The Second Life of Methylarginines as Cardiovascular Targets


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Endogenous Methylarginines as Cardiovascular Risk Factors
3. Metabolism of Endogenous Methylarginines
4. Transport of Endogenous Methylarginines
5. Dimethylarginine Dimethylaminohydrolases
6. DDAH1 and ADMA
7. Controversy 1: DDAH2 and ADMA
8. Non-Enzymatic Function of DDAHs
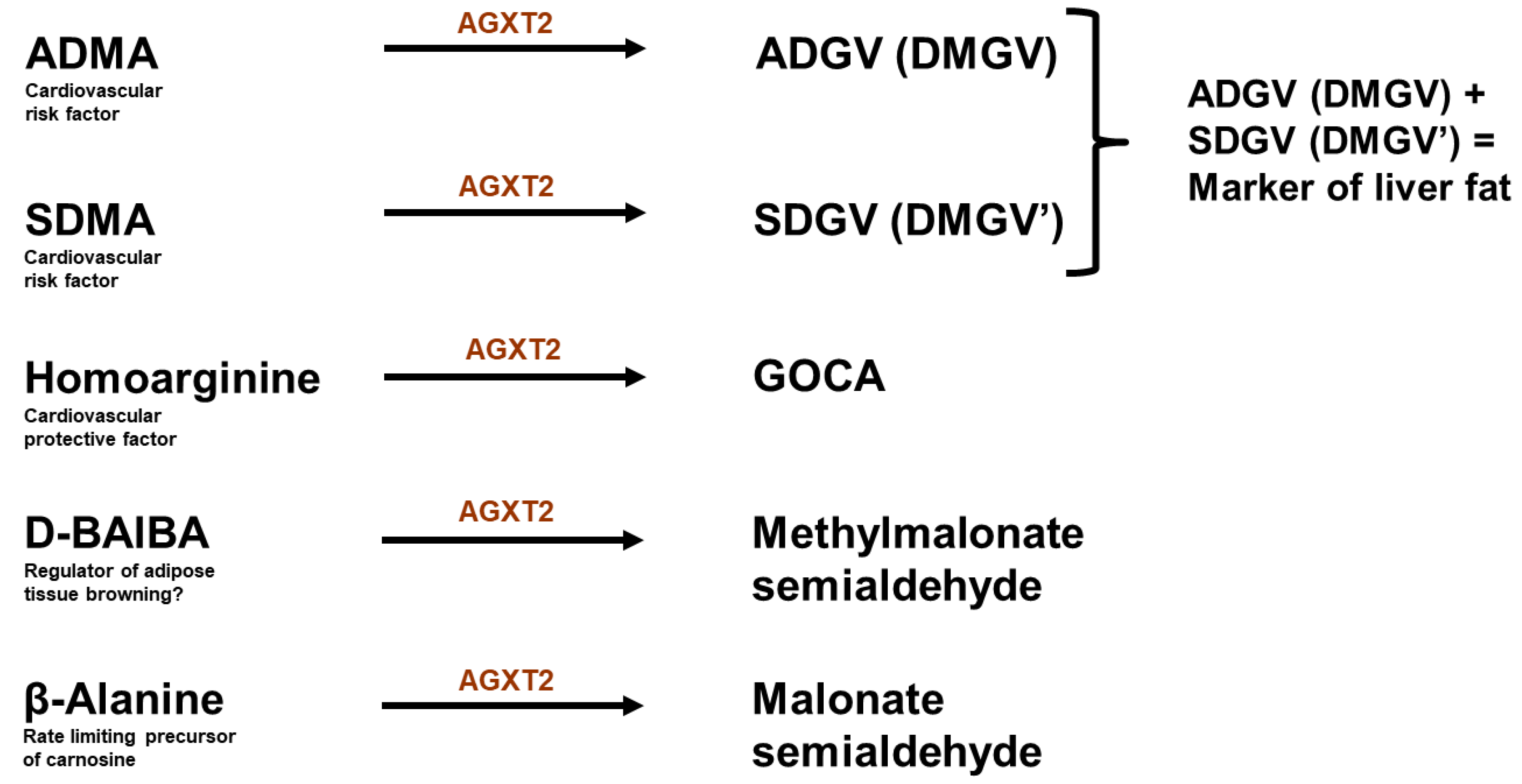
9. Alanine: Glyoxylate Aminotransferase 2
10. Controversy 2: AGXT2 as a Therapeutic Target
11. Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Leiper, J.; Vallance, P. Biological significance of endogenous methylarginines that inhibit nitric oxide synthases. Cardiovasc. Res. 1999, 43, 542–548. [Google Scholar] [CrossRef]
- Rees, D.D.; Palmer, R.M.; Schulz, R.; Hodson, H.F.; Moncada, S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br. J. Pharmacol. 1990, 101, 746–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faraci, F.M.; Brian, J.E., Jr.; Heistad, D.D. Response of cerebral blood vessels to an endogenous inhibitor of nitric oxide synthase. Am. J. Physiol. 1995, 269 Pt 2, H1522–H1527. [Google Scholar] [CrossRef]
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar] [PubMed]
- Ramuschkat, M.; Appelbaum, S.; Atzler, D.; Zeller, T.; Bauer, C.; Ojeda, F.M.; Sinning, C.R.; Hoffmann, B.; Lackner, K.J.; Böger, R.H.; et al. ADMA, subclinical changes and atrial fibrillation in the general population. Int. J. Cardiol. 2016, 203, 640–646. [Google Scholar] [CrossRef]
- Boger, R.H.; Cooke, J.P.; Vallance, P. ADMA: An emerging cardiovascular risk factor. Vasc. Med. 2005, 10 (Suppl. 1), S1–S2. [Google Scholar] [CrossRef] [PubMed]
- Boger, R.H.; Bode-Böger, S.M.; Szuba, A.; Tsao, P.S.; Chan, J.R.; Tangphao, O.; Blaschke, T.F.; Cooke, J.P. Asymmetric dimethylarginine (ADMA): A novel risk factor for endothelial dysfunction: Its role in hypercholesterolemia. Circulation 1998, 98, 1842–1847. [Google Scholar] [CrossRef]
- Surdacki, A.; Nowicki, M.; Sandmann, J.; Tsikas, D.; Boeger, R.H.; Bode-Boeger, S.M.; Kruszelnicka-Kwiatkowska, O.; Kokot, F.; Dubiel, J.S.; Froelich, J.C. Reduced urinary excretion of nitric oxide metabolites and increased plasma levels of asymmetric dimethylarginine in men with essential hypertension. J. Cardiovasc. Pharmacol. 1999, 33, 652–658. [Google Scholar] [CrossRef]
- Abbasi, F.; Asagmi, T.; Cooke, J.P.; Lamendola, C.; McLaughlin, T.; Reaven, G.M.; Stuehlinger, M.; Tsao, P.S. Plasma concentrations of asymmetric dimethylarginine are increased in patients with type 2 diabetes mellitus. Am. J. Cardiol. 2001, 88, 1201–1203. [Google Scholar] [CrossRef]
- Lundman, P.; Eriksson, M.J.; Stühlinger, M.; Cooke, J.P.; Hamsten, A.; Tornvall, P. Mild-to-moderate hypertriglyceridemia in young men is associated with endothelial dysfunction and increased plasma concentrations of asymmetric dimethylarginine. J. Am. Coll. Cardiol. 2001, 38, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Krempl, T.K.; Maas, R.; Sydow, K.; Meinertz, T.; Böger, R.H.; Kähler, J. Elevation of asymmetric dimethylarginine in patients with unstable angina and recurrent cardiovascular events. Eur. Heart J. 2005, 26, 1846–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.H.; Lee, S.C. Elevated levels of plasma homocyst(e)ine and asymmetric dimethylarginine in elderly patients with stroke. Atherosclerosis 2001, 158, 425–430. [Google Scholar] [CrossRef]
