Gentamicin Targets Acid Sphingomyelinase in Cancer: The Case of the Human Gastric Cancer NCI-N87 Cells


 ,
,  , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
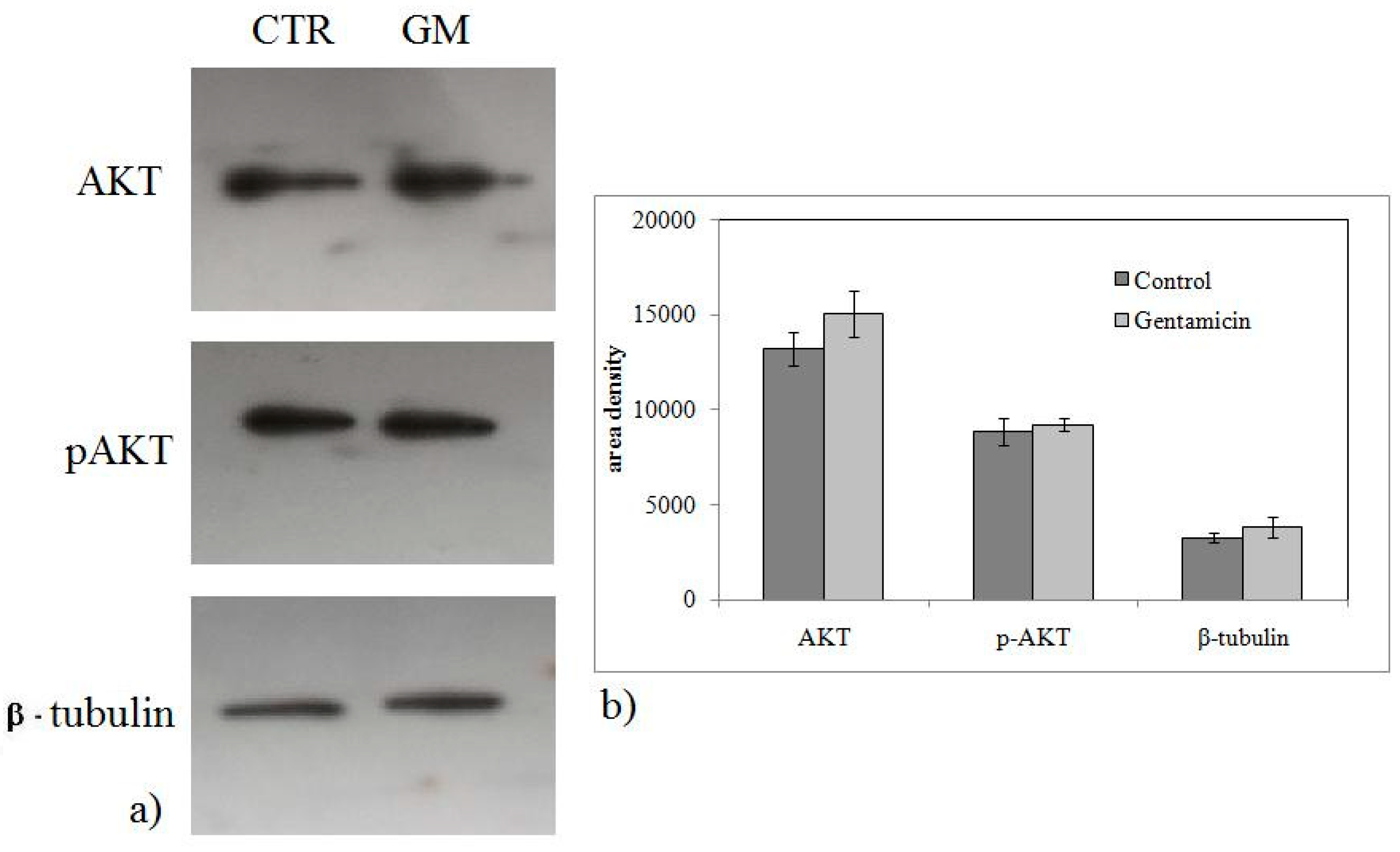{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Advances in Anticancer Action of Gentamicin
2.2. Gentamicin Alters Sphingomyelin Metabolism
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. GM Dose-Dependent Effect
4.4. Flow Cytometry Analysis
4.5. Morphological and Immunohistochemistry Analysis
4.6. Reverse Transcription Quantitative PCR (RTqPCR)
4.7. Western Blot
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Testi, R. Sphingomyelin breakdown and cell fate. Trends Biochem. Sci. 1996, 21, 468–471. [Google Scholar] [CrossRef]
- Albi, E. Role of intranuclear lipids in health and disease. Clin. Lipidol. 2011, 6, 59–69. [Google Scholar] [CrossRef]
- Canals, D.; Perry, D.M.; Jenkins, R.W.; Hannun, Y.A. Drug targeting of sphingolipid metabolism: Sphingomyelinases and ceramidases. Br. J. Pharmacol. 2011, 163, 694–712. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, N.; Luberto, C.; Hannun, Y.A. Biochemical properties of mammalian neutral sphingomyelinase 2 and its role in sphingolipid metabolism. J. Biol. Chem. 2003, 278, 13775–13783. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Murakami, M.; Furuhata, A.; Gao, S.; Yoshida, K.; Sobue, S.; Hagiwara, K.; Takagi, A.; Kojima, T.; Suzuki, M.; et al. Transcriptional regulation of neutral sphingomyelinase 2 gene expression of a human breast cancer cell line, MCF-7, induced by the anti-cancer drug, daunorubicin. Biochim. Biophys. Acta 2009, 1789, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Karakashian, A.A.; Giltiay, N.V.; Smith, G.M.; Nikolova-Karakashian, M.N. Expression of neutral sphingomyelinase-2 (NSMase-2) in primary rat hepatocytes modulates IL-beta-induced JNK activation. FASEB J. 2004, 18, 968–970. [Google Scholar] [CrossRef]
- Zhong, L.; Kong, J.N.; Dinkins, M.B.; Leanhart, S.; Zhu, Z.; Spassieva, S.D.; Qin, H.; Lin, H.P.; Elsherbini, A.; Wang, R.; et al. Increased liver tumor formation in neutral sphingomyelinase-2-deficient mice. J. Lipid Res. 2018, 59, 795–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.J.; Okimoto, R.A.; Purton, L.E.; Goodwin, M.; Haserlat, S.M.; Dayyani, F.; Sweetser, D.A.; McClatchey, A.I.; Bernard, O.A.; Look, A.T.; et al. Mutations in the neutral sphingomyelinase gene SMPD3 implicate the ceramide pathway in human leukemia. Blood 2008, 111, 4716–4722. [Google Scholar] [CrossRef]
- Savic, R.; He, X.; Fiel, I.; Schuchman, E.H. Recombinant human acid sphingomyelinase as an adjuvant to sorafenib treatment of experimental liver cancer. PLoS ONE 2013, 8, e65620. [Google Scholar] [CrossRef]
- Cervia, D.; Assi, E.; De Palma, C.; Giovarelli, M.; Bizzozero, L.; Pambianco, S.; Di Renzo, I.; Zecchini, S.; Moscheni, C.; Vantaggiato, C.; et al. Essential role for acid sphingomyelinase-inhibited autophagy in melanoma response tocisplatin. Oncotarget 2016, 7, 24995–25009. [Google Scholar] [CrossRef]
