Activation of Kv7 Potassium Channels Inhibits Intracellular Ca2+ Increases Triggered By TRPV1-Mediated Pain-Inducing Stimuli in F11 Immortalized Sensory Neurons

 ,
,  ,
,  ,
,  , ,
, ,
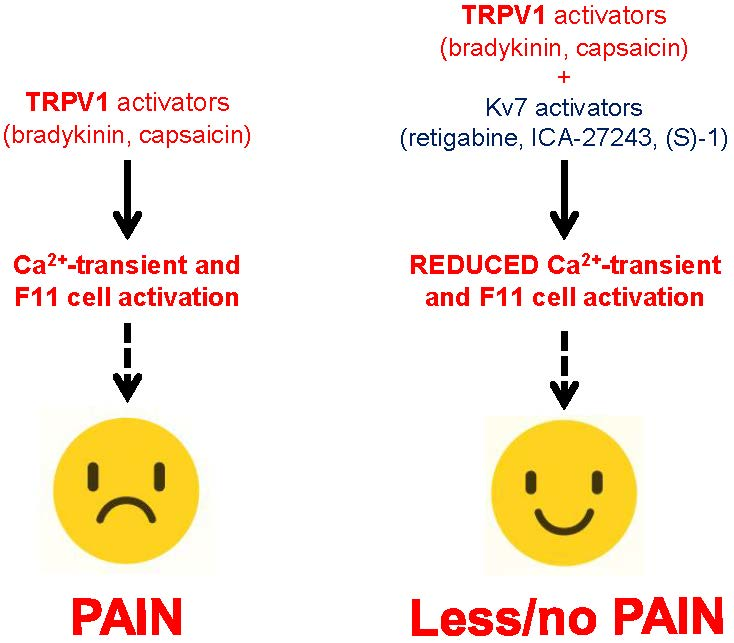
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
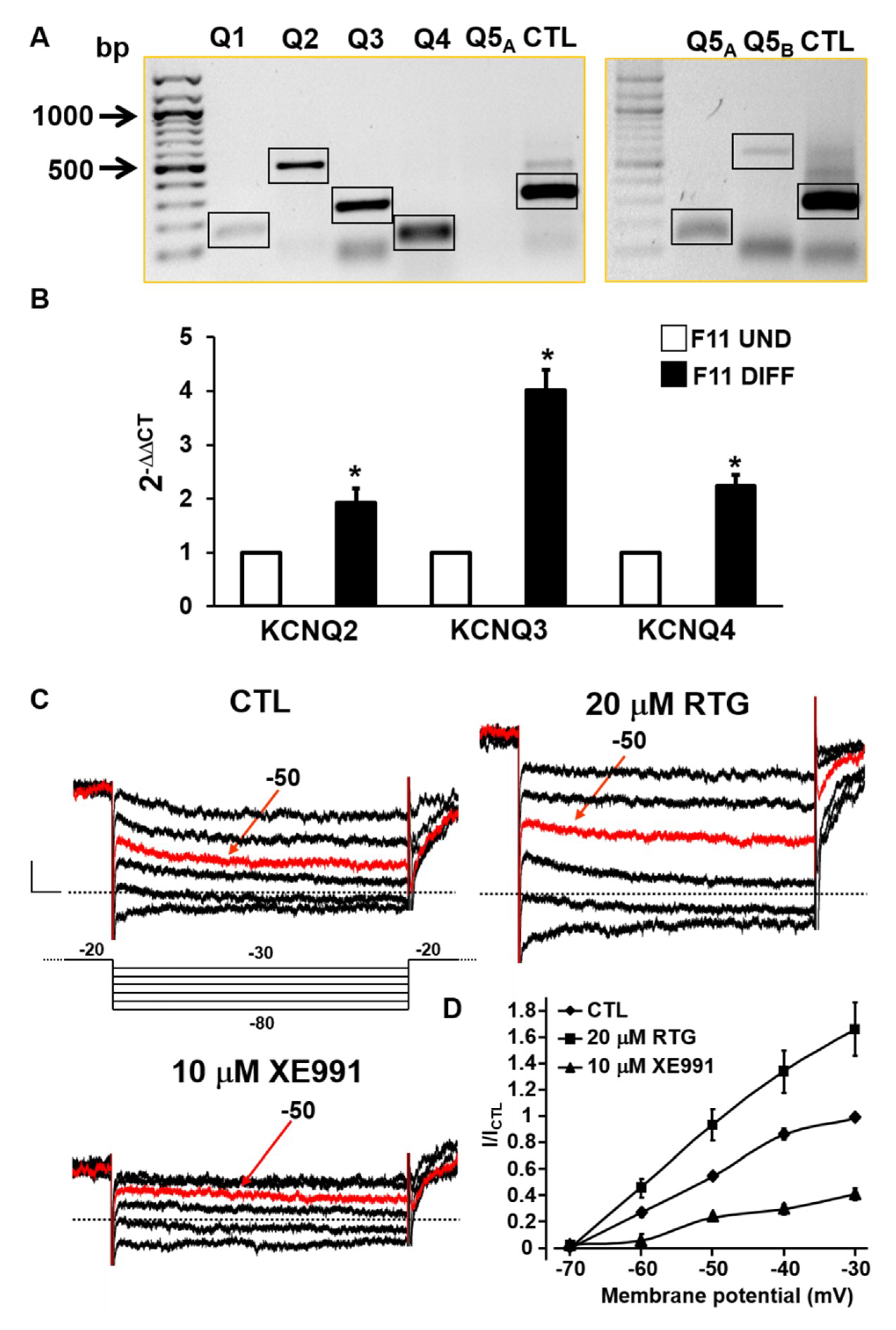
2.1. Biochemical and Functional Evidence for Kv7 Expression in F11 Cells
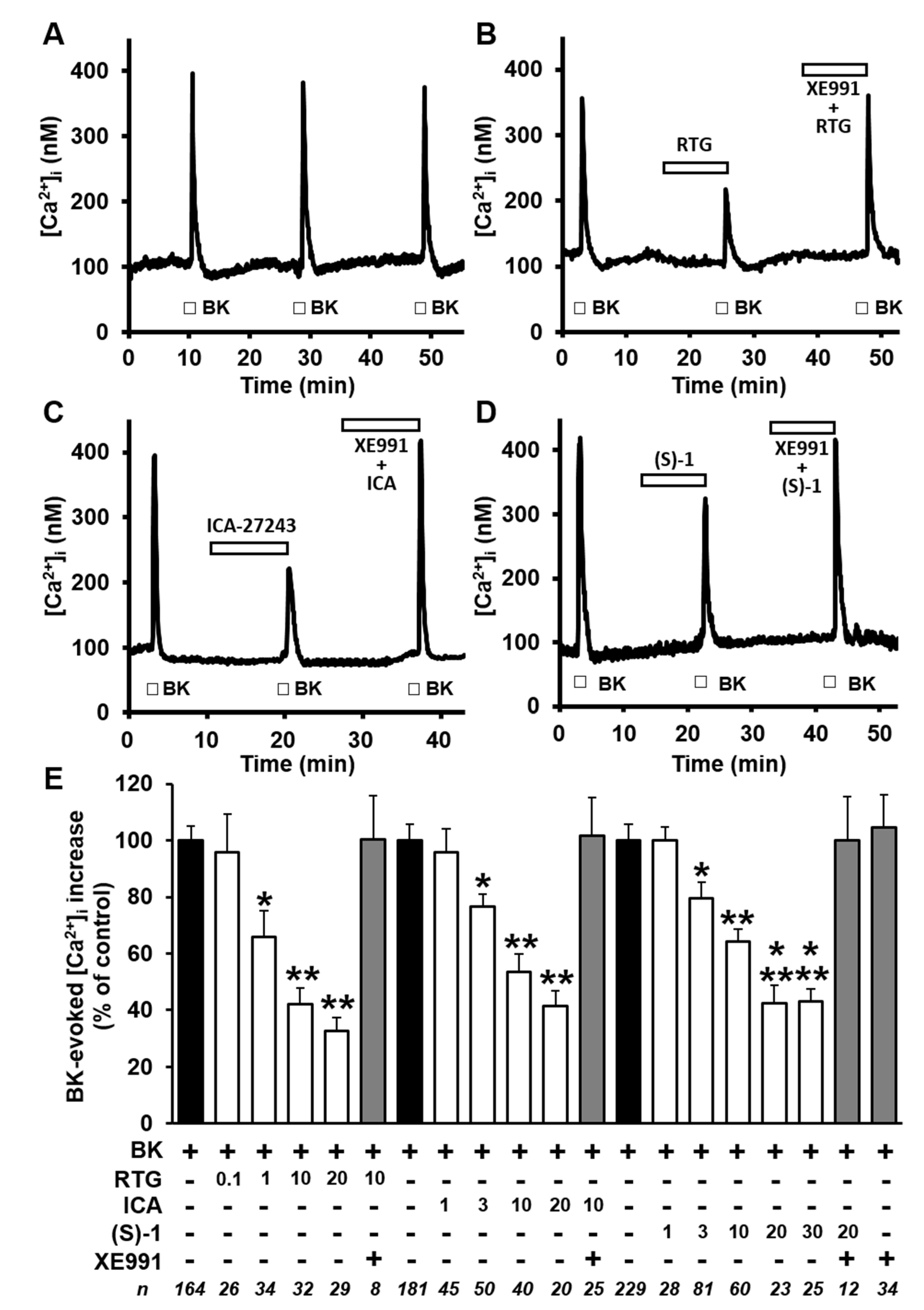
2.2. Kv7 Activators Reduce BK-evoked [Ca2+]i Responses in F11 Cells
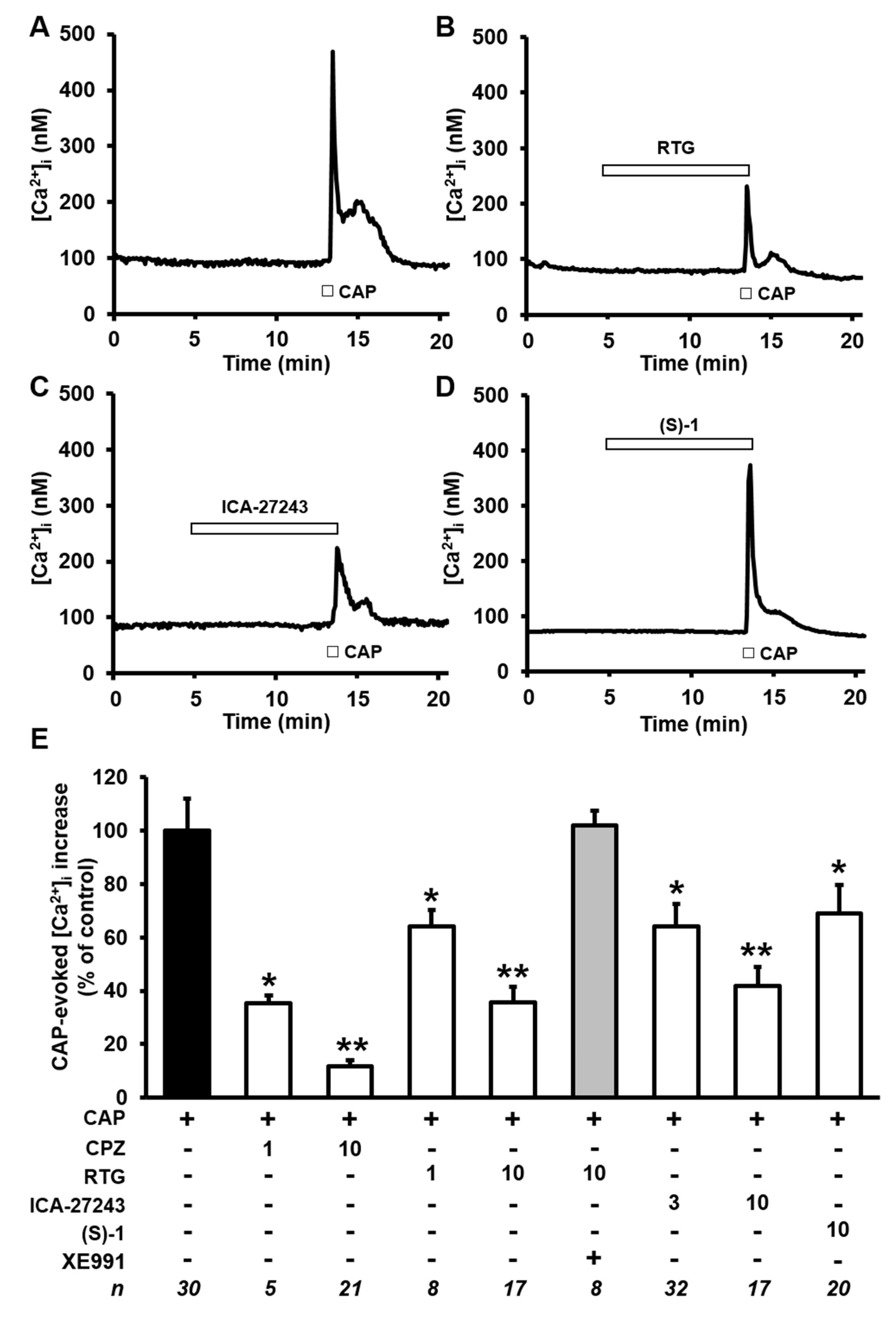
2.3. Kv7 Activators Reduce CAP-evoked [Ca2+]i Responses in F11 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. RNA Extraction and Semiquantitative PCR
4.3. Quantitative Reverse-Transcription PCR (qRT-PCR)
4.4. [Ca2+]i Measurements
4.5. Whole-cell Electrophysiology
4.6. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Soldovieri, M.V.; Miceli, F.; Taglialatela, M. Driving with no brakes: molecular pathophysiology of Kv7 potassium channels. Physiology 2011, 26, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; De Jager, T.; et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996, 12, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.C.; Waldegger, S.; Fehr, S.; Bleich, M.; Warth, R.; Greger, R.; Jentsch, T.J. A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature 2000, 403, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.M.; Mistry, M.; Marsh, S.J.; Brown, D.A.; Delmas, P. Molecular correlates of the M-current in cultured rat hippocampal neurons. J. Physiol. 2002, 544, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.S.; Pan, Z.; Shi, W.; Brown, B.S.; Wymore, R.S.; Cohen, I.S.; Dixon, J.E.; McKinnon, D. KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channel. Science 1998, 282, 1890–1893. [Google Scholar] [CrossRef] [PubMed]
- Marrion, N.V. Control of M-current. Annu. Rev. Physiol. 1997, 59, 483–504. [Google Scholar] [CrossRef] [PubMed]
