Hepatic Tumor Microenvironments and Effects on NK Cell Phenotype and Function

Abstract
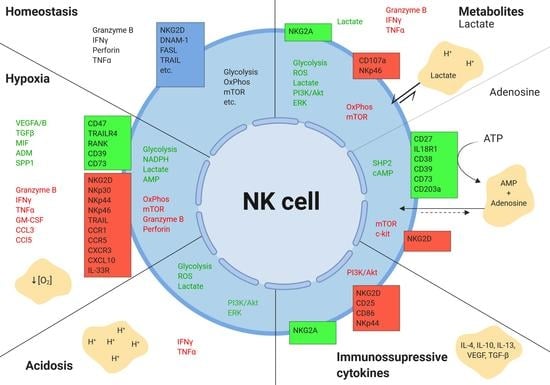
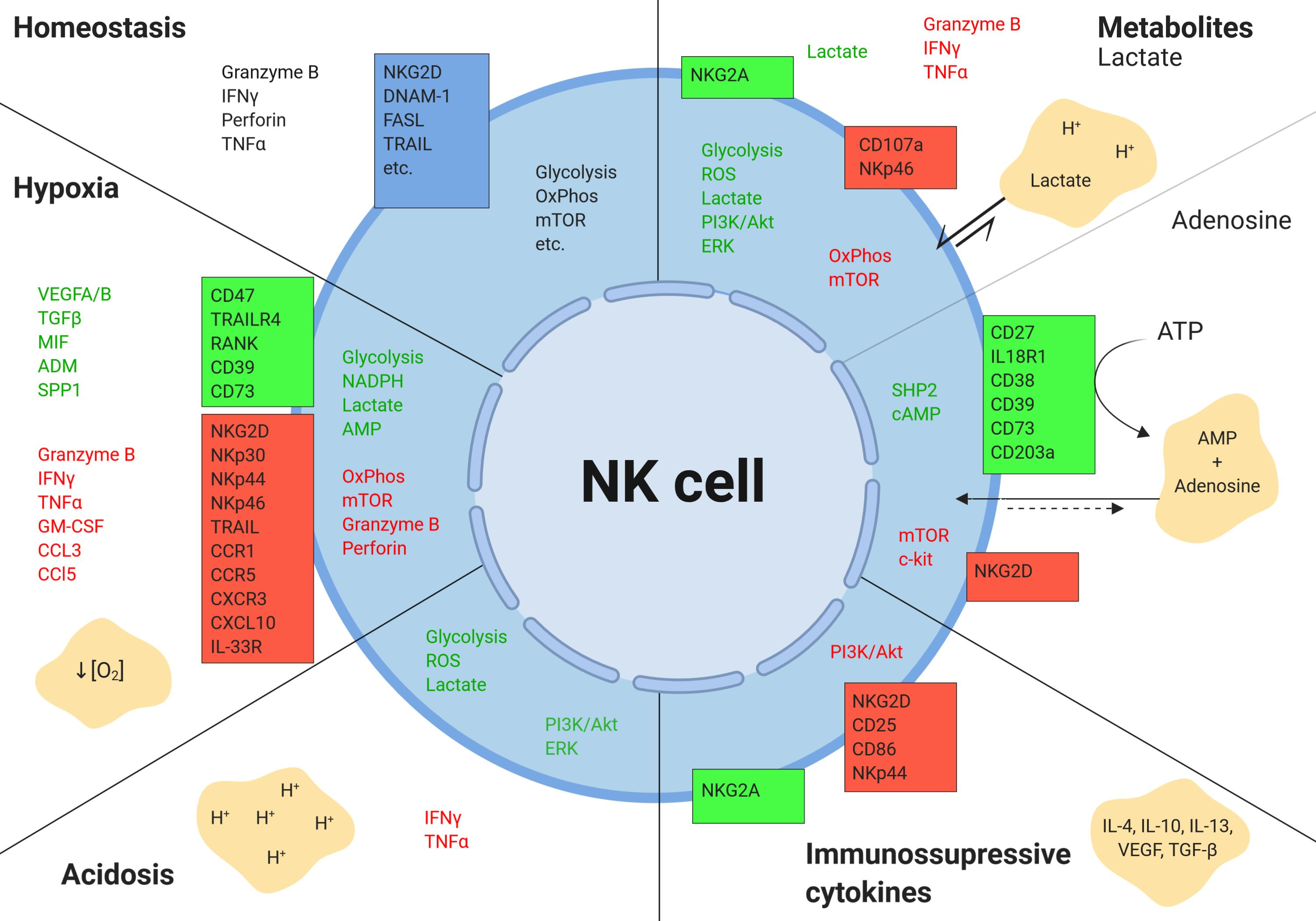
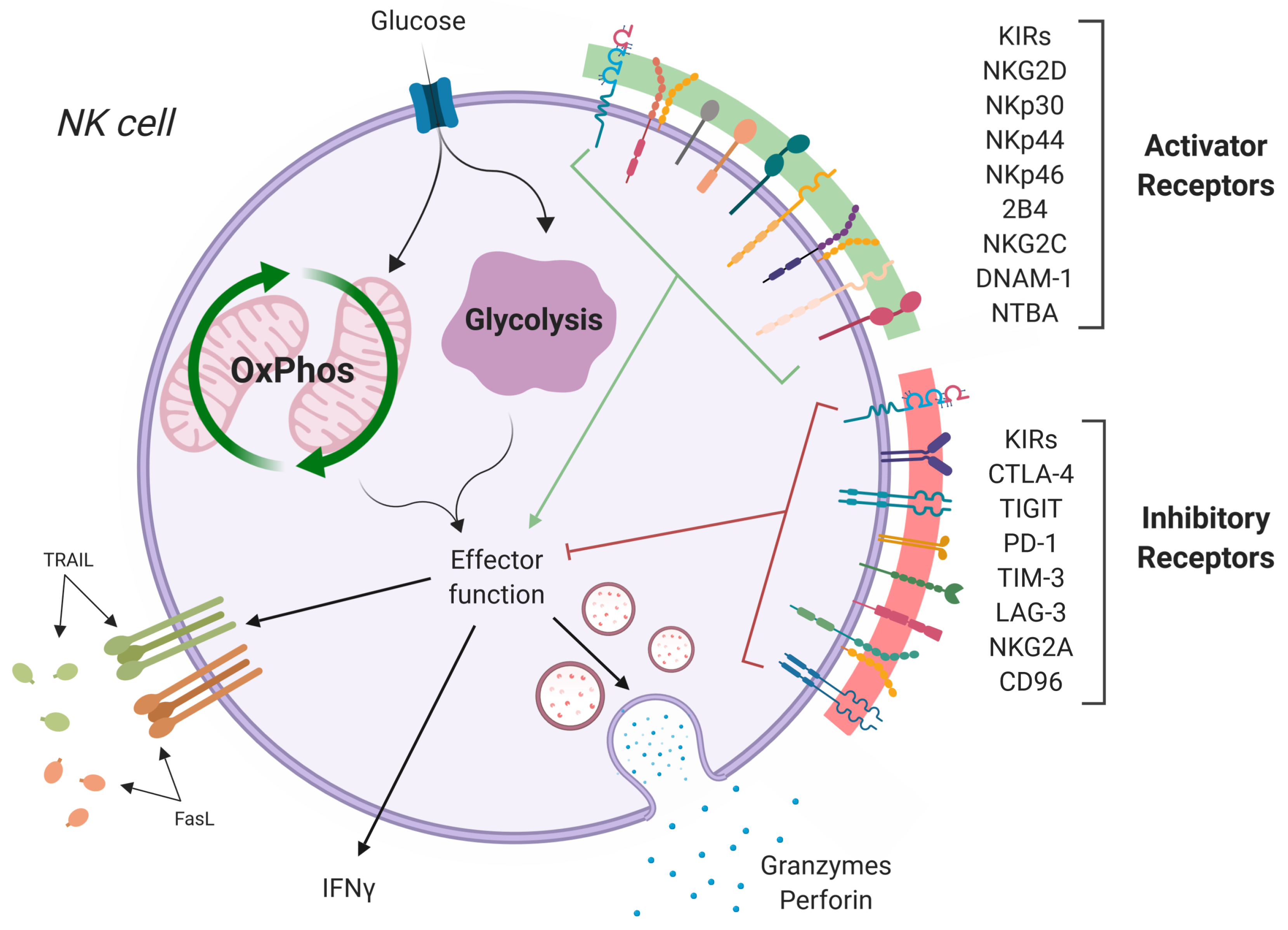
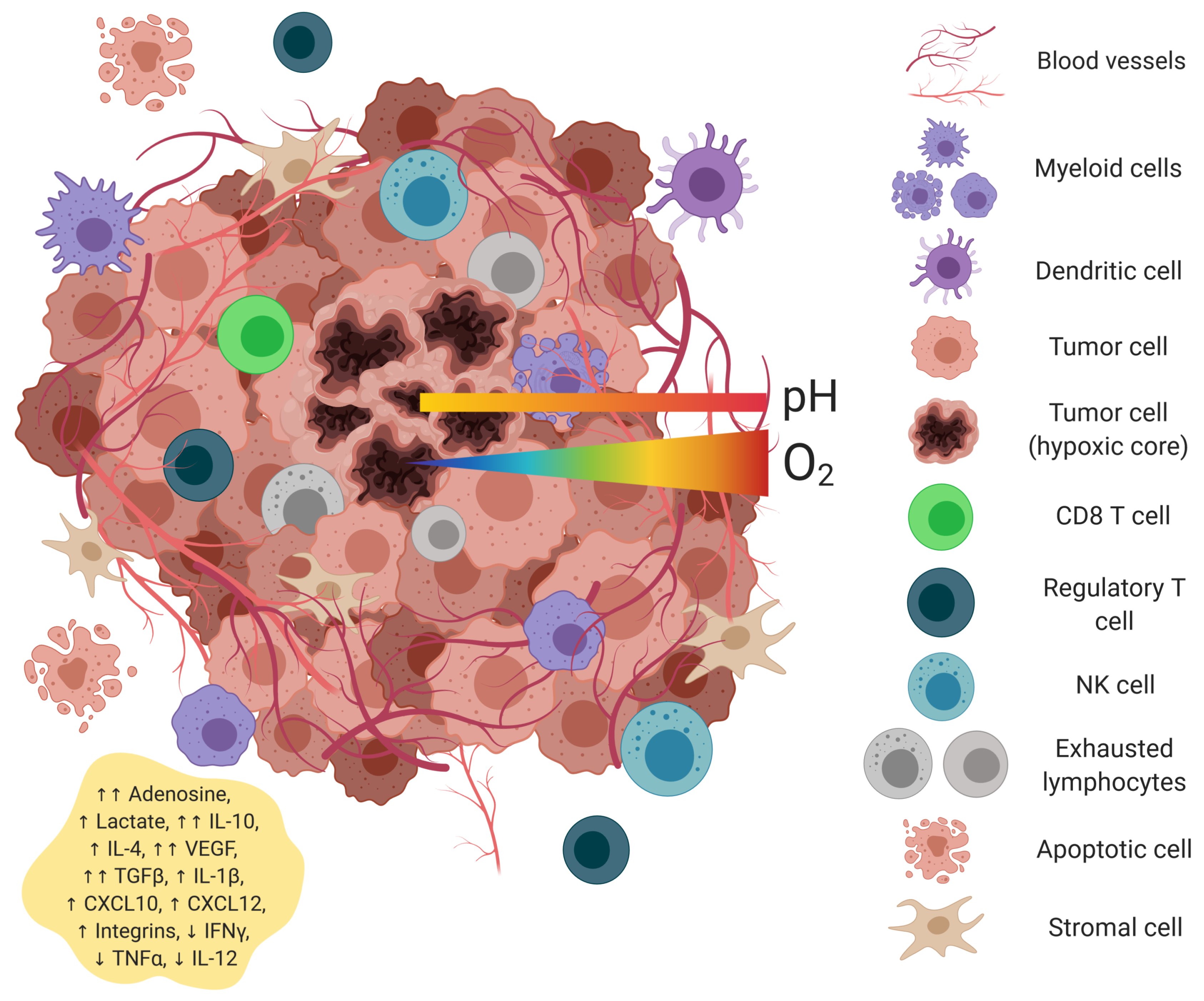
:
{kind=link}
{kind=link}
{kind=link}
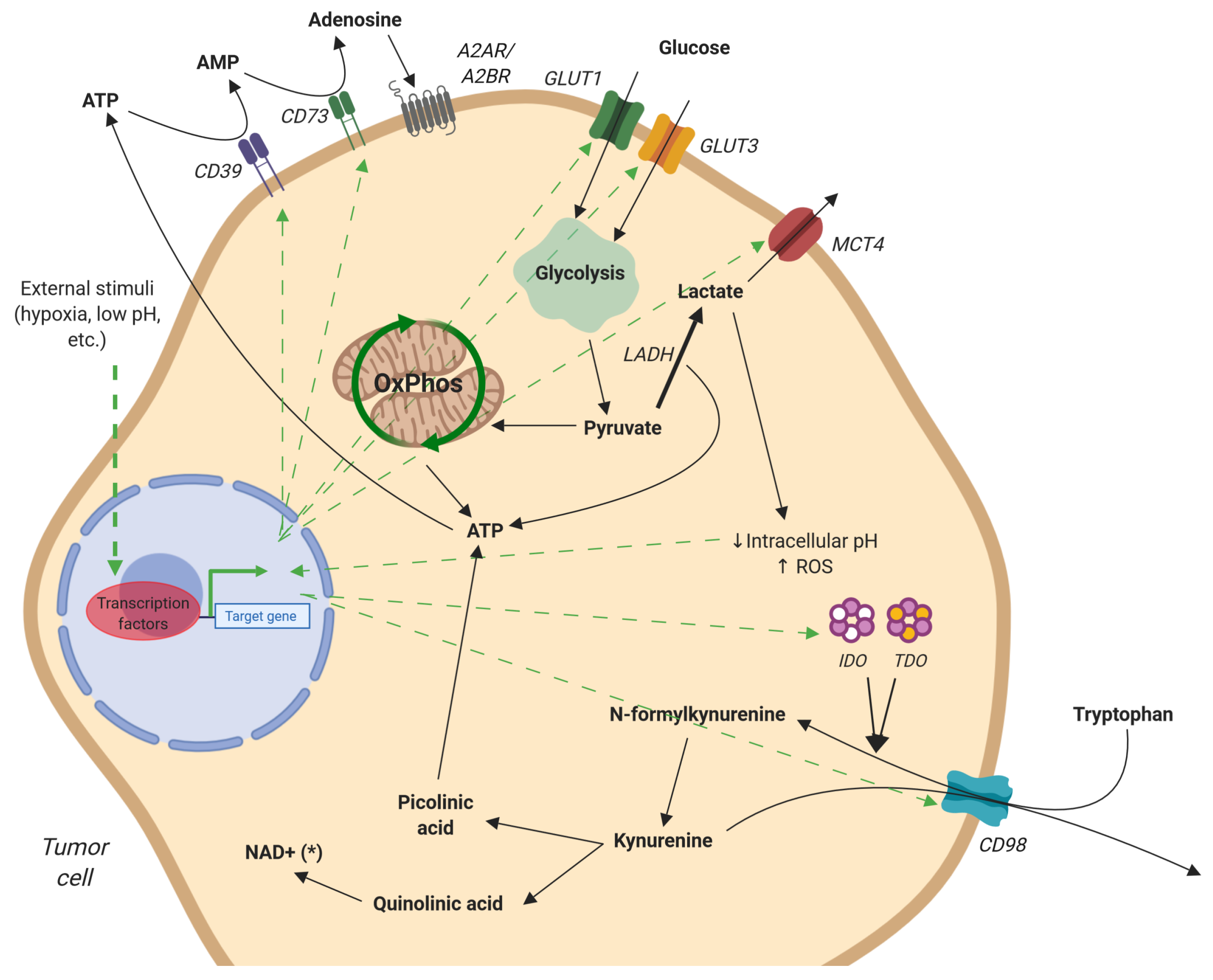{kind=link}
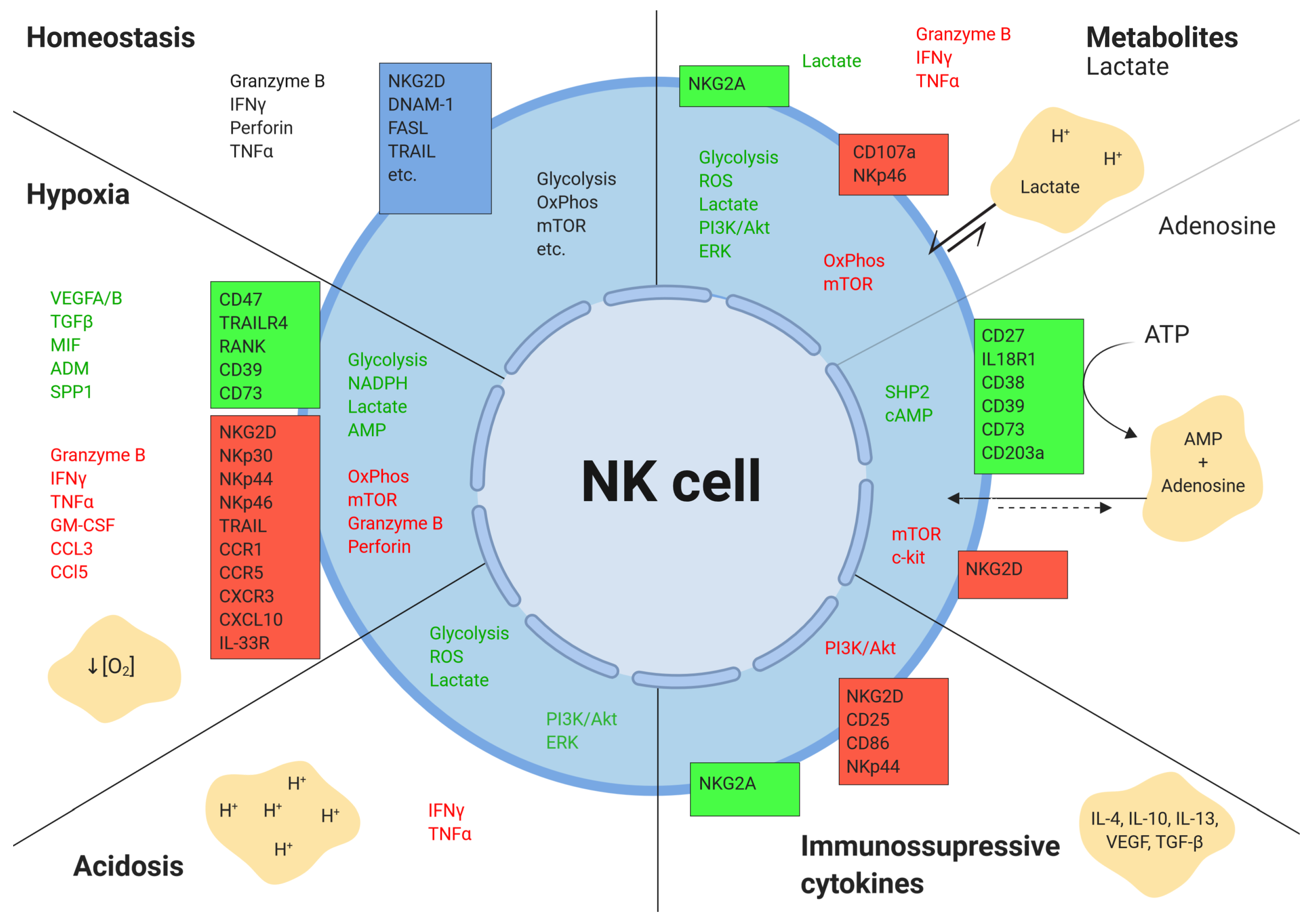{kind=link}
1. Introduction
“The myth of Prometheus means that all the sorrows of the world have their seat in the liver. But it needs a brave man to face so humble a truth.”—François Mauriac, in Le Nœud de vipères (1932)
2. Hepatic Tumor Microenvironment
“Prometheus gave us fire and we use it to light cigarettes.”—Marty Rubin
2.1. Hypoxia
2.2. Tumor Metabolism
2.2.1. Lactate
2.2.2. Adenosine
2.2.3. Tryptophan Catabolism
2.3. Acidic pH
2.4. Cytokines and Growth Factors
3. Conclusions
Funding
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| CRLM | Colorectal liver metastasis |
| HCC | Hepatocellular carcinoma |
| TME | Tumor microenvironment |
| GI | Gastrointestinal |
| NK cell | Natural killer cell |
| NKT cell | Natural killer T cell |
| MAIT cell | Mucosal-associated invariant T cell |
| Treg | Regulatory T cell |
| MDSC | Myeloid-derived suppressor cell |
| CD | Cluster of differentiation |
| IFN | Interferon gamma |
| TNF | Tumor necrosis factor alpha |
| TGF- | Tumor growth factor beta |
| IL | Interleukin |
| NKG2 | Natural killer group 2 receptor |
| OxPhos | Oxidative phosphorylation |
| PPP | Pentose-phosphate pathway |
| mTOR | Mammalian target of Rapamycin |
| ATP | Adenosine triphosphate |
| cAMP | Cyclic adenosine monophosphate |
| LADH | Lactate dehydrogenase |
| IDO | Indoleamine 2,3-dioxygenase |
| TDO | Tryptophan 2,3-dioxygenase |
| NAD | Nicotinamide adenine dinucleotide |
| MCT | Monocarboxylase transporter |
| ADCC | Antibody-dependent cell cytotoxicity |
| PGE2 | Prostaglandin E2 |
| CA | Carbonic anhydrase |
| ROS | Reactive oxygen species |
| FAS | Fatty acid synthase |
| REDD1 | Regulation of DNA damage response 1 |
| HIF-1 | Hypoxia inducible factor 1 |
| MICA | MHC class I polypeptide-related sequence A |
| TRAIL | TNF-related apoptosis-inducing ligand |
| 6PGDH | 6-Phosphogluconate dehydrogenase |
| PI3K | Phosphoinositide 3-kinase |
| PKB | Protein kinase B |
| PDPK1 | 3-phosphoinositide-dependent protein kinase-1 |
References
- Knell, A.J. Liver function and failure: The evolution of liver physiology. J. R. Coll. Physicians Lond. 1980, 14, 205–208. [Google Scholar] [PubMed]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Hua, J. Immune cells in liver regeneration. Oncotarget 2017, 8, 3628–3639. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. Immune tolerance in liver disease. Hepatology 2014, 60, 2109–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundstrom, P.; Szeponik, L.; Ahlmanner, F.; Sundquist, M.; Wong, J.S.B.; Lindskog, E.B.; Gustafsson, B.; Quiding-Jarbrink, M. Tumor-infiltrating mucosal-associated invariant T (MAIT) cells retain expression of cytotoxic effector molecules. Oncotarget 2019, 10, 2810–2823. [Google Scholar] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Wisse, E.; Tian, Z. Liver natural killer cells: Subsets and roles in liver immunity. Cell. Mol. Immunol. 2016, 13, 328–336. [Google Scholar] [CrossRef]
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J. Carcinog. 2017, 16, 1. [Google Scholar]
- Xie, Y. Hepatitis B Virus-Associated Hepatocellular Carcinoma. Adv. Exp. Med. Biol. 2017, 1018, 11–21. [Google Scholar]
