Sorafenib-Induced Apoptosis in Hepatocellular Carcinoma Is Reversed by SIRT1

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
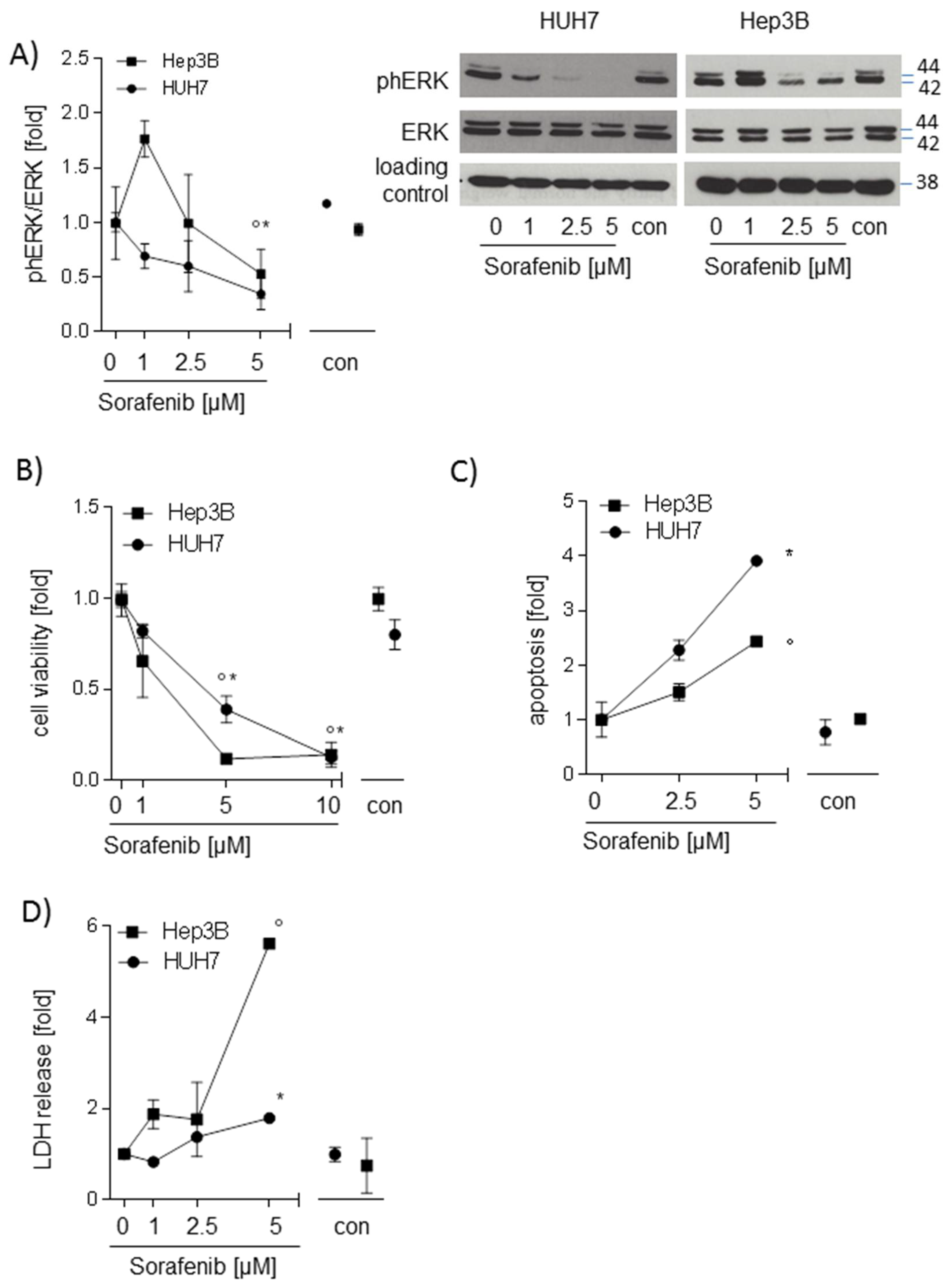
2.1. Sorafenib Decreases Phosphorylation of ERK and Induces Apoptosis in Hepatocarcinoma Cell Lines
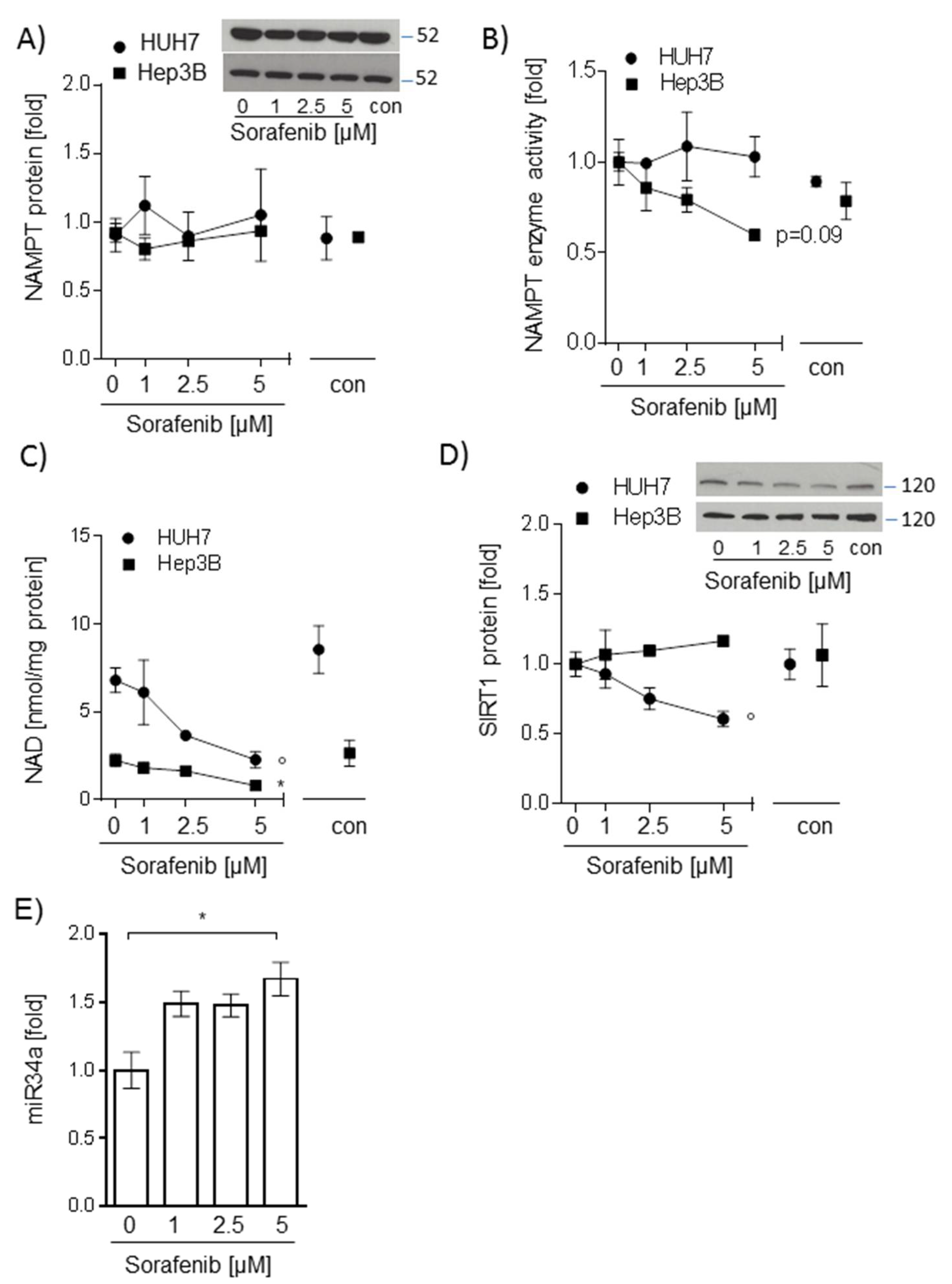
2.2. Sorafenib-Induced Decrease of NAD Levels in Hepatocarcinoma Cells Is Not Mediated by NAMPT or SIRT1
2.3. Sorafenib Leads to Energy Deprivation in HUH7 Cells
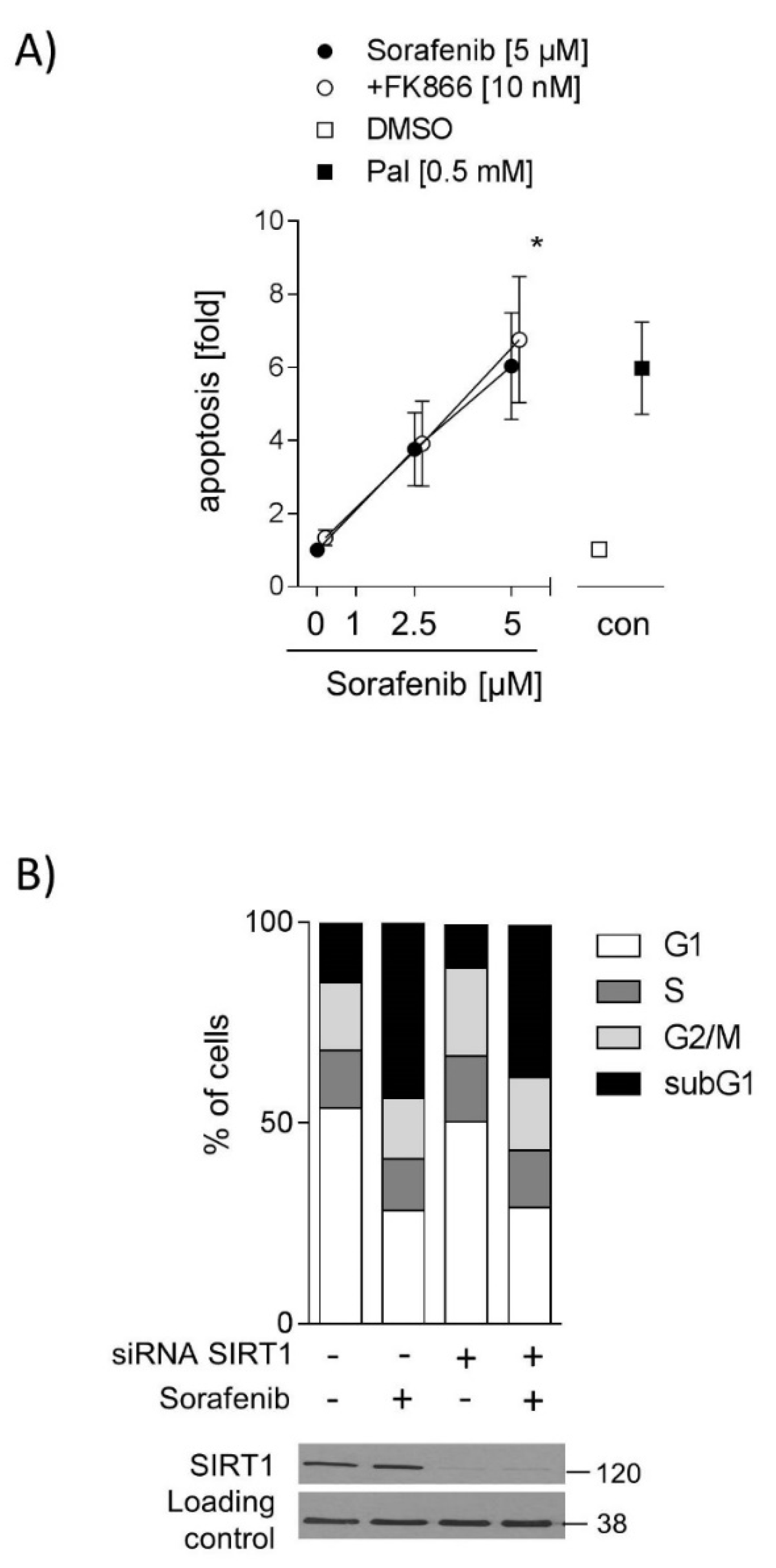
2.4. Inhibition of SIRT1 Does Not Sensitize Hepatocarcinoma Cells to Sorafenib
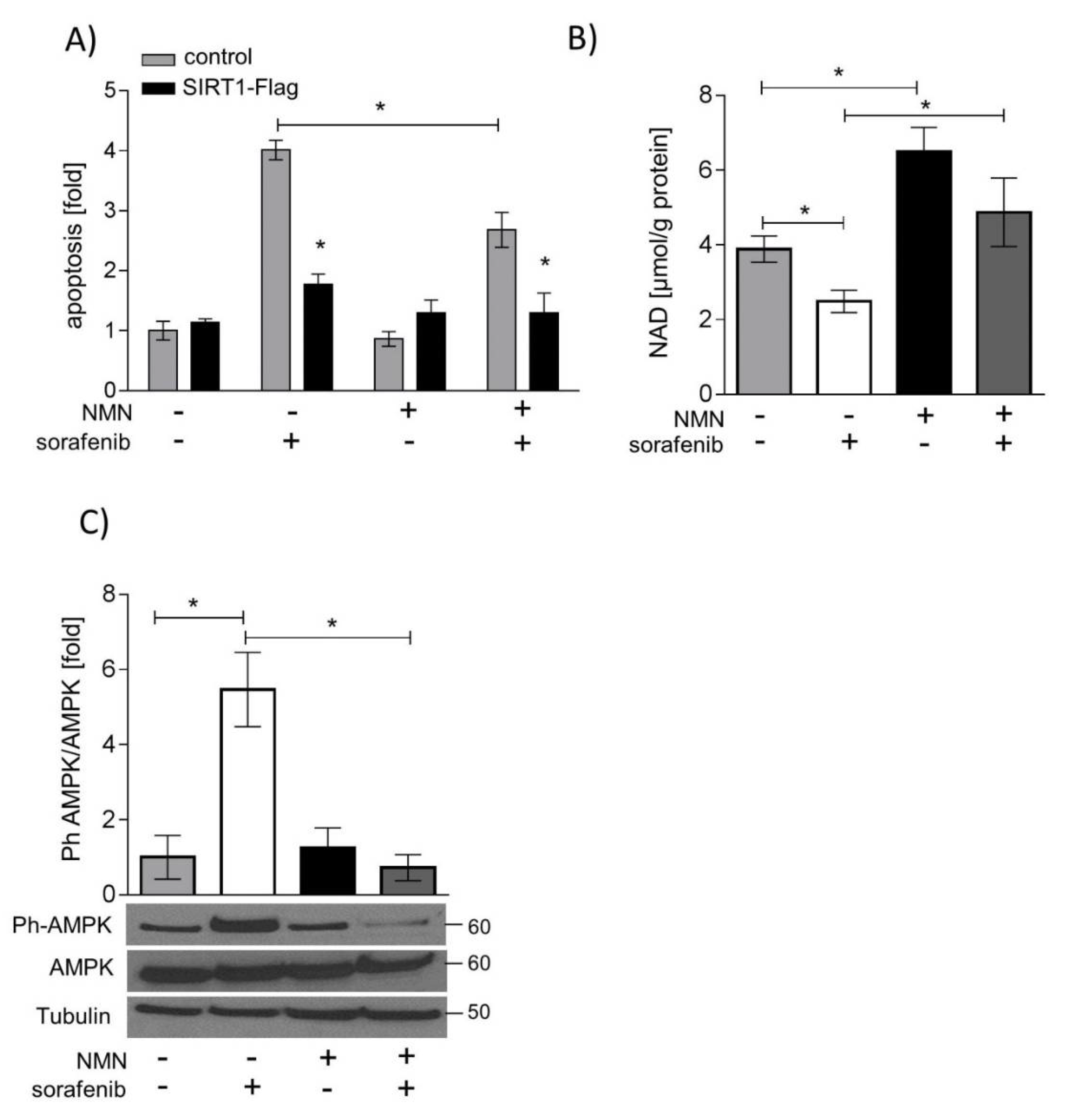
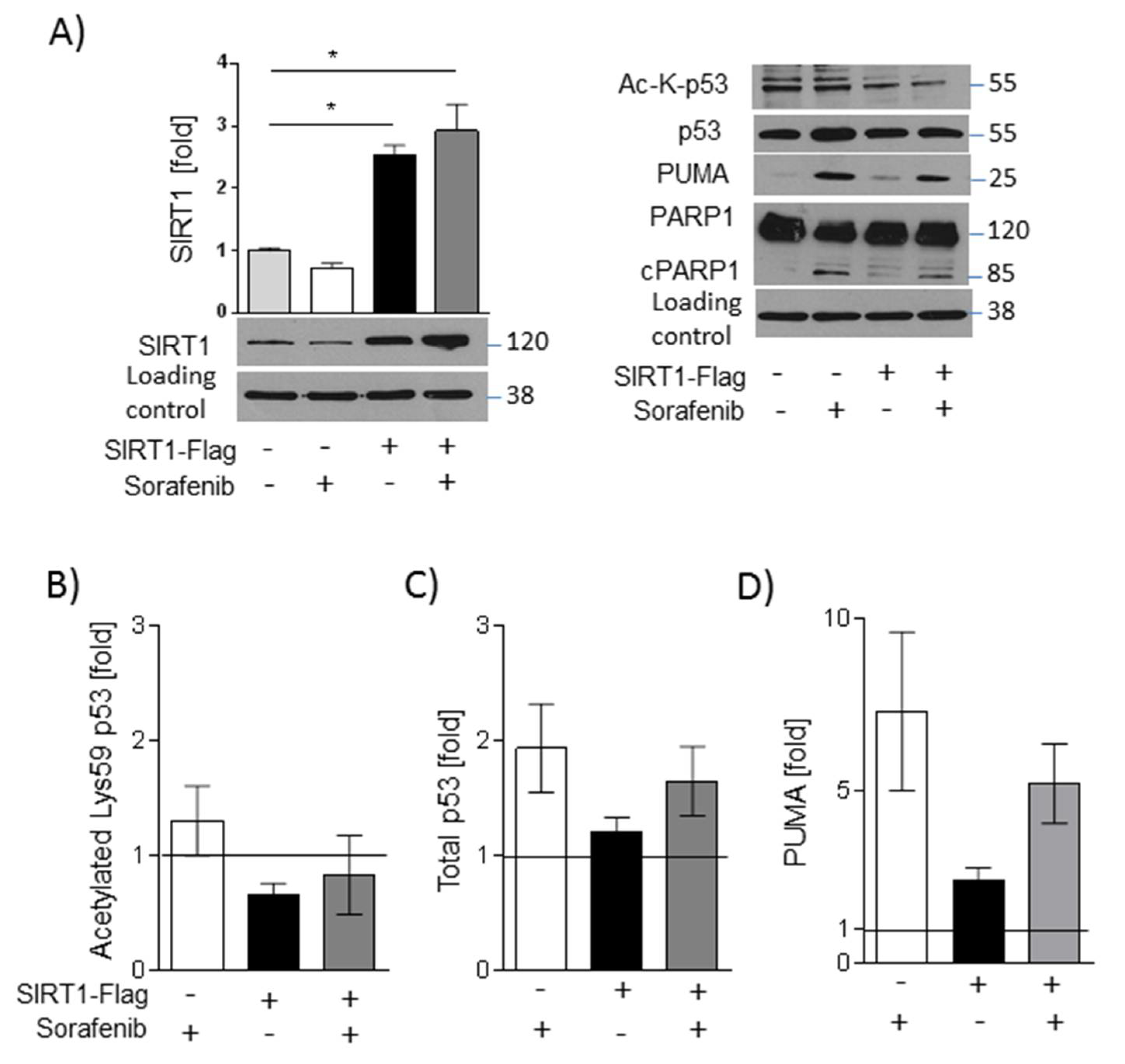
2.5. SIRT1 Overexpression and NMN Supplementation Suppress Apoptosis in HUH7 Cells
3. Discussion
4. Material and Methods
4.1. Cell Culture and Treatment
4.2. Transfection
4.3. Cell Viability and Apoptosis
4.4. Lactate Dehydrogenase (LDH) and Adenosine Triphosphate (ATP) Measurement
4.5. Cell Cycle Analysis
4.6. Protein Extraction and Western Blot Analyses
4.7. NAMPT Enzyme Activity
4.8. Intracellular NAD Levels
4.9. RNA Extraction and Real-Time qPCR
4.10. High-Resolution Respirometry
4.11. Isolation of Mitochondria
4.12. Citrate Synthase Assay
4.13. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Paech, F.; Mingard, C.; Grünig, D.; Abegg, V.F.; Bouitbir, J.; Krähenbühl, S. Mechanisms of mitochondrial toxicity of the kinase inhibitors ponatinib, regorafenib and sorafenib in human hepatic HepG2 cells. Toxicology 2018, 395, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-S.; Lee, Y.-M.; Oh, T.-I.; Shin, D.H.; Kim, G.-H.; Kan, S.