Identification of Structural Variation from NGS-Based Non-Invasive Prenatal Testing

 , , and
, , and
Abstract
:1. Introduction
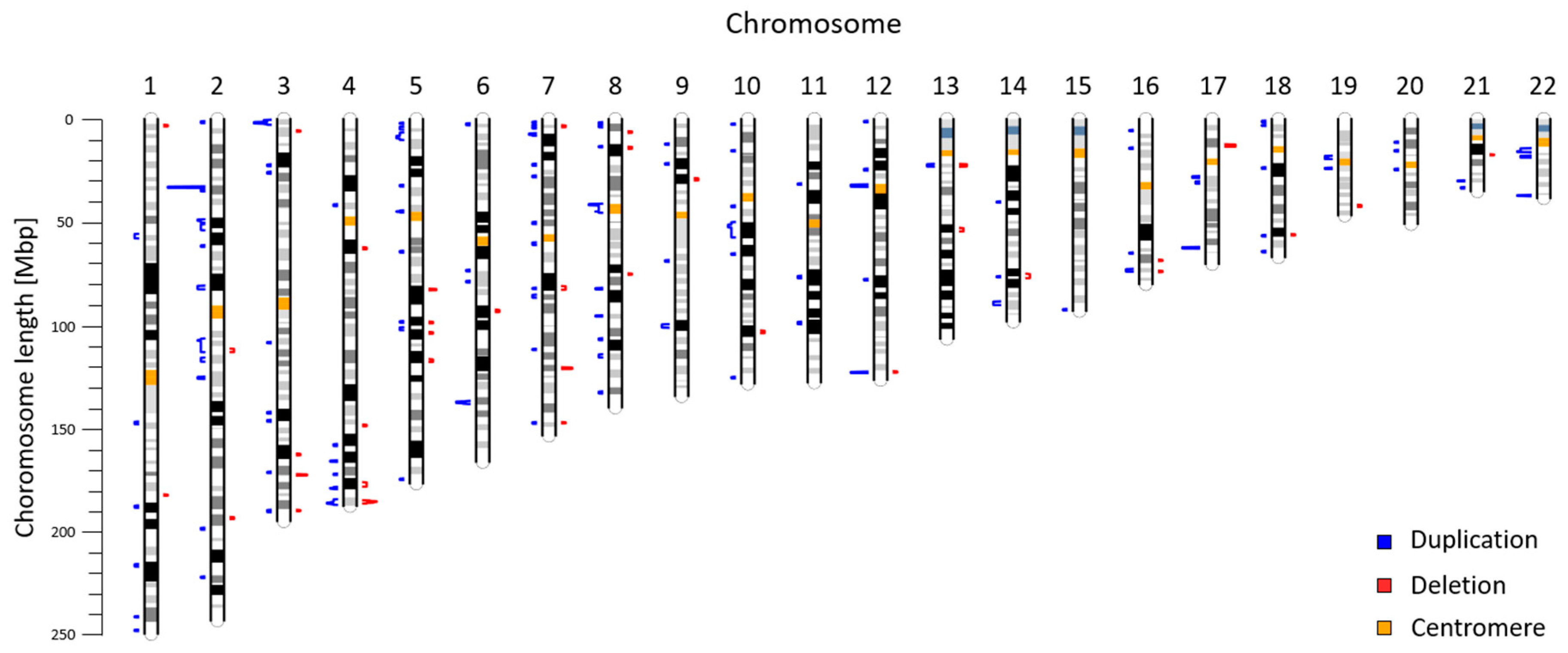
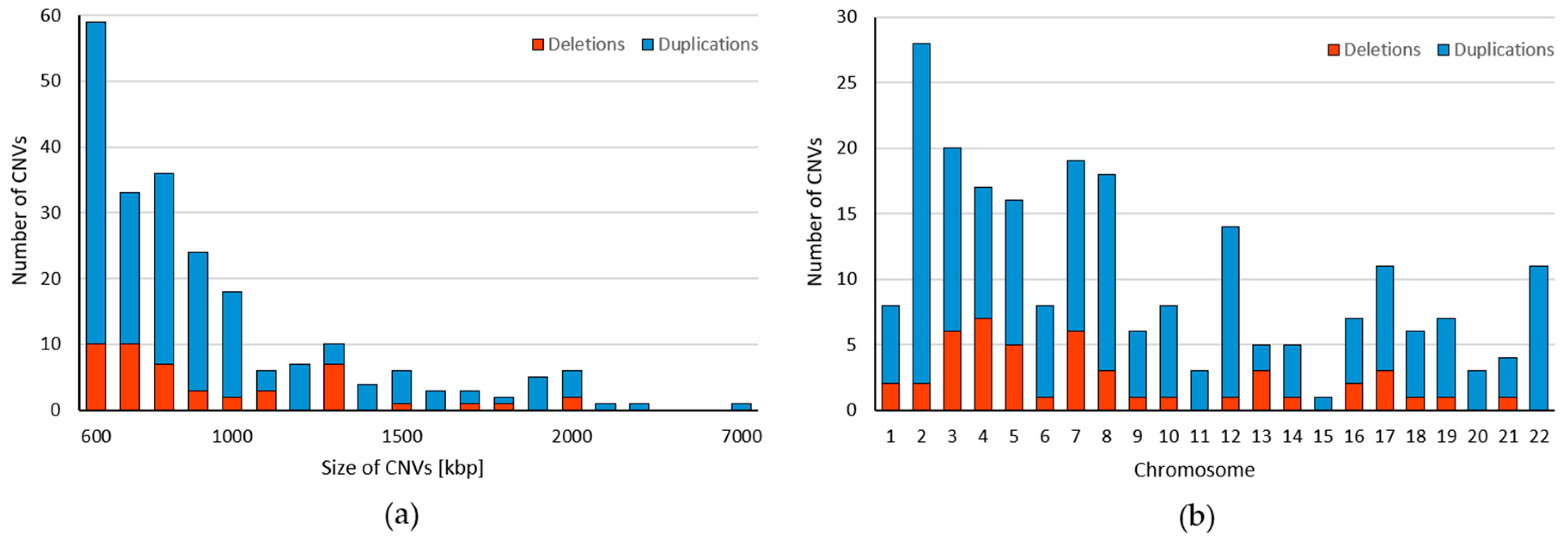
2. Results
3. Discussion
4. Materials and Methods
4.1. Sample Preparation and Sequencing
4.2. Mapping and Read Count Correction
4.3. Segment Identification and CNV Calling
4.4. Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Escaramís, G.; Docampo, E.; Rabionet, R. A decade of structural variants: Description, history and methods to detect structural variation. Brief. Funct. Genom. 2015, 14, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Sudmant, P.H.; Mallick, S.; Nelson, B.J.; Hormozdiari, F.; Krumm, N.; Huddleston, J.; Coe, B.P.; Baker, C.; Nordenfelt, S.; Bamshad, M.; et al. Global diversity, population stratification, and selection of human copy-number variation. Science 2015, 349, aab3761. [Google Scholar] [CrossRef] [PubMed]
- Mikhail, F.M. Copy number variations and human genetic disease. Curr. Opin. Pediatr. 2014, 26, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.L.; Kirkpatrick, B.E.; Ledbetter, D.H. Copy number variants, aneuploidies, and human disease. Clin. Perinatol. 2015, 42, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T. Working group of the American College of Medical Genetics Laboratory quality assurance committee American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, Q.; Wang, Q.; Jia, P.; Zhao, Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: Features and perspectives. BMC Bioinf. 