- Shiani, A.; Narayanan, S.; Pena, L.; Friedman, M. The Role of Diagnosis and Treatment of Underlying Liver Disease for the Prognosis of Primary Liver Cancer. Cancer Control 2017, 24, 1073274817729240. [Google Scholar] [CrossRef] [PubMed]
- Pinero, F.; Pages, J.; Marciano, S.; Fernandez, N.; Silva, J.; Anders, M.; Zerega, A.; Ridruejo, E.; Ameigeiras, B.; D’Amico, C.; et al. Fatty liver disease, an emerging etiology of hepatocellular carcinoma in Argentina. World J. Hepatol. 2018, 10, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306. [Google Scholar] [CrossRef]
- Kim, D.W.; Talati, C.; Kim, R. Hepatocellular carcinoma (HCC): Beyond sorafenib-chemotherapy. J. Gastrointest. Oncol. 2017, 8, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Nam, J.M.; Giaccia, A.J. Hypoxia: Signaling the Metastatic Cascade. Trends Cancer 2016, 2, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Qin, S. Immune Checkpoint Inhibitors in Hepatocellular Carcinoma: Opportunities and Challenges. Oncologist 2019, 24, S3–S10. [Google Scholar] [PubMed] [Green Version]
- Ray, K. Colorectal cancer: Evading the immune response in metastasis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 191. [Google Scholar] [CrossRef]
- Yang, K.R.; Mooney, S.M.; Zarif, J.C.; Coffey, D.S.; Taichman, R.S.; Pienta, K.J. Niche inheritance: A cooperative pathway to enhance cancer cell fitness through ecosystem engineering. J. Cell. Biochem. 2014, 115, 1478–1485. [Google Scholar] [CrossRef]
- Terry, S.; Buart, S.; Chouaib, S. Hypoxic Stress-Induced Tumor and Immune Plasticity, Suppression, and Impact on Tumor Heterogeneity. Front. Immunol. 2017, 8, 1625. [Google Scholar] [CrossRef]
- Giese, M.A.; Hind, L.E.; Huttenlocher, A. Neutrophil plasticity in the tumor microenvironment. Blood 2019, 133, 2159–2167. [Google Scholar] [CrossRef]
- Gonzalez-Gugel, E.; Saxena, M.; Bhardwaj, N. Modulation of innate immunity in the tumor microenvironment. Cancer Immunol. Immunother. 2016, 65, 1261–1268. [Google Scholar] [CrossRef]
- Davoodzadeh Gholami, M.; Kardar, G.A.; Saeedi, Y.; Heydari, S.; Garssen, J.; Falak, R. Exhaustion of T lymphocytes in the tumor microenvironment: Significance and effective mechanisms. Cell. Immunol. 2017, 322, 1–14. [Google Scholar] [CrossRef]
- Hasmim, M.; Messai, Y.; Ziani, L.; Thiery, J.; Bouhris, J.H.; Noman, M.Z.; Chouaib, S. Critical Role of Tumor Microenvironment in Shaping NK Cell Functions: Implication of Hypoxic Stress. Front. Immunol. 2015, 6, 482. [Google Scholar] [CrossRef]
- Lee-Montiel, F.T.; George, S.M.; Gough, A.H.; Sharma, A.D.; Wu, J.; DeBiasio, R.; Vernetti, L.A.; Taylor, D.L. Control of oxygen tension recapitulates zone-specific functions in human liver microphysiology systems. Exp. Biol. Med. (Maywood) 2017, 242, 1617–1632. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Xu, Q.Y.; Wu, H.M.; Hua, J. Effect of lipid-induced macrophage M1/M2 polarization on lipid metabolism in hepatocytes. Zhonghua Gan Zang Bing Za Zhi 2018, 26, 276–281. [Google Scholar]
- Chen, C.; Lou, T. Hypoxia inducible factors in hepatocellular carcinoma. Oncotarget 2017, 8, 46691–46703. [Google Scholar] [CrossRef]
- Loo, J.M.; Scherl, A.; Nguyen, A.; Man, F.Y.; Weinberg, E.; Zeng, Z.; Saltz, L.; Paty, P.B.; Tavazoie, S.F. Extracellular metabolic energetics can promote cancer progression. Cell 2015, 160, 393–406. [Google Scholar] [CrossRef]
- Groulx, I.; Lee, S. Oxygen-dependent ubiquitination and degradation of hypoxia-inducible factor requires nuclear-cytoplasmic trafficking of the von Hippel-Lindau tumor suppressor protein. Mol. Cell. Biol. 2002, 22, 5319–5336. [Google Scholar] [CrossRef]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef]