-Y.; Kang, H.; Kim, J.H.; Kim, B.M.; Yim, W.J.; et al. Emodin Sensitizes Hepatocellular Carcinoma Cells to the Anti-Cancer Effect of Sorafenib through Suppression of Cholesterol Metabolism. Int. J. Mol. Sci. 2018, 19, 3127. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.-J.; Yu, S.J.; Na, J.; Kim, K.; Cho, Y.Y.; Bin Lee, Y.; Cho, E.J.; Lee, J.-H.; Kim, Y.J.; Youn, H.; et al. Hexokinase-II Inhibition Synergistically Augments the Anti-tumor Efficacy of Sorafenib in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 1292. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-C.; Jeng, Y.-M.; Yuan, R.-H.; Hsu, H.-C.; Chen, Y.-L. SIRT1 Promotes Tumorigenesis and Resistance to Chemotherapy in Hepatocellular Carcinoma and its Expression Predicts Poor Prognosis. Ann. Surg. Oncol. 2012, 19, 2011–2019. [Google Scholar] [CrossRef]
- Ling, S.; Li, J.; Shan, Q.; Dai, H.; Lu, D.; Wen, X.; Song, P.; Xie, H.; Zhou, L.; Liu, J.; et al. USP22 mediates the multidrug resistance of hepatocellular carcinoma via the SIRT1/AKT/MRP1 signaling pathway. Mol. Oncol. 2017, 11, 682–695. [Google Scholar] [CrossRef] [Green Version]
- Garten, A.; Schuster, S.; Penke, M.; Gorski, T.; de Giorgis, T.; Kiess, W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 2015, 11, 1–12. [Google Scholar] [CrossRef]
- Revollo, J.R.; Grimm, A.A.; Imai, S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 2004, 279, 50754–50763. [Google Scholar] [CrossRef]
- Schuster, S.; Penke, M.; Gorski, T.; Gebhardt, R.; Weiss, T.S.; Kiess, W.; Garten, A. FK866-induced NAMPT inhibition activates AMPK and downregulates mTOR signaling in hepatocarcinoma cells. Biochem. Biophys. Res. Commun. 2015, 458, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Olmos, Y.; Brosens, J.J.; Lam, E.W.