Growth Hormone Secretagogues and the Regulation of Calcium Signaling in Muscle

 ,
,  , ,
, ,
Abstract
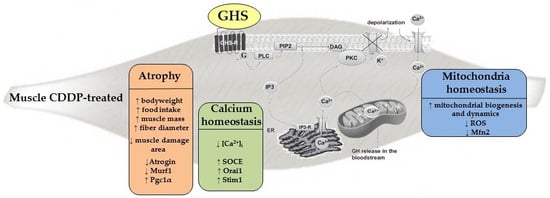
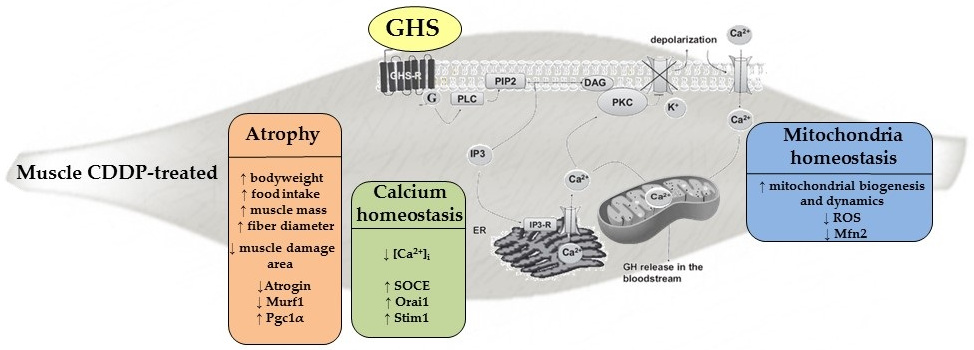
:
1. Introduction
2. Growth Hormone Secretagogues (GHS)
2.1. GHS Discovery and State of Art
2.2. GHS Effects on Muscle
2.2.1. GHS Actions on Cardiac Muscle
2.2.2. GHS Actions on Skeletal Muscle
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATP | adenosine triphosphate |
| Ca2+ | calcium |
| CDDP | cisplatin (cis-dichlorodiamminoplatinum) |
| CRU | Ca2+ release unit |
| D-AG | des-acyl ghrelin |
| DAG | diacylglycerol |
| DHPR | dihydropyridine receptor |
| EDL | extensor digitorum longus muscle |
| GH | growth hormone |
| GHRH | growth hormone-releasing hormone |
| GPCR | G protein-coupled receptor |
| GHRP | growth hormone-releasing peptides |
| GHS | growth hormone secretagogues |
| GHS-R1a | GHS receptor type 1a |
| GHS-R1b | GHS receptor type 1b |
| GOAT | ghrelin O-acyl-transferase |
| ICaL- INa | inward current |
| IGF-I | insulin-like growth factor-I |
| IP | inositol phosphate |
| IP3 | inositol triphosphate |
| IR | ischemia/reperfusion |
| Ito | transient outward current |
| LBM | lean body mass |
| MBOAT | membrane- bound O-acyltrasferase |
| Mfn2 | mitofusin2 |
| Murf-1 | muscle RING-finger protein-1 |
| NO | nitric oxide |
| Orai1 | calcium release-activated calcium modulator 1 |
| PGC-1α | proliferator-activated receptor γ coactivator-1α |
| PNL | phospholamban |
| ROS | reactive oxygen species |
| RyR | ryanodine receptors |
| SERCA | Sarco-Endoplasmic Reticulum Calcium ATPase |
| SERCA2a | sarcoplasmic reticulum Ca2+ loading ability |
| SOCE | store-operated Ca2+ entry |
| Stim1 | stromal interaction molecule |
| Trp | tryptophan |
| TZP-101 | Ulimorelin |
| UAC | unacylated ghrelin |
References
- Gehlert, S.; Bloch, W.; Suhr, F. Ca2+-dependent regulations and signaling in skeletal muscle: From electro-mechanical coupling to adaptation. Int. J. Mol. Sci. 2015, 16, 1066–1095. [Google Scholar] [CrossRef]
- Missiaen, L.; Robberecht, W.; van den Bosch, L.; Callewaert, G.; Parys, JB.; Wuytack, F.; Raeymaekers, L.; Nilius, B.; Eggermont, J.; De Smedt, H. Abnormal intracellular Ca2+ homeostasis and disease. Cell Calcium 2000, 28, 1–21. [Google Scholar] [CrossRef]
- Agrawal, A.; Suryakumar, G.; Rathor, R. Role of defective Ca2+ signaling in skeletal muscle weakness: Pharmacological implications. J. Cell Commun Signal 2018, 12, 645–659. [Google Scholar] [CrossRef]
- Bowers, C.Y. Growth hormone-releasing peptide (GHRP). Cell Mol. Life Sci. 1998, 54, 1316–1329. [Google Scholar] [CrossRef]
- Arvat, E.; Broglio, F.; Aimaretti, G.; Benso, A.; Giordano, R.; Deghenghi, R.; Ghigo, E. Ghrelin and synthetic GH secretagogues. Best Pract. Res. Clin. Endocrinol. Metab. 2002, 116, 505–517. [Google Scholar] [CrossRef]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef]
- Alamri, B.N.; Shin, K.; Chappe, V.; Anini, Y. The role of ghrelin in the regulation of glucose homeostasis. Mol. Biol. Clin. Investig. 2016, 26, 3–11. [Google Scholar] [CrossRef]
- Pradhan, G.; Samson, S.L.; Sun, Y. Ghrelin: Much more than a hunger hormone. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 619–624. [Google Scholar] [CrossRef]