- Boger, R.H.; Bode-Böger, S.M.; Thiele, W.; Junker, W.; Alexander, K.; Frölich, J.C. Biochemical evidence for impaired nitric oxide synthesis in patients with peripheral arterial occlusive disease. Circulation 1997, 95, 2068–2074. [Google Scholar] [CrossRef] [PubMed]
- Fleck, C.; Schweitzer, F.; Karge, E.; Busch, M.; Stein, G. Serum concentrations of asymmetric (ADMA) and symmetric (SDMA) dimethylarginine in patients with chronic kidney diseases. Clin. Chim. Acta 2003, 336, 1–12. [Google Scholar] [CrossRef]
- Pettersson, A.; Hedner, T.; Milsom, I. Increased circulating concentrations of asymmetric dimethyl arginine (ADMA), an endogenous inhibitor of nitric oxide synthesis, in preeclampsia. Acta Obstet. Gynecol. Scand. 1998, 77, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Meinitzer, A.; Seelhorst, U.; Wellnitz, B.; Halwachs-Baumann, G.; Boehm, B.O.; Winkelmann, B.R.; März, W. Asymmetrical dimethylarginine independently predicts total and cardiovascular mortality in individuals with angiographic coronary artery disease (the Ludwigshafen Risk and Cardiovascular Health study). Clin. Chem. 2007, 53, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Leong, T.; Zylberstein, D.; Graham, I.; Lissner, L.; Ward, D.; Fogarty, J.; Bengtsson, C.; Björkelund, C.; Thelle, D. Swedish-Irish-Norwegian Collaboration. Asymmetric dimethylarginine independently predicts fatal and nonfatal myocardial infarction and stroke in women: 24-year follow-up of the population study of women in Gothenburg. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 961–967. [Google Scholar]
- Zoccali, C.; Bode-Böger, S.; Mallamaci, F.; Benedetto, F.; Tripepi, G.; Malatino, L.; Cataliotti, A.; Bellanuova, I.; Fermo, I.; Frölich, J.; et al. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: A prospective study. Lancet 2001, 358, 2113–2117. [Google Scholar] [CrossRef]
- Kielstein, J.T.; Impraim, B.; Simmel, S.; Bode-Böger, S.M.; Tsikas, D.; Frölich, J.C.; Hoeper, M.M.; Haller, H.; Fliser, D. Cardiovascular effects of systemic nitric oxide synthase inhibition with asymmetrical dimethylarginine in humans. Circulation 2004, 109, 172–177. [Google Scholar] [CrossRef]
- Kielstein, J.T.; Impraim, B.; Simmel, S.; Bode-Böger, S.M.; Tsikas, D.; Frölich, J.C.; Hoeper, M.M.; Haller, H.; Fliser, D. ADMA increases arterial stiffness and decreases cerebral blood flow in humans. Stroke 2006, 37, 2024–2029. [Google Scholar] [CrossRef]
- Creager, M.A.; Gallagher, S.J.; Girerd, X.J.; Coleman, S.M.; Dzau, V.J.; Cooke, J.P. l-arginine improves endothelium-dependent vasodilation in hypercholesterolemic humans. J. Clin. Investig. 1992, 90, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Boger, R.H.; Bode-Böger, S.M.; Mügge, A.; Kienke, S.; Brandes, R.; Dwenger, A.; Frölich, J.C. Supplementation of hypercholesterolaemic rabbits with l-arginine reduces the vascular release of superoxide anions and restores NO production. Atherosclerosis 1995, 117, 273–284. [Google Scholar] [CrossRef]