- Perrotta, C.; Cervia, D.; De Palma, C.; Assi, E.; Pellegrino, P.; Bassi, M.T.; Clementi, E. The emerging role of acid sphingomyelinase in autophagy. Apoptosis 2015, 20, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, Y.; Chow, C.S. Pseudouridine modifications influence binding of aminoglycosides to helix 69 ofbacterialribosomes. Org. Biomol. Chem. 2017, 15, 8535–8543. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Q.X.; Xie, X.F.; Ao, Y.; Tie, C.R.; Song, R.J. Differential roles of dihydropyridine calcium antagonist nifedipine, nitrendipine and amlodipine on gentamicin-induced renal tubular toxicity in rats. Eur. J. Pharmacol. 2009, 620, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.R.; Diamantakos, E.A.; Peuler, J.D.; Lamar, P.C.; Prozialeck, W.C. A novel method for the evaluation of proximal tubule epithelial cellular necrosis in the intact rat kidney using ethidium homodimer. BMC Physiol. 2007, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Pessoa, E.A.; Convento, M.B.; Silva, R.G.; Oliveira, A.S.; Borges, F.T.; Schor, N. Gentamicin-induced preconditioning of proximal tubular LLC-PK1 cells stimulates nitric oxide production but not the synthesis of heat shock protein. Braz. J. Med. Biol. Res. 2009, 42, 614–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, P.; Chatterjee, S. Effects of gentamicin on sphingomyelinase activity in cultured human renal proximal tubular cells. J. Biol. Chem. 1987, 262, 12550–12556. [Google Scholar] [PubMed]
- Codini, M.; Cataldi, S.; Ambesi-Impiombato, F.S.; Lazzarini, A.; Floridi, A.; Lazzarini, R.; Curcio, F.; Beccari TAlbi, E. Gentamicin arrests cancer cell growth: The intriguing involvement of nuclear sphingomyelin metabolism. Int. J. Mol. Sci. 2015, 16, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Cuccarese, M.F.; Singh, A.; Amiji, M.; O’Doherty, G.A. A novel use of gentamicin in the ROS-mediated sensitization of NCI-H460 lung cancer cells to various anticancer agents. ACS Chem. Biol. 2013, 8, 2771–2777. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, L.; Bernardini, I.; Pacifico, N.; Peverini, M.; Damaskopoulou, E.; Cataldi, S.; Albi, E. Severe hypocholesterolaemia is often neglected in haematological malignancies. Eur. J. Cancer 2010, 46, 1735–1743. [Google Scholar] [CrossRef]
- Patria, F.F.; Ceccarini, M.R.; Codini, M.; Conte, C.; Perioli, L.; Beccari, T.; Albi, E. A Role for Neutral Sphingomyelinase in Wound Healing Induced by Keratinocyte Proliferation upon 1α, 25-Dihydroxyvitamin D3 Treatment. Int. J. Mol. Sci. 2019, 20, 3634. [Google Scholar] [CrossRef]
- Habberstad, A.H.; Gulati, S.; Torp, S.H. Evaluation of the proliferation markersKi-67/MIB-1, mitosin, survivin, pHH3, and DNA topoisomerase IIa in human anaplastic astrocytomase an immunohistochemical study. Diagn. Pathol. 2011, 6, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Kim, S.; Lee, S.; Jiang, H.L.; Kim, S.B.; Hong, S.H.; Cho, M.H. Knockdown of Importin 7 Inhibits Lung Tumorigenesis in K-rasLA1 Lung Cancer Mice. Anticancer Res. 2017, 37, 2381–2386. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, H.P.; Kim, Y.J.; Oh, D.Y.; Im, S.A.; Lee, D.; Jong, H.S.; Kim, T.Y.; Bang, Y.J. Trastuzumab inhibits the growth of human gastric cancer cell lines with HER2 amplification synergistically with cisplatin. Int. J. Oncol. 2008, 32, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Lordick, F.; Kang, Y.K.; Chung, H.C.; Salman, P.; Oh, S.C.; Bodoky, G.; Kurteva, G.; Volovat, C.; Moiseyenko, V.M.; Gorbunova, V.; et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): A randomized, open-label phase 3 trial. Lancet Oncol. 2013, 14, 490–499. [Google Scholar] [CrossRef]