-F. Interplay between SIRT proteins and tumour suppressor transcription factors in chemotherapeutic resistance of cancer. Drug Resist. Updat. 2011, 14, 35–44. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. MIR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [PubMed]
- Doerrier, C.; Garcia-Souza, L.F.; Krumschnabel, G.; Wohlfarter, Y.; Mészáros, A.T.; Gnaiger, E. High-Resolution FluoRespirometry and OXPHOS Protocols for Human Cells, Permeabilized Fibers from Small Biopsies of Muscle, and Isolated Mitochondria. In Mitochondrial Bioenergetics; Humana Press: New York, NY, USA, 2018; pp. 31–70. Available online: http://www.ncbi.nlm.nih.gov/pubmed/29850993 (accessed on 11 June 2018).
- Pesta, D.; Gnaiger, E. High-Resolution Respirometry: OXPHOS Protocols for Human Cells and Permeabilized Fibers from Small Biopsies of Human Muscle. In Mitochondrial Bioenergetics; Humana Press: New York, NY, USA, 2012; pp. 25–58. Available online: http://link.springer.com/10.1007/978-1-61779-382-0_3 (accessed on 19 November 2019).
- Hasmann, M.; Schemainda, I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar] [PubMed]
- Schuster, S.; Penke, M.; Gorski, T.; Petzold-Quinque, S.; Damm, G.; Gebhardt, R.; Kiess, W.; Garten, A. Resveratrol differentially regulates NAMPT and SIRT1 in hepatocarcinoma cells and primary human hepatocytes. PLoS ONE 2014, 9, e91045. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, W. Emerging Roles of SIRT1 in Cancer Drug Resistance. Genes Cancer 2013, 4, 82–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Zhang, X.; Tao, Y.; Shan, L.; Jiang, Q.; Yu, Y.; Cai, F.; Ma, L. Prognostic and clinicopathologic significance of SIRT1 expression in hepatocellular carcinoma. Oncotarget 2017, 8, 52357–52365. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raou, J.-L.R.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Davis, E.M.; Crabtree, T.R.; Habibi, J.R.; Nguyen, T.K.; Dent, P.; Grant, S. The Kinase Inhibitor Sorafenib Induces Cell Death through a Process Involving Induction of Endoplasmic Reticulum Stress. Mol. Cell Biol. 2007, 27, 5499–5513. [Google Scholar] [CrossRef] [Green Version]
- Lange, M.; Abhari, B.A.; Hinrichs, T.M.; Fulda, S.; Liese, J. Identification of a novel oxidative stress induced cell death by Sorafenib and oleanolic acid in human hepatocellular carcinoma cells. Biochem. Pharmacol. 2016, 118, 9–17. [Google Scholar] [CrossRef]
- Zhu, S.; Dong, Z.; Ke, X.; Hou, J.; Zhao, E.; Zhang, K.; Wang, F.; Yang, L.; Xiang, Z.; Cui, H. The roles of sirtuins family in cell metabolism during tumor development. Semin. Cancer Biol. 2018, 57, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-G.; Zheng, J.-N.; Pei, D.-S. P53/microRNA-34-induced metabolic regulation: New opportunities in anticancer therapy. Mol. Cancer 2014, 13, 115. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Huang, A. Effects of sorafenib on lung metastasis in rats with hepatocellular carcinoma: The role of microRNAs. Tumor Biol. 2015, 36, 8455–8463. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, Q.; Gong, Z.; Zhou, L.; You, N.; Wang, S.; Li, X.L.; Li, J.J.; An, J.Z.; Wang, D.S.; et al. MicroRNA-34a Targets Bcl-2 and Sensitizes Human Hepatocellular Carcinoma Cells to Sorafenib Treatment. Technol. Cancer Res. Treat. 2014, 13, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.V.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef] [PubMed]