2013, 14 (Suppl. 11), S1. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, C.R.; Scharer, G.H.; Shaikh, T.H. Clinical impact of copy number variation analysis using high-resolution microarray technologies: Advantages, limitations and concerns. Genome Med. 2012, 4, 80. [Google Scholar] [CrossRef]
- Russo, C.D.; Di Giacomo, G.; Cignini, P.; Padula, F.; Mangiafico, L.; Mesoraca, A.; D’Emidio, L.; McCluskey, M.R.; Paganelli, A.; Giorlandino, C. Comparative study of aCGH and next generation sequencing (NGS) for chromosomal microdeletion and microduplication screening. J. Prenat. Med. 2014, 8, 57–69. [Google Scholar]
- Wang, H.; Nettleton, D.; Ying, K. Copy number variation detection using next generation sequencing read counts. BMC Bioinf. 2014, 15, 109. [Google Scholar] [CrossRef]
- Minarik, G.; Repiska, G.; Hyblova, M.; Nagyova, E.; Soltys, K.; Budis, J.; Duris, F.; Sysak, R.; Gerykova Bujalkova, M.; Vlkova-Izrael, B.; et al. Utilization of benchtop next generation sequencing platforms ion torrent PGM and MiSeq in noninvasive prenatal testing for chromosome 21 trisomy and testing of impact of in silico and physical size selection on its analytical performance. PLoS ONE 2015, 10, e0144811. [Google Scholar] [CrossRef] [PubMed]