- Chang, L.; Niu, F.; Chen, J.; Cao, X.; Liu, Z.; Bao, X.; Xu, Y. Ghrelin improves muscle function in dystrophin-deficient mdx mice by inhibiting NLRP3 inflammasome activation. Life Sci. 2019, 232, 116654. [Google Scholar] [CrossRef]
- Lucchi, C.; Curia, G.; Vinet, J.; Gualtieri, F.; Bresciani, E.; Locatelli, V.; Torsello, A.; Biagini, G. Protective but not anticonvulsant effects of ghrelin and JMV-1843 in the pilocarpine model of Status epilepticus. PLoS ONE 2013, 88, e72716. [Google Scholar] [CrossRef]
- Seminara, R.S.; Jeet, C.; Biswas, S.; Kanwal, B.; Iftikhar, W.; Sakibuzzaman, M.; Rutkofsky, I.H. The Neurocognitive Effects of Ghrelin-induced Signaling on the Hippocampus: A Promising Approach to Alzheimer’s Disease. Cureus 2018, 10, e3285. [Google Scholar] [CrossRef]
- Müller, D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 44, 437–460. [Google Scholar] [CrossRef]
- Howard, A.D.; Feighner, S.D.; Cully, D.F.; Arena, J.P.; Liberator, P.A.; Rosenblum, C.I.; Hamelin, M.; Hreniuk, DL.; Palyha, O.C.; Anderson, J.; et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science 1996, 273, 974–977. [Google Scholar] [CrossRef]
- Torsello, A.; Rossoni, G.; Locatelli, V.; De Gennaro Colonna, V.; Bernareggi, M.; Francolini, M.; Müller, E.E.; Berti, F. Hexarelin, but not growth hormone, protects heart from damage induced in vitro by calcium deprivation replenishment. Endocrine 2001, 14, 109–112. [Google Scholar] [CrossRef]
- Torsello, A.; Bresciani, E.; Rossoni, G.; Avallone, R.; Tulipano, G.; Cocchi, D.; Bulgarelli, I.; Deghenghi, R.; Berti, F.; Locatelli, V. Ghrelin plays a minor role in the physiological control of cardiac function in the rat. Endocrinology 2003, 144, 1787–1792. [Google Scholar] [CrossRef]
- Xu, X.B.; Pang, J.J.; Cao, J.M.; Ni, C.; Xu, R.K.; Peng, X.Z.; Yu, X.X.; Guo, S.; Chen, M.C.; Chen, C. GH-releasing peptides improve cardiac dysfunction and cachexia and suppress stress-related hormones and cardiomyocyte apoptosis in rats with heart failure. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, 1643–1651. [Google Scholar] [CrossRef]
- Isgaard, J.; Granata, R. Ghrelin in cardiovascular disease and atherogenesis. Mol. Cell Endocrinol. 2011, 340, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Zhang, L.; Launikonis, B.S.; Chen, C. Growth hormone secretagogues preserve the electrophysiological properties of mouse cardiomyocytes isolated from in vitro ischemia/reperfusion heart. Endocrinology 2012, 153, 5480–5490. [Google Scholar] [CrossRef]
- McDonald, H.; Peart, J.; Kurniawan, N.; Galloway, G.; Royce, S.; Samuel, C.; Chen, C. Hexarelin treatment preserves myocardial function and reduces cardiac fibrosis in a mouse model of acute myocardial infarction. Physiol. Rep. 2018, 66, e13699. [Google Scholar] [CrossRef]
- Broglio, F.; Guarracino, F.; Benso, A.; Gottero, C.; Prodam, F.; Granata, R.; Avogadri, E.; Muccioli, G.; Deghenghi, R.; Ghigo, E. Effects of acute hexarelin administration on cardiac performance in patients with coronary artery disease during by-pass surgery. Eur. J. Pharmacol. 2002, 448, 193–200. [Google Scholar] [CrossRef]
- Imazio, M.; Bobbio, M.; Broglio, F.; Benso, A.; Podio, V.; Valetto, M.R.; Bisi, G.; Ghigo, E.; Trevi, G.P. GH-independent cardiotropic activities of hexarelin in patients with severe left ventricular dysfunction due to dilated and ischemic cardiomyopathy. Eur. J. Heart Fail 2002, 44, 185–191. [Google Scholar] [CrossRef]