- Turcios, L.; Vilchez, V.; Acosta, L.F.; Poyil, P.; Butterfield, D.A.; Mitov, M.; Marti, F.; Gedaly, R. Sorafenib and FH535 in combination act synergistically on hepatocellular carcinoma by targeting cell bioenergetics and mitochondrial function. Dig. Liver Dis. 2017, 49, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.; Kumar, S.; Marlowe, T.; Chaudhary, A.K.; Kumar, R.; Wang, J.; O’Malley, J.; Boland, P.M.; Jayanthi, S.; Kumar, T.K.; et al. Oxidative phosphorylation-dependent regulation of cancer cell apoptosis in response to anticancer agents. Cell Death Dis. 2015, 6, e1969. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, Z.; Bunker, E.; Ramírez, A.; Lee, S.; Peng, Y.; Tan, A.-C.; Eckhardt, S.G.; Chapnick, D.A.; Liu, X. Sorafenib targets the mitochondrial electron transport chain complexes and ATP synthase to activate the PINK1–Parkin pathway and modulate cellular drug response. J. Boil. Chem. 2017, 292, 15105–15120. [Google Scholar] [CrossRef]
- Sabatini, D.M. mTOR and cancer: Insights into a complex relationship. Nat. Rev. Cancer. 2006, 6, 729–734. [Google Scholar] [CrossRef]
- Ishijima, N.; Kanki, K.; Shimizu, H.; Shiota, G. Activation of AMP-activated protein kinase by retinoic acid sensitizes hepatocellular carcinoma cells to apoptosis induced by sorafenib. Cancer Sci. 2015, 106, 567–575. [Google Scholar] [CrossRef]
- Grohmann, T.; Penke, M.; Petzold-Quinque, S.; Schuster, S.; Richter, S.; Kiess, W.; Garten, A. Inhibition of NAMPT sensitizes MOLT4 leukemia cells for etoposide treatment through the SIRT2-p53 pathway. Leuk. Res. 2018, 69, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Pinho, A.V.; Mawson, A.; Gill, A.; Arshi, M.; Warmerdam, M.; Giry-Laterriere, M.; Eling, N.; Lie, T.; Kuster, E.; Camargo, S.; et al. Sirtuin 1 stimulates the proliferation and the expression of glycolysis genes in pancreatic neoplastic lesions. Oncotarget 2016, 7, 74768–74778. [Google Scholar] [CrossRef] [Green Version]
- Distelmaier, F.; Valsecchi, F.; Liemburg-Apers, D.C.; Lebiedzinska, M.; Rodenburg, R.J.; Heil, S.; Keijer, J.; Fransen, J.; Imamura, H.; Danhauser, K.; et al. Mitochondrial dysfunction in primary human fibroblasts triggers an adaptive cell survival program that requires AMPK-α. Biochim. Biophys. Acta 2015, 1852, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Tang, W.; Chen, X.; Wang, S.; Wang, X.; Xu, H.; Li, L. The NAMPT/E2F2/SIRT1 axis promotes proliferation and inhibits p53-dependent apoptosis in human melanoma cells. Biochem. Biophys. Res. Commun. 2017, 493, 77–84. [Google Scholar] [CrossRef]
- Sasca, D.; Hahnel, P.S.; Szybinski, J.; Khawaja, K.; Kriege, O.; Pante, S.V.; Bullinger, L.; Strand, S.; Strand, D.; Theobald, M.; et al. SIRT1 prevents genotoxic stress-induced p53 activation in acute myeloid leukemia. Blood 2014, 124, 121–133. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Erster, S.; Mihara, M.; Kim, R.H.; Petrenko, O.; Moll, U.M. In Vivo Mitochondrial p53 Translocation Triggers a Rapid First Wave of Cell Death in Response to DNA Damage That Can Precede p53 Target Gene Activation. Mol. Cell Biol. 2004, 24, 6728–6741. [Google Scholar] [CrossRef] [Green Version]
- Sonntag, R.; Gassler, N.; Bangen, J.-M.; Trautwein, C.; Liedtke, C. Pro-apoptotic Sorafenib signaling in murine hepatocytes depends on malignancy and is associated with PUMA expression in vitro and in vivo. Cell Death Dis. 2014, 5, e1030. [Google Scholar] [CrossRef]