- Boger, R.H. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the “l-arginine paradox” and acts as a novel cardiovascular risk factor. J. Nutr. 2004, 134, 2842S–2847S; discussion 2853S. [Google Scholar] [CrossRef] [PubMed]
- Pollock, J.S.; Förstermann, U.; Mitchell, J.A.; Warner, T.D.; Schmidt, H.H.; Nakane, M.; Murad, F. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc. Natl. Acad. Sci. USA 1991, 88, 10480–10484. [Google Scholar] [CrossRef] [PubMed]
- Bode-Boger, S.M.; Scalera, F.; Kielstein, J.T.; Martens-Lobenhoffer, J.; Breithardt, G.; Fobker, M.; Reinecke, H. Symmetrical dimethylarginine: A new combined parameter for renal function and extent of coronary artery disease. J. Am. Soc. Nephrol. 2006, 17, 1128–1134. [Google Scholar] [CrossRef]
- Kielstein, J.T.; Martens-Lobenhoffer, J.; Vollmer, S.; Bode-Böger, S.M. l-arginine, ADMA, SDMA, creatinine, MDRD formula: Detour to renal function testing. J. Nephrol. 2008, 21, 959–961. [Google Scholar] [PubMed]
- Schulze, F.; Carter, A.M.; Schwedhelm, E.; Ajjan, R.; Maas, R.; von Holten, R.A.; Atzler, D.; Grant, P.J.; Böger, R.H. Symmetric dimethylarginine predicts all-cause mortality following ischemic stroke. Atherosclerosis 2010, 208, 518–523. [Google Scholar] [CrossRef]
- Gore, M.O.; Lüneburg, N.; Schwedhelm, E.; Ayers, C.R.; Anderssohn, M.; Khera, A.; Atzler, D.; de Lemos, J.A.; Grant, P.J.; McGuire, D.K.; et al. Symmetrical dimethylarginine predicts mortality in the general population: Observations from the Dallas heart study. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2682–2688. [Google Scholar] [CrossRef]
- Closs, E.I.; Basha, F.Z.; Habermeier, A.; Förstermann, U. Interference of l-arginine analogues with l-arginine transport mediated by the y+ carrier hCAT-2B. Nitric Oxide 1997, 1, 65–73. [Google Scholar] [CrossRef]
- Speer, T.; Rohrer, L.; Blyszczuk, P.; Shroff, R.; Kuschnerus, K.; Kränkel, N.; Kania, G.; Zewinger, S.; Akhmedov, A.; Shi, Y.; et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity 2013, 38, 754–768. [Google Scholar] [CrossRef]
- Anthony, S.; Leiper, J.; Vallance, P. Endogenous production of nitric oxide synthase inhibitors. Vasc. Med. 2005, 10 (Suppl. 1), S3–S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.K.; Paik, W.K.; Kim, S. Purification and molecular identification of two protein methylases I from calf brain. Myelin basic protein- and histone-specific enzyme. J. Biol. Chem. 1988, 263, 19024–19033. [Google Scholar] [PubMed]
- Rawal, N.; Rajpurohit, R.; Paik, W.K.; Kim, S. Purification and characterization of S-adenosylmethionine-protein-arginine N-methyltransferase from rat liver. Biochem. J. 1994, 300 Pt 2, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, T.; Kimoto, M.; Watanabe, H.; Sasaoka, K. Metabolism of NG, NG-and NG, N’G-dimethylarginine in rats. Arch. Biochem. Biophys. 1987, 252, 526–537. [Google Scholar] [CrossRef]