- Gassmann, M.; Chilov, D.; Wenger, R.H. Regulation of the hypoxia-inducible factor-1 alpha. ARNT is not necessary for hypoxic induction of HIF-1 alpha in the nucleus. Adv. Exp. Med. Biol. 2000, 475, 87–99. [Google Scholar]
- Sheppard, S.; Ferry, A.; Guedes, J.; Guerra, N. The Paradoxical Role of NKG2D in Cancer Immunity. Front. Immunol. 2018, 9, 1808. [Google Scholar] [CrossRef]
- Daskalaki, I.; Gkikas, I.; Tavernarakis, N. Hypoxia and Selective Autophagy in Cancer Development and Therapy. Front. Cell Dev. Biol. 2018, 6, 104. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Paggetti, J.; Moussay, E.; Berchem, G.; Janji, B. Driving Natural Killer cells toward the melanoma tumor battlefield: Autophagy as a valuable therapeutic target. Oncoimmunology 2018, 7, e1452583. [Google Scholar] [CrossRef] [Green Version]
- Parodi, M.; Raggi, F.; Cangelosi, D.; Manzini, C.; Balsamo, M.; Blengio, F.; Eva, A.; Varesio, L.; Pietra, G.; Moretta, L.; et al. Hypoxia Modifies the Transcriptome of Human NK Cells, Modulates Their Immunoregulatory Profile, and Influences NK Cell Subset Migration. Front. Immunol. 2018, 9, 2358. [Google Scholar] [CrossRef] [Green Version]
- Velasquez, S.Y.; Killian, D.; Schulte, J.; Sticht, C.; Thiel, M.; Lindner, H.A. Short Term Hypoxia Synergizes with Interleukin 15 Priming in Driving Glycolytic Gene Transcription and Supports Human Natural Killer Cell Activities. J. Biol. Chem. 2016, 291, 12960–12977. [Google Scholar] [CrossRef] [Green Version]
- Balsamo, M.; Manzini, C.; Pietra, G.; Raggi, F.; Blengio, F.; Mingari, M.C.; Varesio, L.; Moretta, L.; Bosco, M.C.; Vitale, M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur. J. Immunol. 2013, 43, 2756–2764. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008, 68, 1003–1011. [Google Scholar] [CrossRef]
- Chambers, A.M.; Lupo, K.B.; Matosevic, S. Tumor Microenvironment-Induced Immunometabolic Reprogramming of Natural Killer Cells. Front. Immunol. 2018, 9, 2517. [Google Scholar] [CrossRef] [Green Version]
- Weichhart, T.; Hengstschlager, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Tsaih, S.W.; Lemke, A.; Flister, M.J.; Thakar, M.S.; Malarkannan, S. mTORC1 and mTORC2 differentially promote natural killer cell development. eLife 2018, 7, e35619. [Google Scholar] [CrossRef]
- Sarkar, S.; Germeraad, W.T.; Rouschop, K.M.; Steeghs, E.M.; van Gelder, M.; Bos, G.M.; Wieten, L. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 2013, 8, e64835. [Google Scholar] [CrossRef]
- Bruno, A.; Focaccetti, C.; Pagani, A.; Imperatori, A.S.; Spagnoletti, M.; Rotolo, N.; Cantelmo, A.R.; Franzi, F.; Capella, C.; Ferlazzo, G.; et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia 2013, 15, 133–142. [Google Scholar] [CrossRef]
- Cerdeira, A.S.; Rajakumar, A.; Royle, C.M.; Lo, A.; Husain, Z.; Thadhani, R.I.; Sukhatme, V.P.; Karumanchi, S.A.; Kopcow, H.D. Conversion of peripheral blood NK cells to a decidual NK-like phenotype by a cocktail of defined factors. J. Immunol. 2013, 190, 3939–3948. [Google Scholar] [CrossRef]
- Keskin, D.B.; Allan, D.S.; Rybalov, B.; Andzelm, M.M.; Stern, J.N.; Kopcow, H.D.; Koopman, L.A.; Strominger, J.L. TGFbeta promotes conversion of CD16+ peripheral blood NK cells into CD16- NK cells with similarities to decidual NK cells. Proc. Natl. Acad. Sci. USA 2007, 104, 3378–3383. [Google Scholar] [CrossRef]