- Han, M.-K.; Song, E.-K.; Guo, Y.; Ou, X.; Mantel, C.; Broxmeyer, H.E. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell 2008, 2, 241–251. [Google Scholar] [CrossRef]
- Caraglia, M.; Giuberti, G.; Marra, M.; Addeo, R.; Montella, L.; Murolo, M.; Sperlongano, P.; Vincenzi, B.; Naviglio, S.; Del Prete, S.; et al. Oxidative stress and ERK1/2 phosphorylation as predictors of outcome in hepatocellular carcinoma patients treated with sorafenib plus octreotide LAR. Cell Death Dis. 2011, 2, e150. [Google Scholar] [CrossRef]
- Gillissen, B.; Richter, A.; Richter, A.; Preissner, R.; Schulze-Osthoff, K.; Essmann, F.; Daniel, P.T. Bax/Bak-independent mitochondrial depolarization and reactive oxygen species induction by sorafenib overcome resistance to apoptosis in renal cell carcinoma. J. Boil. Chem. 2017, 292, 6478–6492. [Google Scholar] [CrossRef] [Green Version]
- Leipert, J.; Kässner, F.; Schuster, S.; Händel, N.; Körner, A.; Kiess, W.; Garten, A. Resveratrol potentiates growth inhibitory effects of rapamycin in PTEN -deficient lipoma cells by suppressing p70S6 kinase activity. Nutr. Cancer 2016, 68, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Penke, M.; Schuster, S.; Gorski, T.; Gebhardt, R.; Kiess, W.; Garten, A. Oleate ameliorates palmitate-induced reduction of NAMPT activity and NAD levels in primary human hepatocytes and hepatocarcinoma cells. Lipids Health Dis. 2017, 16, 191. [Google Scholar] [CrossRef]