- Budis, J.; Gazdarica, J.; Radvanszky, J.; Szucs, G.; Kucharik, M.; Strieskova, L.; Gazdaricova, I.; Harsanyova, M.; Duris, F.; Minarik, G.; et al. Combining count- and length-based z-scores leads to improved predictions in non-invasive prenatal testing. Bioinformatics 2018, 35, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Pös, O.; Budiš, J.; Szemes, T. Recent trends in prenatal genetic screening and testing. F1000 Res. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Kucharik, M.; Gnip, A.; Hyblova, M.; Budis, J.; Strieskova, L.; Harsanyova, M.; Duris, F.; Radvanszky, J.; Minarik, G.; Szemes, T. Non-invasive prenatal testing by low coverage genomic sequencing: Detection limits of screened chromosomal microdeletions. BioRxiv 2019, 686345. [Google Scholar] [CrossRef] [Green Version]
- Jaroslav, B.; Juraj, G.; Jan, R.; Maria, H.; Iveta, G.; Lucia, S.; Richard, F.; Frantisek, D.; Gabriel, M.; Martina, S.; et al. Non-invasive prenatal testing as a valuable source of population specific allelic frequencies. J. Biotechnol. 2019, 299, 72–78. [Google Scholar]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, E.B.; Kaul, V.; Paschall, J.; Church, D.M.; Bunke, B.; Kunig, D.; Moreno-De-Luca, D.; Moreno-De-Luca, A.; Mulle, J.G.; Warren, S.T.; et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet. Med. 2011, 13, 777–784. [Google Scholar] [CrossRef] [Green Version]
- Bayat, V.; Thiffault, I.; Jaiswal, M.; Tétreault, M.; Donti, T.; Sasarman, F.; Bernard, G.; Demers-Lamarche, J.; Dicaire, M.-J.; Mathieu, J.; et al. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 2012, 10, e1001288. [Google Scholar] [CrossRef]
- Geoffroy, V. AnnotSV. Available online: https://lbgi.fr/AnnotSV/ (accessed on 7 February 2019).