- Allan, D.S.; Rybalov, B.; Awong, G.; Zuniga-Pflucker, J.C.; Kopcow, H.D.; Carlyle, J.R.; Strominger, J.L. TGF-Î2 affects development and differentiation of human natural killer cell subsets. Eur. J. Immunol. 2010, 40, 2289–2295. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to Tumor Angiogenesis From Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Njah, K.; Chakraborty, S.; Qiu, B.; Arumugam, S.; Raju, A.; Pobbati, A.V.; Lakshmanan, M.; Tergaonkar, V.; Thibault, G.; Wang, X.; et al. A Role of Agrin in Maintaining the Stability of Vascular Endothelial Growth Factor Receptor-2 during Tumor Angiogenesis. Cell Rep. 2019, 28, 949–965. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar]
- Vazquez, A.; Kamphorst, J.J.; Markert, E.K.; Schug, Z.T.; Tardito, S.; Gottlieb, E. Cancer metabolism at a glance. J. Cell. Sci. 2016, 129, 3367–3373. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Ko, H.; Yun, M. Cancer Metabolism as a Mechanism of Treatment Resistance and Potential Therapeutic Target in Hepatocellular Carcinoma. Yonsei Med. J. 2018, 59, 1143–1149. [Google Scholar] [CrossRef]
- Jin, F.; Wang, Y.; Zhu, Y.; Li, S.; Liu, Y.; Chen, C.; Wang, X.; Zen, K.; Li, L. The miR-125a/HK2 axis regulates cancer cell energy metabolism reprogramming in hepatocellular carcinoma. Sci. Rep. 2017, 7, 3089. [Google Scholar] [CrossRef]
- Jiao, L.; Zhang, H.L.; Li, D.D.; Yang, K.L.; Tang, J.; Li, X.; Ji, J.; Yu, Y.; Wu, R.Y.; Ravichandran, S.; et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy 2018, 14, 671–684. [Google Scholar] [CrossRef]
- Hu, H.; Zhu, W.; Qin, J.; Chen, M.; Gong, L.; Li, L.; Liu, X.; Tao, Y.; Yin, H.; Zhou, H.; et al. Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatology 2017, 65, 515–528. [Google Scholar] [CrossRef]
- Zarrinpar, A. Metabolic Pathway Inhibition in Liver Cancer. SLAS Technol. 2017, 22, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Kowalik, M.A.; Columbano, A.; Perra, A. Emerging Role of the Pentose Phosphate Pathway in Hepatocellular Carcinoma. Front. Oncol. 2017, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Emerging roles of lipid metabolism in cancer progression. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 254–260. [Google Scholar] [CrossRef]
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 2017, 16, 76. [Google Scholar] [CrossRef]
- Gillies, R.J.; Robey, I.; Gatenby, R.A. Causes and consequences of increased glucose metabolism of cancers. J. Nucl. Med. 2008, 49 (Suppl. 2), 24S–42S. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Kaloyianni, M.; Freedland, R.A. Contribution of several amino acids and lactate to gluconeogenesis in hepatocytes isolated from rats fed various diets. J. Nutr. 1990, 120, 116–122. [Google Scholar] [CrossRef]
- Harmon, C.; Robinson, M.W.; Hand, F.; Almuaili, D.; Mentor, K.; Houlihan, D.D.; Hoti, E.; Lynch, L.; Geoghegan, J.; O’Farrelly, C. Lactate-Mediated Acidification of Tumor Microenvironment Induces Apoptosis of Liver-Resident NK Cells in Colorectal Liver Metastasis. Cancer Immunol. Res. 2019, 7, 335–346. [Google Scholar] [CrossRef]
- Teilhet, C.; Morvan, D.; Joubert-Zakeyh, J.; Biesse, A.S.; Pereira, B.; Massoulier, S.; Dechelotte, P.; Pezet, D.; Buc, E.; Lamblin, G.; et al. Specificities of Human Hepatocellular Carcinoma Developed on Non-Alcoholic Fatty Liver Disease in Absence of Cirrhosis Revealed by Tissue Extracts 1H-NMR Spectroscopy. Metabolites 2017, 7, 49. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L.; et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef]