- Ogawa, T.; Kimoto, M.; Sasaoka, K. Purification and properties of a new enzyme, NG, NG-dimethylarginine dimethylaminohydrolase, from rat kidney. J. Biol. Chem. 1989, 264, 10205–10209. [Google Scholar]
- Bassareo, P.P.; Fanos, V.; Puddu, M.; Flore, G.; Mercuro, G. Advanced intrauterine growth restriction is associated with reduced excretion of asymmetric dimethylarginine. Early Hum. Dev. 2014, 90, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Kimoto, M.; Sasaoka, K. Dimethylarginine: Pyruvate aminotransferase in rats. Purification, properties, and identity with alanine: Glyoxylate aminotransferase 2. J. Biol. Chem. 1990, 265, 20938–20945. [Google Scholar] [PubMed]
- Sasaoka, K.; Ogawa, T.; Kimoto, M. N-Acetyl conjugates of basic amino acids newly identified in rat urine. Arch. Biochem. Biophys. 1982, 219, 454–458. [Google Scholar] [CrossRef]
- Martens-Lobenhoffer, J.; Rodionov, R.N.; Bode-Boger, S.M. Determination of asymmetric Nalpha-acetyldimethylarginine in humans: A phase II metabolite of asymmetric dimethylarginine. Anal. Biochem. 2014, 452, 25–30. [Google Scholar] [CrossRef]
- Martens-Lobenhoffer, J.; Bode-Boger, S.M. Amino acid N-acetylation: Metabolic elimination of symmetric dimethylarginine as symmetric N(alpha)-acetyldimethylarginine, determined in human plasma and urine by LC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 975, 59–64. [Google Scholar] [CrossRef]
- Strobel, J.; Müller, F.; Zolk, O.; Endreß, B.; König, J.; Fromm, M.F.; Maas, R. Transport of asymmetric dimethylarginine (ADMA) by cationic amino acid transporter 2 (CAT2), organic cation transporter 2 (OCT2) and multidrug and toxin extrusion protein 1 (MATE1). Amino Acids 2013, 45, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, V.; Longo, A.; Palmieri, L.; Closs, E.I.; Palmieri, F. Asymmetric dimethylarginine is transported by the mitochondrial carrier SLC25A2. Amino Acids 2016, 48, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Leiper, J.M.; Santa Maria, J.; Chubb, A.; MacAllister, R.J.; Charles, I.G.; Whitley, G.S.; Vallance, P. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem. J. 1999, 343 Pt 1, 209–214. [Google Scholar] [CrossRef]
- Tran, C.T.; Fox, M.F.; Vallance, P.; Leiper, J.M. Chromosomal localization, gene structure, and expression pattern of DDAH1: Comparison with DDAH2 and implications for evolutionary origins. Genomics 2000, 68, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gill, P.S.; Chabrashvili, T.; Onozato, M.L.; Raggio, J.; Mendonca, M.; Dennehy, K.; Li, M.; Modlinger, P.; Leiper, J.; et al. Isoform-Specific Regulation by NG-NG-Dimethylarginine Dimethylaminohydrolase of Rat Serum Asymmetric Dimethylarginine and Vascular Endothelium-Derived Relaxing Factor/NO. Circ. Res. 2007, 101, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Dowsett, L.; Piper, S.; Slaviero, A.; Dufton, N.; Wang, Z.; Boruc, O.; Delahaye, M.; Colman, L.; Kalk, E.; Tomlinson, J.; et al. Endothelial Dimethylarginine Dimethylaminohydrolase 1 Is an Important Regulator of Angiogenesis but Does Not Regulate Vascular Reactivity or Hemodynamic Homeostasis. Circulation 2015, 131, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Hu, X.; Xu, X.; Chen, Y.; Bache, R.J. Dimethylarginine dimethylaminohydrolase 1 modulates endothelial cell growth through nitric oxide and Akt. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Schwedhelm, E.; Leitner, E.; Atzler, D.; Schmitz, C.; Jacobi, J.; Meinertz, T.; Münzel, T.; Baldus, S.; Cooke, J.P.; Böger, R.H.; et al. Extensive characterization of the human DDAH1 transgenic mice. Pharmacol. Res. 2009, 60, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Dayoub, H.; Achan, V.; Adimoolam, S.; Jacobi, J.; Stuehlinger, M.C.; Wang, B.Y.; Tsao, P.S.; Kimoto, M.; Vallance, P.; Patterson, A.J.; et al. Dimethylarginine dimethylaminohydrolase regulates nitric oxide synthesis: Genetic and physiological evidence. Circulation 2003, 108, 3042–3047. [Google Scholar] [CrossRef]
- Dayoub, H.; Rodionov, R.N.; Lynch, C.; Cooke, J.P.; Arning, E.; Bottiglieri, T.; Lentz, S.R.; Faraci, F.M. Overexpression of dimethylarginine dimethylaminohydrolase inhibits asymmetric dimethylarginine-induced endothelial dysfunction in the cerebral circulation. Stroke 2008, 39, 180–184. [Google Scholar] [CrossRef]
- Leiper, J.; Nandi, M.; Torondel, B.; Murray-Rust, J.; Malaki, M.; O’Hara, B.; Rossiter, S.; Anthony, S.; Madhani, M.; Selwood, D.; et al. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat. Med. 2007, 13, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Atzler, D.; Xu, X.; Zhang, P.; Guo, H.; Lu, Z.; Fassett, J.; Schwedhelm, E.; Böger, R.H.; Bache, R.J.; et al. Dimethylarginine dimethylaminohydrolase-1 is the critical enzyme for degrading the cardiovascular risk factor asymmetrical dimethylarginine. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1540–1546. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, J.; Sydow, K.; von Degenfeld, G.; Zhang, Y.; Dayoub, H.; Wang, B.; Patterson, A.J.; Kimoto, M.; Blau, H.M.; Cooke, J.P. Overexpression of dimethylarginine dimethylaminohydrolase reduces tissue asymmetric dimethylarginine levels and enhances angiogenesis. Circulation 2005, 111, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Stuhlinger, M.C.; Conci, E.; Haubner, B.J.; Stocker, E.M.; Schwaighofer, J.; Cooke, J.P.; Tsao, P.S.; Pachinger, O.; Metzler, B. Asymmetric dimethyl l-arginine (ADMA) is a critical regulator of myocardial reperfusion injury. Cardiovasc. Res. 2007, 75, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Ueda, S.; Yamagishi, S.; Obara, N.; Taguchi, K.; Ando, R.; Kaida, Y.; Iwatani, R.; Kaifu, K.; Yokoro, M.; et al. Asymmetric dimethylarginine accumulates in the kidney during ischemia/reperfusion injury. Kidney Int. Suppl. 2014, 85, 570–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobi, J.; Maas, R.; Cardounel, A.J.; Arend, M.; Pope, A.J.; Cordasic, N.; Heusinger-Ribeiro, J.; Atzler, D.; Strobel, J.; Schwedhelm, E.; et al. Dimethylarginine dimethylaminohydrolase overexpression ameliorates atherosclerosis in apolipoprotein E-deficient mice by lowering asymmetric dimethylarginine. Am. J. Pathol. 2010, 176, 2559–2570. [Google Scholar] [CrossRef]