- Yus-Nájera, E.; Santana-Castro, I.; Villarroel, A. The identification and characterization of a noncontinuous calmodulin-binding site in noninactivating voltage-dependent KCNQ potassium channels. J. Biol. Chem. 2002, 277, 28545–28553. [Google Scholar] [CrossRef] [PubMed]
- Regev, N.; Degani-Katzav, N.; Korngreen, A.; Etzioni, A.; Siloni, S.; Alaimo, A.; Chikvashvili, D.; Villarroel, A.; Attali, B.; Lotan, I. Selective interaction of syntaxin 1A with KCNQ2: Possible implications for specific modulation of presynaptic activity. PLoS ONE 2009, 4, e6586. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z. A Common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. 2006, 26, 2599–2613. [Google Scholar] [CrossRef]
- Hoshi, N.; Zhang, J.S.; Omaki, M.; Takeuchi, T.; Yokoyama, S.; Wanaverbecq, N.; Langeberg, L.K.; Yoneda, Y.; Scott, J.D.; Brown, D.A.; et al. AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat. Neurosci. 2003, 6, 564–571. [Google Scholar] [CrossRef] [Green Version]
- Ekberg, J.; Schuetz, F.; Boase, N.A.; Conroy, S.J.; Manning, J.; Kumar, S.; Poronnik, P.; Adams, D.J. Regulation of the voltage-gated K+ channels KCNQ2/3 and KCNQ3/5 by ubiquitination: Novel role for Nedd4-2. J. Biol. Chem. 2007, 282, 12135–12142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Craciun, L.C.; Mirshahi, T.; Rohács, T.; Lopes, C.M.B.; Jin, T.; Logothetis, D.E. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 2003, 37, 963–975. [Google Scholar] [CrossRef]
- Rose, K.; Ooi, L.; Dalle, C.; Robertson, B.; Wood, I.C.; Gamper, N. Transcriptional repression of the M channel subunit Kv7.2 in chronic nerve injury. Pain 2011, 152, 742–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanaumi, T.; Takashima, S.; Iwasaki, H.; Itoh, M.; Mitsudome, A.; Hirose, S. Developmental changes in KCNQ2 and KCNQ3 expression in human brain: Possible