- Chambers, A.M.; Wang, J.; Lupo, K.B.; Yu, H.; Atallah Lanman, N.M.; Matosevic, S. Adenosinergic Signaling Alters Natural Killer Cell Functional Responses. Front. Immunol. 2018, 9, 2533. [Google Scholar] [CrossRef]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Hasko, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.Y.; Ni, X.C.; Yi, Y.; He, H.W.; Wang, J.X.; Fu, Y.P.; Sun, J.; Zhou, J.; Cheng, Y.F.; Jin, J.J.; et al. Overexpression of CD39 in hepatocellular carcinoma is an independent indicator of poor outcome after radical resection. Medicine (Baltimore) 2016, 95, e4989. [Google Scholar] [CrossRef]
- Land, S.C.; Tee, A.R. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef]
- Morandi, F.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Chiesa, S.; Imperatori, A.; Zanellato, S.; Mortara, L.; Gattorno, M.; Pistoia, V.; et al. CD56brightCD16- NK Cells Produce Adenosine through a CD38-Mediated Pathway and Act as Regulatory Cells Inhibiting Autologous CD4+ T Cell Proliferation. J. Immunol. 2015, 195, 965–972. [Google Scholar] [CrossRef]
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F.; et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-Î2-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011. [Google Scholar] [CrossRef]
- Kaku, H.; Cheng, K.F.; Al-Abed, Y.; Rothstein, T.L. A novel mechanism of B cell-mediated immune suppression through CD73 expression and adenosine production. J. Immunol. 2014, 193, 5904–5913. [Google Scholar] [CrossRef]
- Sakowicz-Burkiewicz, M.; Kocbuch, K.; Grden, M.; Maciejewska, I.; Szutowicz, A.; Pawelczyk, T. Impact of adenosine receptors on immunoglobulin production by human peripheral blood B lymphocytes. J. Physiol. Pharmacol. 2012, 63, 661–668. [Google Scholar]
- Wang, J.; Matosevic, S. Adenosinergic signaling as a target for natural killer cell immunotherapy. J. Mol. Med. 2018, 96, 903–913. [Google Scholar] [CrossRef]
- Barrow, A.D.; Colonna, M. Tailoring Natural Killer cell immunotherapy to the tumour microenvironment. Semin. Immunol. 2017, 31, 30–36. [Google Scholar] [CrossRef]
- Strasser, B.; Gostner, J.M.; Fuchs, D. Mood, food, and cognition: Role of tryptophan and serotonin. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 55–61. [Google Scholar] [CrossRef]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Lee, J.R.; Dalton, R.R.; Messina, J.L.; Sharma, M.D.; Smith, D.M.; Burgess, R.E.; Mazzella, F.; Antonia, S.J.; Mellor, A.L.; Munn, D.H. Pattern of recruitment of immunoregulatory antigen-presenting cells in malignant melanoma. Lab. Investig. 2003, 83, 1457–1466. [Google Scholar] [CrossRef]
- Platten, M.; Wick, W.; Van den Eynde, B.J. Tryptophan catabolism in cancer: Beyond IDO and tryptophan depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef]
- Badawy, A.A. Targeting tryptophan availability to tumors: The answer to immune escape? Immunol. Cell Biol. 2018, 96, 1026–1034. [Google Scholar] [CrossRef]
- Sucher, R.; Fischler, K.; Oberhuber, R.; Kronberger, I.; Margreiter, C.; Ollinger, R.; Schneeberger, S.; Fuchs, D.; Werner, E.R.; Watschinger, K.; et al. IDO and regulatory T cell support are critical for cytotoxic T lymphocyte-associated Ag-4 Ig-mediated long-term solid organ allograft survival. J. Immunol. 2012, 188, 37–46. [Google Scholar] [CrossRef]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef]
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef]
- Muller, A.J.; DuHadaway, J.B.; Donover, P.S.; Sutanto-Ward, E.; Prendergast, G.C. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 2005, 11, 312–319. [Google Scholar] [CrossRef]
- Finkelmeier, F.; Canli, O.; Peiffer, K.H.; Walter, D.; Tal, A.; Koch, C.; Pession, U.; Vermehren, J.; Trojan, J.; Zeuzem, S.; et al. Circulating hypoxia marker carbonic anhydrase IX (CA9) in patients with hepatocellular carcinoma and patients with cirrhosis. PLoS ONE 2018, 13, e0200855. [Google Scholar] [CrossRef]