- Ilson, D.H. Advances in the treatment of gastric cancer. Curr. Opin. Gastroenterol. 2017, 33, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Pento, J.T. Monoclonal Antibodies for the Treatment of Cancer. Anticancer Res. 2017, 37, 5935–5939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, C.S.; Hou, Y.C.; Pai, M.H.; Lin, M.T.; Yeh, S.L. Effects of quercetin combined with anticancer drugs on metastasis-associated factors of gastric cancer cells: In vitro and in vivo studies. J. Nutr. Biochem. 2017, 51, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Frumkin, J. Gentamicin, a read-through agent for the treatment of rectal cancer. Colorectal Dis. 2017, 19, 864. [Google Scholar] [CrossRef] [PubMed]
- Newbold, A.; Salmon, J.M.; Martin, B.P.; Stanley, K.; Johnstone, R.W. The role of p21waf1/cip1 and p27Kip1 in HDACi-mediated tumor cell death and cell cycle arrest in the Eμ-myc model of B-cell lymphoma. Oncogene 2013, 33, 5415–5423. [Google Scholar] [CrossRef]
- Bustany, S.; Tchakarska, G.; Sola, B. Cyclin D1 regulates p27Kip1 stability in B cells. Cell Signal. 2011, 23, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Moskalev, A.A.; Smit-McBride, Z.; Shaposhnikov, M.V.; Plyusnina, E.N.; Zhavoronkov, A.; Budovsky, A.; Tacutu, R.; Fraifeld, V.E. Gadd45 proteins: Relevance to aging, longevity and age-related pathologies. Ageing Res. Rev. 2012, 11, 51–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, T. Mechanisms by which thiazolidinediones induce anti-cancer effects in cancers in digestive organs. J. Gastroenterol. 2010, 45, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Anantharaju, P.G.; Gowda, P.C.; Vimalambike, M.G.; Madhunapantula, S.V. An overview on the role of dietary phenolics for the treatment of cancers. Nutr. J. 2016, 15, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Matusiak, D.; Murillo, G.; Carroll, R.E.; Mehta, R.G.; Benya, R.V. Expression of vitamin D receptor and 25-hydroxyvitamin D3-1{alpha}-hydroxylase in normal and malignant human colon. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2370–2376. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bi, L.; Wang, Q.; Wen, M.; Li, C.; Ren, Y.; Jiao, Q.; Mao, J.H.; Wang, C.; Wei, G.; et al. miR-1204 targets VDR to promotes epithelial-mesenchymal transition and metastasis inbreast cancer. Oncogene 2018, 37, 3426–3439. [Google Scholar] [CrossRef]
- Jeon, S.M.; Shin, E.A. Exploring vitamin D metabolism and function in cancer. Exp. Mol. Med. 2018, 50, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Warabi, E. Mechanism of Rapid Nuclear Factor-E2-Related Factor 2 (Nrf2) Activation via Membrane-Associated Estrogen Receptors: Roles of NADPH Oxidase 1, Neutral Sphingomyelinase 2 and Epidermal Growth Factor Receptor (EGFR). Antioxidants 2019, 8, 69. [Google Scholar] [CrossRef]