contribution to the age-dependent etiology of benign familial neonatal convulsions. Brain Dev. 2008, 30, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Barrese, V.; Miceli, F.; Soldovieri, M.V.; Ambrosino, P.; Iannotti, F.A.; Cilio, M.R.; Taglialatela, M. Neuronal potassium channel openers in the management of epilepsy: Role and potential of retigabine. Clin. Pharm. Adv. Appl. 2010, 2, 225–236. [Google Scholar]
- Du, X.; Gao, H.; Jaffe, D.; Zhang, H.; Gamper, N. M-type K+ channels in peripheral nociceptive pathways. Br. J. Pharm. 2018, 2158–2172. [Google Scholar] [CrossRef]
- Devulder, J. Flupirtine in pain management: Pharmacological properties and clinical use. CNS Drugs 2010, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Miceli, F.; Soldovieri, M.V.; Ambrosino, P.; Manocchio, L.; Mosca, I.; Taglialatela, M. Pharmacological targeting of neuronal Kv7.2/3 channels: a focus on chemotypes and receptor sites. Curr. Med. Chem. 2018, 25, 2637–2660. [Google Scholar] [CrossRef]
- Linley, J.E.; Rose, K.; Patil, M.; Robertson, B.; Akopian, A.N.; Gamper, N. Inhibition of M current in sensory neurons by exogenous proteases: a signaling pathway mediating inflammatory nociception. J. Neurosci. 2008, 28, 11240–11249. [Google Scholar] [CrossRef]
- Liu, B.; Linley, J.E.; Du, X.; Zhang, X.; Ooi, L.; Zhang, H.; Gamper, N. The acute nociceptie signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl- channels. J. Clin. Invest. 2010, 120, 1240–1252. [Google Scholar] [CrossRef]
- Passmore, G.M.; Selyanko, A.A.; Mistry, M.; Al-Qatari, M.; Marsh, S.J.; Matthews, E.A.; Dickenson, A.H.; Brown, T.A.; Burbidge, S.A.; Main, M.; et al. KCNQ/M currents in sensory neurons: Significance for pain therapy. J. Neurosci. 2003, 23, 7227–7236. [Google Scholar] [CrossRef] [PubMed]