- Bresciani, E.; Rizzi, L.; Molteni, L.; Ravelli, M.; Liantonio, A.; Ben Haj Salah, K.; Fehrentz, J.A.; Martinez, J.; Omeljaniuk, RJ.; Biagini, G.; et al. JMV2894, a novel growth hormone secretagogue, accelerates body mass recovery in an experimental model of cachexia. Endocrine 2017, 58, 106–114. [Google Scholar] [CrossRef]
- Conte, E.; Camerino, G.M.; Mele, A.; De Bellis, M.; Pierno, S.; Rana, F.; Fonzino, A.; Caloiero, R.; Rizzi, L.; Bresciani, E.; et al. Growth hormone secretagogues prevent dysregulation of skeletal muscle calcium homeostasis in a rat model of cisplatin-induced cachexia. J. Cachexia Sarcopenia Muscle 2017, 88, 386–404. [Google Scholar] [CrossRef]
- Virdis, A.; Lerman, L.O.; Regoli, F.; Ghiadoni, L.; Lerman, A.; Taddei, S. Human Ghrelin: A Gastric Hormone with Cardiovascular Properties. Curr. Pharm. Des. 2016, 22, 52–58. [Google Scholar] [CrossRef]
- Chen, J.; Splenser, A.; Guillory, B.; Luo, J.; Mendiratta, M.; Belinova, B.; Halder, T.; Zhang, G.; Li, Y.P.; Garcia, J.M. Ghrelin prevents tumour- and cisplatin-induced muscle wasting: Characterization of multiple mechanisms involved. J. Cachexia Sarcopenia Muscle 2015, 66, 132–143. [Google Scholar] [CrossRef]
- Rivier, J.; Spiess, J.; Thorner, M.O.; Vale, W. Characterization of a growth hormone-releasing factor from a human pancreatic islet tumor. Nature 1982, 300, 276–278. [Google Scholar] [CrossRef]
- Bowers, C.Y.; Momany, F.A.; Reynolds, G.A.; Hong, A. On the in vitro and in vivo activity of a new synthetic hexapeptide that acts on the pituitary to specifically release growth hormone. Endocrinology 1984, 114, 1537–1545. [Google Scholar] [CrossRef]
- Gaylinn, B.D.; Harrison, J.K.; Zysk, J.R.; Lyons, C.E.; Lynch, K.R.; Thorner, M.O. Molecular cloning and expression of a human anterior pituitary receptor for growth hormone-releasing hormone. Mol. Endocrinol. 1993, 7, 77–84. [Google Scholar]
- Cheng, K.; Chan, W.W.S.; Barreto, A.; Convey, E.M.; Smith, R.G. The synergistic effects of His-D-Trp-Ala-Trp-D-Phe-Lys-NH2 on growth hormone (GH)-releasing factor-stimulated GH release and intracellular adenosine 3’,5’-monophosphate accumulation in rat primary pituitary cell culture. Endocrinology 1989, 124, 2791–2798. [Google Scholar] [CrossRef]
- Bowers, C.Y.; Sartor, A.O.; Reynolds, G.A.; Badger, T.M. On the actions of the growth hormone-releasing hexapeptide, GHRP. Endocrinology 1991, 128, 2027–2035. [Google Scholar] [CrossRef]
- Massoud, A.F.; Hindmarsh, P.C.; Matthews, D.R.; Brook, C.G. The effect of repeated administration of hexarelin, a growth hormone releasing peptide, and growth hormone releasing hormone on growth hormone responsivity. Clin. Endocrinol. 1996, 44, 555–562. [Google Scholar] [CrossRef]
- Smith, R.G. Development of growth hormone secretagogues. Endocr. Rev. 2005, 26, 346–360. [Google Scholar] [CrossRef]
- Huhn, W.C.; Hartman, M.L.; Pezzoli, S.S.; Thorner, M.O. Twenty-four-hour growth hormone (GH)-releasing peptide (GHRP) infusion enhances pulsatile GH secretion and specifically attenuates the response to a subsequent GHRP bolus. J. Clin. Endocrinol. Metab. 1993, 76, 1202–1208. [Google Scholar]