- Carrasco-Ramiro, F.; Peiró-Pastor, R.; Aguado, B. Human genomics projects and precision medicine. Gene Ther. 2017, 24, 551–561. [Google Scholar] [CrossRef]
- Beyene, J.; Pare, G. Statistical genetics with application to population-based study design: A primer for clinicians. Eur. Heart, J. 2014, 35, 495–500. [Google Scholar] [CrossRef]
- Valsesia, A.; Macé, A.; Jacquemont, S.; Beckmann, J.S.; Kutalik, Z. The growing importance of CNVs: New insights for detection and clinical interpretation. Front. Genet. 2013, 4, 92. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M.; Coe, B.P.; Girirajan, S.; Rosenfeld, J.A.; Vu, T.H.; Baker, C.; Williams, C.; Stalker, H.; Hamid, R.; Hannig, V.; et al. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011, 43, 838–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietiläinen, O.P.H.; Rehnström, K.; Jakkula, E.; Service, S.K.; Congdon, E.; Tilgmann, C.; Hartikainen, A.-L.; Taanila, A.; Heikura, U.; Paunio, T.; et al. Phenotype mining in CNV carriers from a population cohort. Hum. Mol. Genet. 2011, 20, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Guyatt, A.L.; Stergiakouli, E.; Martin, J.; Walters, J.; O’Donovan, M.; Owen, M.; Thapar, A.; Kirov, G.; Rodriguez, S.; Rai, D.; et al. Association of copy number variation across the genome with neuropsychiatric traits in the general population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Männik, K.; Mägi, R.; Macé, A.; Cole, B.; Guyatt, A.L.; Shihab, H.A.; Maillard, A.M.; Alavere, H.; Kolk, A.; Reigo, A.; et al. Copy number variations and cognitive phenotypes in unselected populations. JAMA 2015, 313, 2044–2054. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Millson, A.; Lagrave, D.; Willis, M.J.H.; Rowe, L.R.; Lyon, E.; South, S.T. Chromosomal loss of 3q26.3-3q26.32, involving a partial neuroligin 1 deletion, identified by genomic microarray in a child with microcephaly, seizure disorder, and severe intellectual disability. Am. J. Med. Genet. A 2012, 158, 159–165. [Google Scholar] [CrossRef]
- Aller, E.; Jaijo, T.; García-García, G.; Aparisi, M.J.; Blesa, D.; Díaz-Llopis, M.; Ayuso, C.; Millán, J.M. Identification of large rearrangements of the PCDH15 gene by combined MLPA and a CGH: Large duplications are responsible for Usher syndrome. Invest. Ophthalmol. Vis. Sci. 2010, 51, 5480–5485. [Google Scholar] [CrossRef]
- OMIM ENTRY—# 601067—USHER SYNDROME, TYPE ID.; USH1D. Available online: https://www.omim.org/entry/601067?search=usher%20syndrome%20pcdh15&highlight=%28syndrome%7Csyndromic%29 (accessed on 31 July 2019).
- Brison, N.; Van Den Bogaert, K.; Dehaspe, L.; van den Oever, J.M.E.; Janssens, K.; Blaumeiser, B.; Peeters, H.; Van Esch, H.; Van Buggenhout, G.; Vogels, A.; et al. Accuracy and clinical value of maternal incidental findings during noninvasive prenatal testing for fetal aneuploidies. Genet. Med. 2017, 19, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Imbert-Bouteille, M.; Chiesa, J.; Gaillard, J.-B.; Dorvaux, V.; Altounian, L.; Gatinois, V.; Mousty, E.; Finge, S.; Bourquard, P.; Vermeesch, J.R.; et al. An incidental finding of maternal multiple myeloma by non invasive prenatal testing. Prenat. Diagn. 2017, 37, 1257–1260. [Google Scholar] [CrossRef] [Green Version]