- Kluckova, K.; Sticha, M.; Černý, J.; Mracek, T.; Dong, L.; Drahota, Z.; Gottlieb, E.; Neuzil, J.; Rohlena, J. Ubiquinone-binding site mutagenesis reveals the role of mitochondrial complex II in cell death initiation. Cell Death Dis. 2015, 6, e1749. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Scorrano, L. Organelle isolation: Functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007, 2, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Horscroft, J.A.; Kotwica, A.O.; Laner, V.; West, J.A.; Hennis, P.J.; Levett, D.Z.H.; Howard, D.J.; Fernandez, B.O.; Burgess, S.L.; Ament, Z.; et al. Metabolic basis to Sherpa altitude adaptation. Proc. Natl. Acad. Sci. USA 2017, 114, 6382–6387. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garten, A.; Grohmann, T.; Kluckova, K.; Lavery, G.G.; Kiess, W.; Penke, M. Sorafenib-Induced Apoptosis in Hepatocellular Carcinoma Is Reversed by SIRT1. Int. J. Mol. Sci. 2019, 20, 4048. https://doi.org/10.3390/ijms20164048
Garten A, Grohmann T, Kluckova K, Lavery GG, Kiess W, Penke M. Sorafenib-Induced Apoptosis in Hepatocellular Carcinoma Is Reversed by SIRT1. International Journal of Molecular Sciences. 2019; 20(16):4048. https://doi.org/10.3390/ijms20164048
Chicago/Turabian StyleGarten, Antje, Theresa Grohmann, Katarina Kluckova, Gareth G. Lavery, Wieland Kiess, and Melanie Penke. 2019. "Sorafenib-Induced Apoptosis in Hepatocellular Carcinoma Is Reversed by SIRT1" International Journal of Molecular Sciences 20, no. 16: 4048. https://doi.org/10.3390/ijms20164048
APA StyleGarten, A., Grohmann, T., Kluckova, K., Lavery, G. G., Kiess, W., & Penke, M. (2019). Sorafenib-Induced Apoptosis in Hepatocellular Carcinoma Is Reversed by SIRT1. International Journal of Molecular Sciences, 20(16), 4048. https://doi.org/10.3390/ijms20164048