- Sydow, K.; Schmitz, C.; von Leitner, E.C.; von Leitner, R.; Klinke, A.; Atzler, D.; Krebs, C.; Wieboldt, H.; Ehmke, H.; Schwedhelm, E.; et al. Dimethylarginine dimethylaminohydrolase1 is an organ-specific mediator of end organ damage in a murine model of hypertension. PLoS ONE 2012, 7, e48150. [Google Scholar] [CrossRef] [PubMed]
- Nandi, M.; Kelly, P.; Torondel, B.; Wang, Z.; Starr, A.; Ma, Y.; Cunningham, P.; Stidwill, R.; Leiper, J. Genetic and pharmacological inhibition of dimethylarginine dimethylaminohydrolase 1 is protective in endotoxic shock. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2589–2597. [Google Scholar] [CrossRef] [PubMed]
- Ghebremariam, Y.T.; Erlanson, D.A.; Yamada, K.; Cooke, J.P. Development of a dimethylarginine dimethylaminohydrolase (DDAH) assay for high-throughput chemical screening. J. Biomol. Screen. 2012, 17, 651–661. [Google Scholar] [CrossRef]
- Linsky, T.W.; Fast, W. Discovery of structurally-diverse inhibitor scaffolds by high-throughput screening of a fragment library with dimethylarginine dimethylaminohydrolase. Bioorg. Med. Chem. 2012, 20, 5550–5558. [Google Scholar] [CrossRef] [Green Version]
- Hartzoulakis, B.; Rossiter, S.; Gill, H.; O’Hara, B.; Steinke, E.; Gane, P.J.; Hurtado-Guerrero, R.; Leiper, J.M.; Vallance, P.; Rust, J.M.; et al. Discovery of inhibitors of the pentein superfamily protein dimethylarginine dimethylaminohydrolase (DDAH), by virtual screening and hit analysis. Bioorg. Med. Chem. Lett. 2007, 17, 3953–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kami Reddy, K.R.; Dasari, C.; Vandavasi, S.; Natani, S.; Supriya, B.; Jadav, S.S.; Sai Ram, N.; Kumar, J.M.; Ummanni, R. Novel Cellularly Active Inhibitor Regresses DDAH1 Induced Prostate Tumor Growth by Restraining Tumor Angiogenesis through Targeting DDAH1/ADMA/NOS Pathway. ACS Comb. Sci. 2019, 21, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Hulin, J.A.; Tommasi, S.; Elliot, D.; Mangoni, A.A. Small molecule inhibition of DDAH1 significantly attenuates triple negative breast cancer cell vasculogenic mimicry in vitro. Biomed. Pharmacother. 2019, 111, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Pope, A.J.; Karrupiah, K.; Kearns, P.N.; Xia, Y.; Cardounel, A.J. Role of dimethylarginine dimethylaminohydrolases in the regulation of endothelial nitric oxide production. J. Biol. Chem. 2009, 284, 35338–35347. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Wakino, S.; Tatematsu, S.; Yoshioka, K.; Homma, K.; Sugano, N.; Kimoto, M.; Hayashi, K.; Itoh, H. Role of asymmetric dimethylarginine in vascular injury in transgenic mice overexpressing dimethylarginie dimethylaminohydrolase 2. Circ. Res. 2007, 101, e2–e10. [Google Scholar] [CrossRef] [PubMed]
- Lambden, S.; Kelly, P.; Ahmetaj-Shala, B.; Wang, Z.; Lee, B.; Nandi, M.; Torondel, B.; Delahaye, M.; Dowsett, L.; Piper, S.; et al. Dimethylarginine dimethylaminohydrolase 2 regulates nitric oxide synthesis and hemodynamics and determines outcome in polymicrobial sepsis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Tokuo, H.; Yunoue, S.; Feng, L.; Kimoto, M.; Tsuji, H.; Ono, T.; Saya, H.; Araki, N. Phosphorylation of neurofibromin by cAMP-dependent protein kinase is regulated via a cellular association of N(G), N(G)-dimethylarginine dimethylaminohydrolase. FEBS Lett. 2001, 494, 48–53. [Google Scholar] [CrossRef]