- Linley, J.E.; Pettinger, L.; Huang, D.; Gamper, N. M channel enhancers and physiological M channel block. J. Physiol. 2012, 590, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.M.; Fleckenstein, J.; Passmore, G.M.; Brown, D.A.; Grafe, P. Retigabine reduces the excitability of unmyelinated peripheral human axons. Neuropharmacology 2008, 54, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-F.; Han, P.; Neelands, T.R.; McGaraughty, S.; Honore, P.; Surowy, C.S.; Zhang, D. Coexpression and activation of TRPV1 suppress the activity of the KCNQ2/3 channel. J. Gen. Physiol. 2011, 138, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardín, I.; López, J.J.; Diez, R.; Sánchez-Collado, J.; Cantonero, C.; Albarrán, L.; Woodard, G.E.; Redondo, P.C.; Salido, G.M.; Smani, T.; et al. TRPs in pain sensation. Front. Physiol. 2017, 392. [Google Scholar] [CrossRef] [PubMed]
- Jow, F.; He, L.; Kramer, A.; Hinson, J.; Bowlby, M.R.; Dunlop, J.; Wang, K. Validation of DRG-like F11 cells for evaluation of KCNQ/M-channel modulators. Assay Drug Dev. Technol. 2006, 4, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, P.; Soldovieri, M.V.; Russo, C.; Taglialatela, M. Activation and desensitization of TRPV1 channels in sensory neurons by the PPARα agonist palmitoylethanolamide. Br. J. Pharm. 2013, 168, 1430–1444. [Google Scholar] [CrossRef] [PubMed]
- Wickenden, A.D.; Krajewski, J.L.; London, B.; Wagoner, P.K.; Wilson, W.A.; Clark, S.; Roeloffs, R.; McNaughton-Smith, G.; Rigdon, G.C. n-(6-Chloro-pyridin-3-yl)-3,4-difluoro-benzamide (ICA-27243): A novel, selective KCNQ2/Q3 potassium channel activator. Mol. Pharm. 2008, 73, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Bentzen, B.H.; Schmitt, N.; Calloe, K.; Dalby Brown, W.; Grunnet, M.; Olesen, S.P. The acrylamide (S)-1 differentially affects Kv7 (KCNQ) potassium channels. Neuropharmacology 2006, 51, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Zaczek, R.; Chorvat, R.J.; Saye, J.A.; Pierdomenico, M.E.; Maciag, C.M.; Logue, A.R.; Fisher, B.N.; Rominger, D.H.; Earl, R.A. Two new potent neurotransmitter release enhancers, 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone and 10,10-bis(2-fluoro-4-pyridinylmethyl)-9(10H)-anthracenone: Comparison to linopirdine. J. Pharm. Exp. Ther. 1998, 285, 724–730. [Google Scholar]