- Navarro, G.; Aguinaga, D.; Angelats, E.; Medrano, M.; Moreno, E.; Mallol, J.; Cortés, A.; Canela, EI.; Casadó, V.; McCormick, P.J.; et al. A Significant Role of the Truncated Ghrelin Receptor GHS-R1b in Ghrelin-induced Signaling in Neurons. J. Biol. Chem. 2016, 291, 13048–13062. [Google Scholar] [CrossRef] [Green Version]
- Delhanty, P.J.; Neggers, S.J.; van der Lely, A.J. Should we consider des-acyl ghrelin as a separate hormone and if so, what does it do? Front. Horm. Res. 2014, 42, 163–174. [Google Scholar]
- Currow, D.C.; Maddocks, M.; Cella, D.; Muscaritoli, M. Efficacy of Anamorelin, a Novel Non-Peptide Ghrelin Analogue, in Patients with Advanced Non-Small Cell Lung Cancer (NSCLC) and Cachexia-Review and Expert Opinion. Int. J. Mol. Sci. 2018, 19, 3471. [Google Scholar] [CrossRef]
- Shaw, M.; Pediconi, C.; McVey, D.; Mondou, E.; Quinn, J.; Chamblin, B.; Rousseau, F. Safety and efficacy of ulimorelin administered postoperatively to accelerate recovery of gastrointestinal motility following partial bowel resection: Results of two randomized, placebo-controlled phase 3 trials. Dis. Colon. Rectum. 2013, 56, 888–897. [Google Scholar] [CrossRef]
- Lee, M.R.; Tapocik, J.D.; Ghareeb, M.; Schwandt, M.L.; Dias, A.A.; Le, A.N.; Cobbina, E.; Farinelli, L.A.; Bouhlal, S.; Farokhnia, M.; et al. The novel ghrelin receptor inverse agonist PF-5190457 administered with alcohol: Preclinical safety experiments and a phase 1b human laboratory study. Mol. Psychiatry 2018, 1. [Google Scholar] [CrossRef]
- Stangl, M.K.; Böcker, W.; Chubanov, V.; Ferrari, U.; Fischereder, M.; Gudermann, T.; Hesse, E.; Meinke, P.; Reincke, M.; Reisch, N.; et al. Sarcopenia-Endocrinological and Neurological Aspects. Exp. Clin. Endocrinol. Diabetes 2019, 127, 8–22. [Google Scholar]
- Martin, L.; Birdsell, L.; Macdonald, N.; Reiman, T.; Clandinin, M.T.; McCargar, L.J.; Murphy, R.; Ghosh, S.; Sawyer, MB.; Baracos, VE. Cancer cachexia in the age of obesity: Skeletal muscle depletion is a powerful prognostic factor, independent of body mass index. J. Clin. Oncol. 2013, 31, 1539–1547. [Google Scholar] [CrossRef]
- Sun, Q.; Ma, Y.; Zhang, L.; Zhao, Y.F.; Zang, W.J.; Chen, C. Effects of GH secretagogues on contractility and Ca2+ homeostasis of isolated adult rat ventricular myocytes. Endocrinology 2010, 151, 4446–4454. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, L.; Edwards, J.N.; Launikonis, B.S.; Chen, C. Growth hormone secretagogues protect mouse cardiomyocytes from in vitro ischemia/reperfusion injury through regulation of intracellular calcium. PLoS ONE 2012, 77, e35265. [Google Scholar] [CrossRef]
- Shimokawa, H.; Yasuda, S. Myocardial ischemia: Current concepts and future perspectives. J. Cardiol. 2008, 52, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; He, G.W.; Underwood, M.J.; Yu, C.M. Cellular and molecular mechanisms of endothelial ischemia/reperfusion injury: Perspectives and implications for postischemic myocardial protection. Am. J. Transl. Res. 2016, 88, 765–777. [Google Scholar]
- Bisi, G.; Podio, V.; Valetto, M.R.; Broglio, F.; Bertuccio, G.; Del Rio, G.; Arvat, E.; Boghen, M.F.; Deghenghi, R.; Muccioli, G.; et al. Acute cardiovascular and hormonal effects of GH and hexarelin, a synthetic GH-releasing peptide, in humans. J. Endocrinol. Invest. 1999, 22, 266–272. [Google Scholar] [CrossRef]
- Mao, Y.; Tokudome, T.; Kishimoto, I. The cardiovascular action of hexarelin. J. Geriatr. Cardiol. 2014, 11, 253–258. [Google Scholar]