- Huang, W.J.; Jeng, Y.M.; Lai, H.S.; Fong, I.U.; Sheu, F.Y.; Lai, P.L.; Yuan, R.H. Expression of hypoxic marker carbonic anhydrase IX predicts poor prognosis in resectable hepatocellular carcinoma. PLoS ONE 2015, 10, e0119181. [Google Scholar] [CrossRef]
- Fischer, B.; Muller, B.; Fischer, K.G.; Baur, N.; Kreutz, W. Acidic pH inhibits non-MHC-restricted killer cell functions. Clin. Immunol. 2000, 96, 252–263. [Google Scholar] [CrossRef]
- Motallebnezhad, M.; Jadidi-Niaragh, F.; Qamsari, E.S.; Bagheri, S.; Gharibi, T.; Yousefi, M. The immunobiology of myeloid-derived suppressor cells in cancer. Tumor Biol. 2016, 37, 1387–1406. [Google Scholar] [CrossRef]
- Selleri, S.; Bifsha, P.; Civini, S.; Pacelli, C.; Dieng, M.M.; Lemieux, W.; Jin, P.; Bazin, R.; Patey, N.; Marincola, F.M.; et al. Human mesenchymal stromal cell-secreted lactate induces M2-macrophage differentiation by metabolic reprogramming. Oncotarget 2016, 7, 30193–30210. [Google Scholar] [CrossRef]
- El-Kenawi, A.; Gatenbee, C.; Robertson-Tessi, M.; Bravo, R.; Dhillon, J.; Balagurunathan, Y.; Berglund, A.; Visvakarma, N.; Ibrahim-Hashim, A.; Choi, J.; et al. Acidity promotes tumor progression by altering macrophage phenotype in prostate cancer. bioRxiv 2018. [Google Scholar] [CrossRef]
- Martinez, D.; Vermeulen, M.; von Euw, E.; Sabatte, J.; Maggini, J.; Ceballos, A.; Trevani, A.; Nahmod, K.; Salamone, G.; Barrio, M.; et al. Extracellular acidosis triggers the maturation of human dendritic cells and the production of IL-12. J. Immunol. 2007, 179, 1950–1959. [Google Scholar] [CrossRef]
- Potzl, J.; Roser, D.; Bankel, L.; Homberg, N.; Geishauser, A.; Brenner, C.D.; Weigand, M.; Rocken, M.; Mocikat, R. Reversal of tumor acidosis by systemic buffering reactivates NK cells to express IFN-Î3 and induces NK cell-dependent lymphoma control without other immunotherapies. Int. J. Cancer 2017, 140, 2125–2133. [Google Scholar] [CrossRef]
- Lv, L.H.; Yu, J.D.; Li, G.L.; Long, T.Z.; Zhang, W.; Chen, Y.J.; Min, J.; Wan, Y.L. Functional distinction of rat liver natural killer cells from spleen natural killer cells under normal and acidic conditions in vitro. Hepatobiliary Pancreat. Dis. Int. 2012, 11, 285–293. [Google Scholar] [CrossRef]
- Robey, I.F.; Baggett, B.K.; Kirkpatrick, N.D.; Roe, D.J.; Dosescu, J.; Sloane, B.F.; Hashim, A.I.; Morse, D.L.; Raghunand, N.; Gatenby, R.A.; et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009, 69, 2260–2268. [Google Scholar] [CrossRef]
- Islam, A.; Li, S.S.; Oykhman, P.; Timm-McCann, M.; Huston, S.M.; Stack, D.; Xiang, R.F.; Kelly, M.M.; Mody, C.H. An acidic microenvironment increases NK cell killing of Cryptococcus neoformans and Cryptococcus gattii by enhancing perforin degranulation. PLoS Pathog. 2013, 9, e1003439. [Google Scholar] [CrossRef]
- Ramadori, G.; Armbrust, T. Cytokines in the liver. Eur. J. Gastroenterol. Hepatol. 2001, 13, 777–784. [Google Scholar] [CrossRef]
- Li, P.; He, K.; Li, J.; Liu, Z.; Gong, J. The role of Kupffer cells in hepatic diseases. Mol. Immunol. 2017, 85, 222–229. [Google Scholar] [CrossRef]
- Tsutsui, H.; Nishiguchi, S. Importance of Kupffer cells in the development of acute liver injuries in mice. Int. J. Mol. Sci. 2014, 15, 7711–7730. [Google Scholar] [CrossRef]
- Sudo, K.; Yamada, Y.; Saito, K.; Shimizu, S.; Ohashi, H.; Kato, T.; Moriwaki, H.; Ito, H.; Seishima, M. TNF-alpha and IL-6 signals from the bone marrow derived cells are necessary for normal murine liver regeneration. Biochim. Biophys. Acta 2008, 1782, 671–679. [Google Scholar] [CrossRef]