- Fan, S.F.; Shen, K.F.; Scheideler, M.A.; Crain, S.M. F11 neuroblastoma × DRG neuron hybrid cells express inhibitory μ- and δ-opioid receptors which increase voltage-dependent K+ currents upon activation. Brain Res. 1992, 590, 329–333. [Google Scholar] [CrossRef]
- Vetter, I.; Lewis, R.J. Characterization of endogenous calcium responses in neuronal cell lines. Biochem. Pharm. 2010, 79, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Francel, P.C.; Harris, K.; Smith, M.; Fishman, M.C.; Miller, R.J. Neurochemical characteristics of a novel dorsal root ganglion X neuroblastoma hybrid cell line. J. Neurochem. 1987, 1624–1631. [Google Scholar] [CrossRef] [PubMed]
- Raymon, H.K.; Thode, S.; Zhou, J.; Friedman, G.C.; Pardinas, J.R.; Barrere, C.; Johnson, R.M.; Sah, D.W. Immortalized human dorsal root ganglion cells differentiate into neurons with nociceptive properties. J. Neurosci. 1999, 19, 5420–5428. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Adams, P.R. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 1980, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, P.; Soldovieri, M.V.; De Maria, M.; Russo, C.; Taglialatela, M. Functional and biochemical interaction between PPARα receptors and TRPV1 channels: Potential role in PPARα agonists-mediated analgesia. Pharm. Res. 2014, 87, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Roeloffs, R.; Wickenden, A.D.; Crean, C.; Werness, S.; Mcnaughton-smith, G.; Stables, J.; Mcnamara, J.O.; Ghodadra, N.; Rigdon, G.C. In Vivo Profile of ICA-27243 [n-(6-Chloro-pyridin-3-yl)-3,4- Kv7.3) Activator in Rodent Anticonvulsant Models. J. Pharm. Exp. Ther. 2008, 326, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef]