- Giles, M.E.; Murphy, L.; Krstić, N.; Sullivan, C.; Hashmi, S.S.; Stevens, B. Prenatal cfDNA screening results indicative of maternal neoplasm: Survey of current practice and management needs. Prenatal. Diagnosis 2017, 37, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Zarrei, M.; MacDonald, J.R.; Merico, D.; Scherer, S.W. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Database of Genomic Variants. Available online: http://dgv.tcag.ca/v103_20131106/app/statistics (accessed on 14 May 2019).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, C.; Yin, A.-H.; Peng, C.-F.; Fu, F.; Yang, J.-X.; Li, R.; Chen, Y.-Y.; Luo, D.-H.; Zhang, Y.-L.; Ou, Y.-M.; et al. Noninvasive prenatal diagnosis of common aneuploidies by semiconductor sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, 7415–7420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Tynan, J.; Ehrich, M.; Hannum, G.; McCullough, R.; Saldivar, J.-S.; Oeth, P.; van den Boom, D.; Deciu, C. Detection of fetal subchromosomal abnormalities by sequencing circulating cell-free DNA from maternal plasma. Clin. Chem. 2015, 61, 608–616. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J. AnnotSV: An integrated tool for structural variations annotation. Bioinformatics 2018, 34, 3572–3574. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Koster, J.; Rahmann, S. Snakemake—A scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Variant Type | Location | Identifier | Phenotype | Events | Reference |
|---|---|---|---|---|---|
| Duplication | 1q21.1-21.2 | dbVar: nsv531885 | Developmental delay AND/OR other significant developmental or morphological phenotypes, Global developmental delay | 1 | [16,17] |
| Duplication | 2q33.1 | OMIM: 609728.0002 | Autosomal Recessive Spastic Ataxia with Leukoencephalopathy | 1 | [18] |
| Duplication | 7q11.23 | dbVar: nsv532240 | Encephalopathy, Global developmental delay, Muscular hypotonia | 1 | [17] |
| Deletion | 13q12.12 | dbVar: nsv491643 | Developmental delay AND/OR other significant developmental or morphological phenotypes, Seizures, Intellectual disability, Intrauterine growth retardation | 2 | [16] |
| Duplication | 17q12 | dbVar: nsv2775541 | Developmental delay AND/OR other significant developmental or morphological phenotypes, Behavioral abnormality | 1 | [16,17] |
| Duplication | 22q11.21 | dbVar: nssv577068 nsv530653 | Global developmental delay | 3 | [16,17] |
| Duplication | 22q11.21 | dbVar: nssv578923 nsv531796 | Developmental delay AND/OR other significant developmental or morphological phenotypes | 1 | [16,17] |
| Duplication | 22q11.23 | dbVar: nssv13653977 nsv2769497 | Short stature, Macrocephalus, Abnormality of the face, Intellectual disability | 2 | [16] |
| Type of Variant | Number of CNVs | Total Sequence (Mbp) | Coding Regions (Mbp) | Non-Coding Regions (Mbp) |
|---|---|---|---|---|
| CNV gain | 178 | 191.54 | 3.27 | 188.27 |
| CNV loss | 47 | 46.98 | 0.44 | 46.54 |
| Sum | 225 | 238.52 | 3.71 | 234.81 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pös, O.; Budis, J.; Kubiritova, Z.; Kucharik, M.; Duris, F.; Radvanszky, J.; Szemes, T. Identification of Structural Variation from NGS-Based Non-Invasive Prenatal Testing. Int. J. Mol. Sci. 2019, 20, 4403. https://doi.org/10.3390/ijms20184403
Pös O, Budis J, Kubiritova Z, Kucharik M, Duris F, Radvanszky J, Szemes T. Identification of Structural Variation from NGS-Based Non-Invasive Prenatal Testing. International Journal of Molecular Sciences. 2019; 20(18):4403. https://doi.org/10.3390/ijms20184403
Chicago/Turabian StylePös, Ondrej, Jaroslav Budis, Zuzana Kubiritova, Marcel Kucharik, Frantisek Duris, Jan Radvanszky, and Tomas Szemes. 2019. "Identification of Structural Variation from NGS-Based Non-Invasive Prenatal Testing" International Journal of Molecular Sciences 20, no. 18: 4403. https://doi.org/10.3390/ijms20184403
APA StylePös, O., Budis, J., Kubiritova, Z., Kucharik, M., Duris, F., Radvanszky, J., & Szemes, T. (2019). Identification of Structural Variation from NGS-Based Non-Invasive Prenatal Testing. International Journal of Molecular Sciences, 20(18), 4403. https://doi.org/10.3390/ijms20184403