- Albi, E.; Ambesi-Impiombato, S.; Villani, M.; de Pol, I.; Spelat, R.; Lazzarini, R.; Perrella, G. Thyroid cell growth: Sphingomyelin metabolism as non-invasive marker for cell damage acquired during spaceflight. Astrobiology 2010, 10, 811–820. [Google Scholar] [CrossRef]
- Albi, E.; Cataldi, S.; Ferri, I.; Sidoni, A.; Traina, G.; Fettucciari, K.; Ambesi-Impiombato, F.S.; Lazzarini, A.; Curcio, F.; Ceccarini, M.R.; et al. VDR independent induction of acid-sphingomyelinase by 1,23(OH)2 D3 in gastric cancer cells: Impact on apoptosis and cell morphology. Biochimie 2018, 146, 35–42. [Google Scholar] [CrossRef]
- Lazzarini, A.; Macchiarulo, A.; Floridi, A.; Coletti, A.; Cataldi, S.; Codini, M.; Lazzarini, R.; Bartoccini, E.; Cascianelli, G.; Ambesi-Impiombato, F.S.; et al. Very-long-chain fatty acid sphingomyelin in nuclear lipid microdomains of hepatocytes and hepatoma cells: Can the exchange from C24:0 to C16:0 affect signal proteins and vitamin D receptor? Mol. Biol. Cell. 2015, 26, 2418–2425. [Google Scholar] [CrossRef]
- Ceccarini, M.R.; Codini, M.; Cataldi, S.; Vannini, S.; Lazzarini, A.; Floridi, A.; Moretti, M.; Villarini, M.; Fioretti, B.; Beccari, T.; et al. Acid sphingomyelinase as target of LyciumChinense: Promising new action for cell health. Lipids Health Dis. 2016, 15, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Albi, E.; Curcio, F.; Lazzarini, A.; Floridi, A.; Cataldi, S.; Lazzarini, R.; Loreti, E.; Ferri, I.; Ambesi-Impiombato, F.S. How microgravity changes galectin-3 in thyroid follicles. Biomed. Res. Int. 2014, 2014, 652863–652867. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Bartoccini, E.; Marini, F.; Damaskopoulou, E.; Lazzarini, R.; Cataldi, S.; Cascianelli, G.; Gil Garcia, M.; Albi, E. Nuclear lipid microdomains regulate nuclear vitamin D3 uptake and influence embryonic hippocampal cell differentiation. Mol. Biol. Cell 2011, 17, 3022–3031. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albi, E.; Cataldi, S.; Ceccarini, M.R.; Conte, C.; Ferri, I.; Fettucciari, K.; Patria, F.F.; Beccari, T.; Codini, M. Gentamicin Targets Acid Sphingomyelinase in Cancer: The Case of the Human Gastric Cancer NCI-N87 Cells. Int. J. Mol. Sci. 2019, 20, 4375. https://doi.org/10.3390/ijms20184375
Albi E, Cataldi S, Ceccarini MR, Conte C, Ferri I, Fettucciari K, Patria FF, Beccari T, Codini M. Gentamicin Targets Acid Sphingomyelinase in Cancer: The Case of the Human Gastric Cancer NCI-N87 Cells. International Journal of Molecular Sciences. 2019; 20(18):4375. https://doi.org/10.3390/ijms20184375
Chicago/Turabian StyleAlbi, Elisabetta, Samuela Cataldi, Maria Rachele Ceccarini, Carmela Conte, Ivana Ferri, Katia Fettucciari, Federica Filomena Patria, Tommaso Beccari, and Michela Codini. 2019. "Gentamicin Targets Acid Sphingomyelinase in Cancer: The Case of the Human Gastric Cancer NCI-N87 Cells" International Journal of Molecular Sciences 20, no. 18: 4375. https://doi.org/10.3390/ijms20184375
APA StyleAlbi, E., Cataldi, S., Ceccarini, M. R., Conte, C., Ferri, I., Fettucciari, K., Patria, F. F., Beccari, T., & Codini, M. (2019). Gentamicin Targets Acid Sphingomyelinase in Cancer: The Case of the Human Gastric Cancer NCI-N87 Cells. International Journal of Molecular Sciences, 20(18), 4375. https://doi.org/10.3390/ijms20184375