- Knolle, P.; Schlaak, J.; Uhrig, A.; Kempf, P.; Meyer zum Buschenfelde, K.H.; Gerken, G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J. Hepatol. 1995, 22, 226–229. [Google Scholar] [CrossRef]
- Bishayee, A. The role of inflammation and liver cancer. Adv. Exp. Med. Biol. 2014, 816, 401–435. [Google Scholar]
- Lasry, A.; Zinger, A.; Ben-Neriah, Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016, 17, 230–240. [Google Scholar] [CrossRef]
- Ueda, T.; Shimada, E.; Urakawa, T. Serum levels of cytokines in patients with colorectal cancer: Possible involvement of interleukin-6 and interleukin-8 in hematogenous metastasis. J. Gastroenterol. 1994, 29, 423–429. [Google Scholar] [CrossRef]
- Hammerich, L.; Tacke, F. Interleukins in chronic liver disease: Lessons learned from experimental mouse models. Clin. Exp. Gastroenterol. 2014, 7, 297–306. [Google Scholar]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef]
- Spolski, R.; Leonard, W.J. Interleukin-21: A double-edged sword with therapeutic potential. Nat. Rev. Drug Discov. 2014, 13, 379–395. [Google Scholar] [CrossRef]
- Santegoets, S.J.; Turksma, A.W.; Suhoski, M.M.; Stam, A.G.; Albelda, S.M.; Hooijberg, E.; Scheper, R.J.; van den Eertwegh, A.J.; Gerritsen, W.R.; Powell, D.J.; et al. IL-21 promotes the expansion of CD27+ CD28+ tumor infiltrating lymphocytes with high cytotoxic potential and low collateral expansion of regulatory T cells. J. Transl. Med. 2013, 11, 37. [Google Scholar] [CrossRef]
- Skak, K.; Frederiksen, K.S.; Lundsgaard, D. Interleukin-21 activates human natural killer cells and modulates their surface receptor expression. Immunology 2008, 123, 575–583. [Google Scholar] [CrossRef]
- Burgess, S.J.; Marusina, A.I.; Pathmanathan, I.; Borrego, F.; Coligan, J.E. IL-21 down-regulates NKG2D/DAP10 expression on human NK and CD8+ T cells. J. Immunol. 2006, 176, 1490–1497. [Google Scholar] [CrossRef]
- Carson, W.E.; Ross, M.E.; Baiocchi, R.A.; Marien, M.J.; Boiani, N.; Grabstein, K.; Caligiuri, M.A. Endogenous production of interleukin 15 by activated human monocytes is critical for optimal production of interferon-gamma by natural killer cells in vitro. J. Clin. Investig. 1995, 96, 2578–2582. [Google Scholar] [CrossRef]
- Yoshikai, Y.; Nishimura, H. The role of interleukin 15 in mounting an immune response against microbial infections. Microbes Infect. 2000, 2, 381–389. [Google Scholar] [CrossRef]
- Golden-Mason, L.; Kelly, A.M.; Doherty, D.G.; Traynor, O.; McEntee, G.; Kelly, J.; Hegarty, J.E.; O’Farrelly, C. Hepatic interleuklin 15 (IL-15) expression: Implications for local NK/NKT cell homeostasis and development. Clin. Exp. Immunol. 2004, 138, 94–101. [Google Scholar] [CrossRef]
- Easom, N.J.W.; Stegmann, K.A.; Swadling, L.; Pallett, L.J.; Burton, A.R.; Odera, D.; Schmidt, N.; Huang, W.C.; Fusai, G.; Davidson, B.; et al. IL-15 Overcomes Hepatocellular Carcinoma-Induced NK Cell Dysfunction. Front. Immunol. 2018, 9, 1009. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Zhang, J.; Liu, H.; Wan, L.; Zhang, H.; Huang, Q.; Xu, E.; Lai, M. IL-13/STAT6 signaling plays a critical role in the epithelial-mesenchymal transition of colorectal cancer cells. Oncotarget 2016, 7, 61183–61198. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Munker, S.; Mullenbach, R.; Weng, H.L. IL-13 Signaling in Liver Fibrogenesis. Front. Immunol. 2012, 3, 116. [Google Scholar] [CrossRef] [Green Version]
- Kusmartsev, S.; Gabrilovich, D.I. Effect of tumor-derived cytokines and growth factors on differentiation and immune suppressive features of myeloid cells in cancer. Cancer Metastasis Rev. 2006, 25, 323–331. [Google Scholar] [CrossRef]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef]