- Pang, J.J.; Xu, R.K.; Xu, X.B.; Cao, J.M.; Ni, C.; Zhu, W.L.; Asotra, K.; Chen, M.C.; Chen, C. Hexarelin protects rat cardiomyocytes from angiotensin II-induced apoptosis in vitro. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1063–H1069. [Google Scholar] [CrossRef] [Green Version]
- Filigheddu, N.; Fubini, A.; Baldanzi, G.; Cutrupi, S.; Ghè, C.; Catapano, F.; Broglio, F.; Bosia, A.; Papotti, M.; Muccioli, G.; et al. Hexarelin protects H9c2 cardiomyocytes from doxorubicin-induced cell death. Endocrine 2001, 14, 113–119. [Google Scholar] [CrossRef]
- De Gennaro-Colonna, V.; Rossoni, G.; Cocchi, D.; Rigamonti, A.E.; Berti, F.; Muller, E.E. Endocrine, metabolic and cardioprotective effects of hexarelin in obese Zucker rats. J. Endocrinol. 2000, 166, 529–536. [Google Scholar] [CrossRef] [Green Version]
- Berti, F.; Müller, E.; De Gennaro Colonna, V.; Rossoni, G. Hexarelin exhibits protective activity against cardiac ischaemia in hearts from growth hormone-deficient rats. Growth Horm. IGF Res. 1998, 8, 149–152. [Google Scholar] [CrossRef]
- Xu, X.; Ding, F.; Pang, J.; Gao, X.; Xu, R.K.; Hao, W.; Cao, J.M.; Chen, C. Chronic administration of hexarelin attenuates cardiac fibrosis in the spontaneously hypertensive rat. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H703–H711. [Google Scholar] [CrossRef] [Green Version]
- Mosa, R.M.; Zhang, Z.; Shao, R.; Deng, C.; Chen, J.; Chen, C. Implications of ghrelin and hexarelin in diabetes and diabetes-associated heart diseases. Endocrine 2015, 49, 307–323. [Google Scholar] [CrossRef] [Green Version]
- Avallone, R.; Demers, A.; Rodrigue-Way, A.; Bujold, K.; Harb, D.; Anghel, S.; Wahli, W.; Marleau, S.; Ong, H.; Tremblay, A. A growth hormone-releasing peptide that binds scavenger receptor CD36 and ghrelin receptor up-regulates sterol transporters and cholesterol efflux in macrophages through a peroxisome proliferator-activated receptor gamma-dependent pathway. Mol. Endocrinol. 2006, 20, 3165–3178. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003, 44, 566–577. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef]
- Sakuma, K.; Aoi, W.; Yamaguchi, A. Molecular mechanism of sarcopenia and cachexia: Recent research advances. Pflug. Arch. 2017, 469, 573–591. [Google Scholar] [CrossRef]
- Mantovani, G.; Madeddu, C. Cancer cachexia: Medical management. Support Care Cancer 2010, 18, 1–9. [Google Scholar] [CrossRef]
- Madeddu, C.; Mantovani, G.; Gramignano, G.; Astara, G.; Macciò, A. Muscle wasting as main evidence of energy impairment in cancer cachexia: Future therapeutic approaches. Future Oncol. 2015, 11, 2697–2710. [Google Scholar] [CrossRef]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer. 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Colldén, G.; Tschöp, M.H.; Müller, T.D. Therapeutic Potential of Targeting the Ghrelin Pathway. Int. J. Mol. Sci. 2017, 18, 798. [Google Scholar] [CrossRef]
- Le Bricon, T.; Gugins, S.; Cynober, L.; Baracos, V.E. Negative impact of cancer chemotherapy on protein metabolism in healthy and tumor-bearing rats. Metabolism 1995, 44, 1340–1348. [Google Scholar] [CrossRef]