- Docherty, R.J.; Yeats, J.C.; Piper, A.S. Capsazepine block of voltage-activated calcium channels in adult rat dorsal root ganglion neurones in culture. Br. J. Pharm. 1997, 121, 1461–1467. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef]
- Trimmer, J.S. Ion channels and pain: Important steps towards validating a new therapeutic target for neuropathic pain. Exp. Neurol. 2014, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Platika, D.; Boulos, M.H.; Baizer, L.; Fishman, M.C. Neuronal traits of clonal cell lines derived by fusion of dorsal root ganglia neurons with neuroblastoma cells. Proc. Natl. Acad. Sci. USA 1985, 82, 3499–3503. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.H.; Prescott, E.D.; Kong, H.; Shields, S.; Jordt, S.E.; Basbaum, A.I.; Chao, M.V.; Julius, D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 2001, 411, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Tatulian, L.; Delmas, P.; Abogadie, F.C.; Brown, D.A. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J. Neurosci. 2001, 21, 5535–5545. [Google Scholar] [CrossRef] [PubMed]
- Padilla, K.; Wickenden, A.D.; Gerlach, A.C.; McCormack, K. The KCNQ2/3 selective channel opener ICA-27243 binds to a novel voltage-sensor domain site. Neurosci. Lett. 2009, 465, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Boissard, C.G.; Greco, C.; Gribkoff, V.K.; Harden, D.G.; He, H.; L’Heureux, A.; Kang, S.H.; Kinney, G.G.; Knox, R.J.; et al. (S)-n-[1-(3-morpholin-4-ylphenyl)ethyl]-3-phenylacrylamide: an orally bioavailable KCNQ2 opener with significant activity in a cortical spreading depression model of migraine. J. Med. Chem. 2003, 46, 3197–3200. [Google Scholar] [CrossRef] [PubMed]