- Lassen, M.G.; Lukens, J.R.; Dolina, J.S.; Brown, M.G.; Hahn, Y.S. Intrahepatic IL-10 maintains NKG2A+Ly49- liver NK cells in a functionally hyporesponsive state. J. Immunol. 2010, 184, 2693–2701. [Google Scholar] [CrossRef]
- Sun, C.; Xu, J.; Huang, Q.; Huang, M.; Wen, H.; Zhang, C.; Wang, J.; Song, J.; Zheng, M.; Sun, H.; et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology 2017, 6, e1264562. [Google Scholar] [CrossRef]
- Viel, S.; Marcais, A.; Guimaraes, F.S.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-Î2 inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal 2016, 9, ra19. [Google Scholar] [CrossRef]
- Otegbeye, F.; Ojo, E.; Moreton, S.; Mackowski, N.; Lee, D.A.; de Lima, M.; Wald, D.N. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS ONE 2018, 13, e0191358. [Google Scholar]
- Sun, C.; Sun, H.Y.; Xiao, W.H.; Zhang, C.; Tian, Z.G. Natural killer cell dysfunction in hepatocellular carcinoma and NK cell-based immunotherapy. Acta Pharmacol. Sin. 2015, 36, 1191–1199. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.J.; Kim, Y.J. Hepatitis B viral load affects prognosis of hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 12039–12044. [Google Scholar] [CrossRef]
- Yoon, J.C.; Yang, C.M.; Song, Y.; Lee, J.M. Natural killer cells in hepatitis C: Current progress. World J. Gastroenterol. 2016, 22, 1449–1460. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, X.M.; Li, S.R.; Twelkmeyer, T.; Wang, W.H.; Zhang, S.Y.; Wang, S.F.; Chen, J.Z.; Jin, X.; Wu, Y.Z.; et al. NKG2A is a NK cell exhaustion checkpoint for HCV persistence. Nat. Commun. 2019, 10, 1507. [Google Scholar] [CrossRef]
- Strunz, B.; Hengst, J.; Deterding, K.; Manns, M.P.; Cornberg, M.; Ljunggren, H.G.; Wedemeyer, H.; Bjorkstrom, N.K. Chronic hepatitis C virus infection irreversibly impacts human natural killer cell repertoire diversity. Nat. Commun. 2018, 9, 2275. [Google Scholar] [CrossRef]
- Vitale, M.; Cantoni, C.; Pietra, G.; Mingari, M.C.; Moretta, L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 2014, 44, 1582–1592. [Google Scholar] [CrossRef]
- Bernardini, G.; Antonangeli, F.; Bonanni, V.; Santoni, A. Dysregulation of Chemokine/Chemokine Receptor Axes and NK Cell Tissue Localization during Diseases. Front. Immunol. 2016, 7, 402. [Google Scholar] [CrossRef]
- Sungur, C.M.; Murphy, W.J. Positive and negative regulation by NK cells in cancer. Crit. Rev. Oncog. 2014, 19, 57–66. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piñeiro Fernández, J.; Luddy, K.A.; Harmon, C.; O’Farrelly, C. Hepatic Tumor Microenvironments and Effects on NK Cell Phenotype and Function. Int. J. Mol. Sci. 2019, 20, 4131. https://doi.org/10.3390/ijms20174131
Piñeiro Fernández J, Luddy KA, Harmon C, O’Farrelly C. Hepatic Tumor Microenvironments and Effects on NK Cell Phenotype and Function. International Journal of Molecular Sciences. 2019; 20(17):4131. https://doi.org/10.3390/ijms20174131
Chicago/Turabian StylePiñeiro Fernández, Julián, Kimberly A. Luddy, Cathal Harmon, and Cliona O’Farrelly. 2019. "Hepatic Tumor Microenvironments and Effects on NK Cell Phenotype and Function" International Journal of Molecular Sciences 20, no. 17: 4131. https://doi.org/10.3390/ijms20174131
APA StylePiñeiro Fernández, J., Luddy, K. A., Harmon, C., & O’Farrelly, C. (2019). Hepatic Tumor Microenvironments and Effects on NK Cell Phenotype and Function. International Journal of Molecular Sciences, 20(17), 4131. https://doi.org/10.3390/ijms20174131