- Torsello, A.; Luoni, M.; Schweiger, F.; Grilli, R.; Guidi, M.; Bresciani, E.; Deghenghi, R.; Müller, EE.; Locatelli, V. Novel hexarelin analogs stimulate feeding in the rat through a mechanism not involving growth hormone release. Eur. J. Pharmacol. 1998, 360, 123–129. [Google Scholar] [CrossRef]
- Liantonio, A.; Gramegna, G.; Carbonara, G.; Sblendorio, V.T.; Pierno, S.; Fraysse, B.; Giannuzzi, V.; Rizzi, L.; Torsello, A.; Camerino, D.C. Growth hormone secretagogues exert differential effects on skeletal muscle calcium homeostasis in male rats depending on the peptidyl/nonpeptidyl structure. Endocrinology 2013, 154, 3764–3775. [Google Scholar] [CrossRef]
- Moulin, A.; Demange, L.; Bergé, G.; Gagne, D.; Ryan, J.; Mousseaux, D.; Heitz, A.; Perrissoud, D.; Locatelli, V.; Torsello, A.; et al. Toward potent ghrelin receptor ligands based on trisubstituted 1,2,4-triazole structure. 2. Synthesis and pharmacological in vitro and in vivo evaluations. J. Med. Chem. 2007, 50, 5790–5806. [Google Scholar] [CrossRef]
- Demange, L.; Boeglin, D.; Moulin, A.; Mousseaux, D.; Ryan, J.; Bergé, G.; Gagne, D.; Heitz, A.; Perrissoud, D.; Locatelli, V.; et al. Synthesis and pharmacological in vitro and in vivo evaluations of novel triazole derivatives as ligands of the ghrelin receptor. J. Med. Chem. 2007, 50, 1939–1957. [Google Scholar] [CrossRef]
- Marks, A.R. Calcium cycling proteins and heart failure: Mechanisms and therapeutics. J. Clin. Invest. 2013, 123, 46–52. [Google Scholar] [CrossRef]
- Bellinger, A.M.; Reiken, S.; Dura, M.; Murphy, P.W.; Deng, S.X.; Landry, D.W.; Nieman, D.; Lehnart, S.E.; Samaru, M.; LaCampagne, A.; et al. Remodeling of ryanodine receptor complex causes “leaky” channels: A molecular mechanism for decreased exercise capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 2198–2202. [Google Scholar] [CrossRef]
- Andersson, D.C.; Betzenhauser, M.J.; Reiken, S.; Meli, A.C.; Umanskaya, A.; Xie, W.; Shiomi, T.; Zalk, R.; Lacampagne, A.; Marks, A.R. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011, 14, 196–207. [Google Scholar] [CrossRef]
- Sirago, G.; Conte, E.; Fracasso, F.; Cormio, A.; Fehrentz, J.A.; Martinez, J.; Musicco, C.; Camerino, G.M.; Fonzino, A.; Rizzi, L.; et al. Growth hormone secretagogues hexarelin and JMV2894 protect skeletal muscle from mitochondrial damages in a rat model of cisplatin-induced cachexia. Sci. Rep. 2017, 7, 13017. [Google Scholar] [CrossRef]
- Romanello, V.; Guadagnin, E.; Gomes, L.; Roder, I.; Sandri, C.; Petersen, Y.; Milan, G.; Masiero, E.; Del Piccolo, P.; Foretz, M.; et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010, 29, 1774–1785. [Google Scholar] [CrossRef]
- Fontes-Oliveira, C.C.; Busquets, S.; Toledo, M.; Penna, F.; Paz Aylwin, M.; Sirisi, S.; Silva, A.P.; Orpí, M.; García, A.; Sette, A.; et al. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: Altered energetic efficiency? Biochim. Biophys. Acta 2013, 1830, 2770–2778. [Google Scholar] [CrossRef]
- Rossi, A.E.; Boncompagni, S.; Dirksen, R.T. Sarcoplasmic Reticulum-Mitochondrial Symbiosis: Bidirectional Signaling in Skeletal Muscle. Exerc. Sport Sci. Rev. 2009, 37, 29–35. [Google Scholar] [CrossRef]
- Pietrangelo, L.; D’Incecco, A.; Ainbinder, A.; Michelucci, A.; Kern, H.; Dirksen, R.T.; Boncompagni, S.; Protasi, F. Age-dependent uncoupling of mitochondria from Ca2+ release units in skeletal muscle. Oncotarget 2015, 6, 35358–35371. [Google Scholar] [CrossRef]