- Iannotti, F.A.; Panza, E.; Barrese, V.; Viggiano, D.; Soldovieri, M.V.; Taglialatela, M. Expression, localization, and pharmacological role of Kv7 potassium channels in skeletal muscle proliferation, differentiation, and survival after myotoxic insults. J. Pharm. Exp. Ther. 2010, 332, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambrosino, P.; Soldovieri, M.V.; Di Zazzo, E.; Paventi, G.; Iannotti, F.A.; Mosca, I.; Miceli, F.; Franco, C.; Canzoniero, L.M.T.; Taglialatela, M. Activation of Kv7 Potassium Channels Inhibits Intracellular Ca2+ Increases Triggered By TRPV1-Mediated Pain-Inducing Stimuli in F11 Immortalized Sensory Neurons. Int. J. Mol. Sci. 2019, 20, 4322. https://doi.org/10.3390/ijms20184322
Ambrosino P, Soldovieri MV, Di Zazzo E, Paventi G, Iannotti FA, Mosca I, Miceli F, Franco C, Canzoniero LMT, Taglialatela M. Activation of Kv7 Potassium Channels Inhibits Intracellular Ca2+ Increases Triggered By TRPV1-Mediated Pain-Inducing Stimuli in F11 Immortalized Sensory Neurons. International Journal of Molecular Sciences. 2019; 20(18):4322. https://doi.org/10.3390/ijms20184322
Chicago/Turabian StyleAmbrosino, Paolo, Maria Virginia Soldovieri, Erika Di Zazzo, Gianluca Paventi, Fabio Arturo Iannotti, Ilaria Mosca, Francesco Miceli, Cristina Franco, Lorella Maria Teresa Canzoniero, and Maurizio Taglialatela. 2019. "Activation of Kv7 Potassium Channels Inhibits Intracellular Ca2+ Increases Triggered By TRPV1-Mediated Pain-Inducing Stimuli in F11 Immortalized Sensory Neurons" International Journal of Molecular Sciences 20, no. 18: 4322. https://doi.org/10.3390/ijms20184322
APA StyleAmbrosino, P., Soldovieri, M. V., Di Zazzo, E., Paventi, G., Iannotti, F. A., Mosca, I., Miceli, F., Franco, C., Canzoniero, L. M. T., & Taglialatela, M. (2019). Activation of Kv7 Potassium Channels Inhibits Intracellular Ca2+ Increases Triggered By TRPV1-Mediated Pain-Inducing Stimuli in F11 Immortalized Sensory Neurons. International Journal of Molecular Sciences, 20(18), 4322. https://doi.org/10.3390/ijms20184322