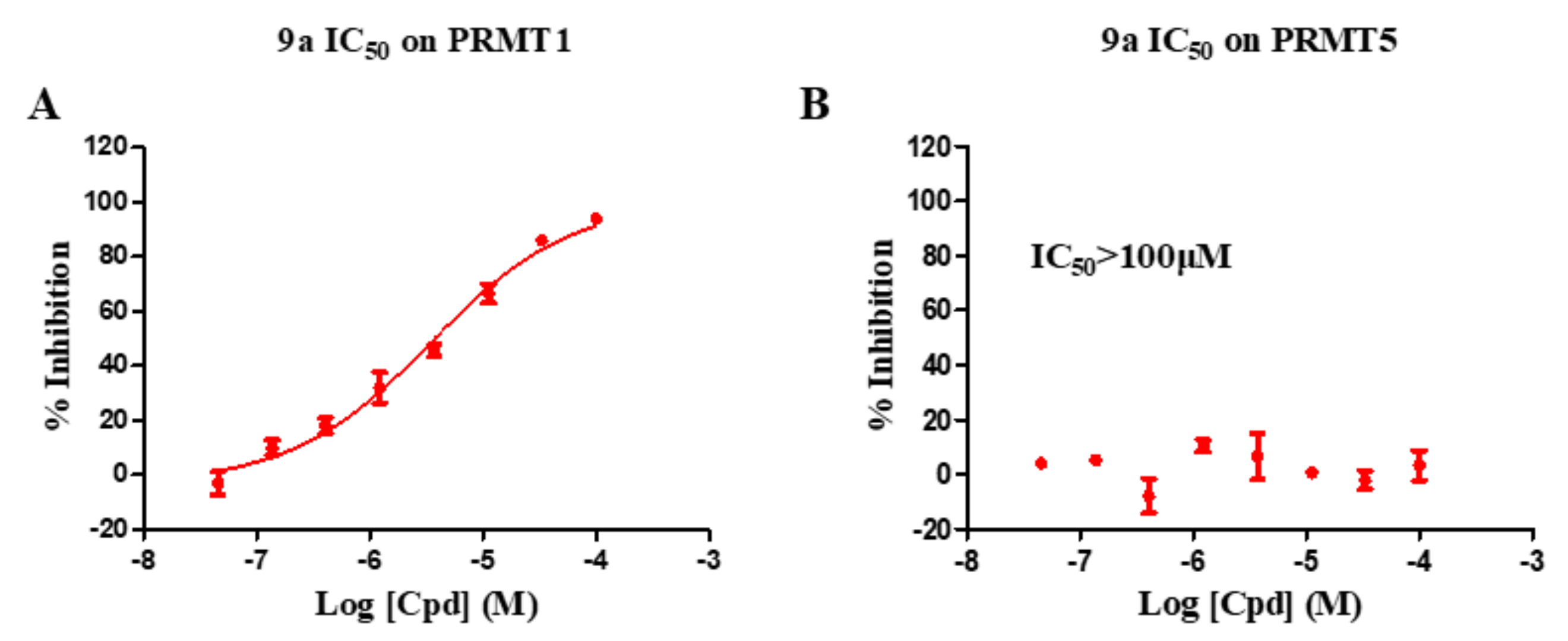{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






