- Ainbinder, A.; Boncompagni, S.; Protasi, F.; Dirksen, R.T. Role of Mitofusin-2 in mitochondrial localization and calcium uptake in skeletal muscle. Cell Calcium 2015, 57, 14–24. [Google Scholar] [CrossRef]
- Argilés, J.M.; López-Soriano, F.X.; Stemmler, B.; Busquets, S. Therapeutic strategies against cancer cachexia. Eur. J. Transl. Myol. 2019, 29, 7960. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-García, V.; López-Briz, E.; Carbonell-Sanchis, R.; Bort-Martí, S.; Gonzálvez-Perales, J.L. Megestrol acetate for cachexia–anorexia syndrome. A systematic review. J. Cachexia Sarcopenia Muscle 2018, 9, 444–452. [Google Scholar] [CrossRef]

{kind=link}
| GHS | Human Physiological Effects |
|---|---|
| Macrilen (JMV1843) (Macimorelin) | FDA approved for diagnostic test for adult GH deficiency (AGHD). |
| Anamorelin | Phase 3 clinical trial in on small cell lung cancer: increased lean body mass and bodyweight; improved some symptoms of anorexia/cachexia. |
| Ulimorelin (TZP-101) | Phase 3 clinical trial in diabetic patients: accelerated gastric emptying and improvd upper gastrointestinal symptoms. |
| PF-5190457 | Phase 1b clinical trial in heavy drinkers: reduced alcohol-primed craving behavior. |
| MK-0677 – IBUTAMOREN MESYLATE (L-163,191) | Catabolic states: slowed nitrogen wasting and increased fat free mass and bodyweight. GH-deficient adults and hemodialysis patients: increased GH, IGF-1 and prolactin levels throughout the night and ACTH and cortisol levels during the first half of the night. |
| ARD-07 (EP01572) | Healthy male volunteers: increased GH and IGF-1 release. |
| Hexarelin | Healthy male volunteers and GH-deficient subject: increased GH and IGF-1 secretion; stimulated food intake. Patients with cardiac dysfunction: cardioprotective effects. |
| GHRP-2 | GH-deficient subject: increased GH and IGF-1 secretion. Healthy male volunteers and obese subjects: stimulated food intake. Anorexia nervosa patients: improved bodyweight and hypoglycemia. |
| EP80317 | Preclinical studies: anti-atherosclerotic activity; anticonvulsivant activity in rat models of status epilepticus; in vitro ACE-inhibiting activity |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bresciani, E.; Rizzi, L.; Coco, S.; Molteni, L.; Meanti, R.; Locatelli, V.; Torsello, A. Growth Hormone Secretagogues and the Regulation of Calcium Signaling in Muscle. Int. J. Mol. Sci. 2019, 20, 4361. https://doi.org/10.3390/ijms20184361
Bresciani E, Rizzi L, Coco S, Molteni L, Meanti R, Locatelli V, Torsello A. Growth Hormone Secretagogues and the Regulation of Calcium Signaling in Muscle. International Journal of Molecular Sciences. 2019; 20(18):4361. https://doi.org/10.3390/ijms20184361
Chicago/Turabian StyleBresciani, Elena, Laura Rizzi, Silvia Coco, Laura Molteni, Ramona Meanti, Vittorio Locatelli, and Antonio Torsello. 2019. "Growth Hormone Secretagogues and the Regulation of Calcium Signaling in Muscle" International Journal of Molecular Sciences 20, no. 18: 4361. https://doi.org/10.3390/ijms20184361
APA StyleBresciani, E., Rizzi, L., Coco, S., Molteni, L., Meanti, R., Locatelli, V., & Torsello, A. (2019). Growth Hormone Secretagogues and the Regulation of Calcium Signaling in Muscle. International Journal of Molecular Sciences, 20(18), 4361. https://doi.org/10.3390/ijms20184361