- Li, F.; Munchhof, A.M.; White, H.A.; Mead, L.E.; Krier, T.R.; Fenoglio, A.; Chen, S.; Wu, X.; Cai, S.; Yang, F.C.; et al. Neurofibromin is a novel regulator of RAS-induced signals in primary vascular smooth muscle cells. Hum. Mol. Genet. 2006, 15, 1921–1930. [Google Scholar] [CrossRef] [Green Version]
- Boult, J.K.; Walker-Samuel, S.; Jamin, Y.; Leiper, J.M.; Whitley, G.S.; Robinson, S.P. Active site mutant dimethylarginine dimethylaminohydrolase 1 expression confers an intermediate tumour phenotype in C6 gliomas. J. Pathol. 2011, 225, 344–352. [Google Scholar] [CrossRef]
- Pullamsetti, S.S.; Savai, R.; Dumitrascu, R.; Dahal, B.K.; Wilhelm, J.; Konigshoff, M.; Zakrzewicz, D.; Ghofrani, H.A.; Weissmann, N.; Eickelberg, O.; et al. The role of dimethylarginine dimethylaminohydrolase in idiopathic pulmonary fibrosis. Sci. Transl. Med. 2011, 3, 87ra53. [Google Scholar] [CrossRef]
- Rodionov, R.N.; Dayoub, H.; Lynch, C.M.; Wilson, K.M.; Stevens, J.W.; Murry, D.J.; Kimoto, M.; Arning, E.; Bottiglieri, T.; Cooke, J.P.; et al. Overexpression of dimethylarginine dimethylaminohydrolase protects against cerebral vascular effects of hyperhomocysteinemia. Circ. Res. 2010, 106, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Wakino, S.; Tanaka, T.; Kimoto, M.; Tatematsu, S.; Kanda, T.; Yoshioka, K.; Homma, K.; Sugano, N.; Kurabayashi, M.; et al. Dimethylarginine dimethylaminohydrolase 2 increases vascular endothelial growth factor expression through Sp1 transcription factor in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.F.; Morningstar, J.E.; Yang, Q.; Zheng, B.; Gao, Y.; Jeanfavre, S.; Scott, J.; Fernandez, C.; Zheng, H.; O’Connor, S.; et al. Dimethylguanidino valeric acid is a marker of liver fat and predicts diabetes. J. Clin. Investig. 2017, 127, 4394–4402. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.S.; Nishikimi, M.; Inoue, M.; Muragaki, Y.; Ooshima, A. Specific expression of alanine-glyoxylate aminotransferase 2 in the epithelial cells of Henle’s loop. Nephron 1999, 83, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Kittel, A.; Maas, R.; König, J.; Mieth, M.; Weiss, N.; Jarzebska, N.; Hohenstein, B.; Martens-Lobenhoffer, J.; Bode-Böger, S.M.; Rodionov, R.N. In vivo evidence that Agxt2 can regulate plasma levels of dimethylarginines in mice. Biochem. Biophys. Res. Commun. 2013, 430, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Minatogawa, Y.; Takada, Y.; Okuno, E.; Kido, R. Subcellular distribution of pyruvate (glyoxylate) aminotransferases in rat liver. Biochem. J. 1978, 170, 173–175. [Google Scholar] [CrossRef] [Green Version]
- Cellini, B.; Lorenzetto, A.; Montioli, R.; Oppici, E.; Voltattorni, C.B. Human liver peroxisomal alanine: Glyoxylate aminotransferase: Different stability under chemical stress of the major allele, the minor allele, and its pathogenic G170R variant. Biochimie 2010, 92, 1801–1811. [Google Scholar] [CrossRef]
- Kakimoto, Y.; Taniguchi, K.; Sano, I. D-beta-aminoisobutyrate: Pyruvate aminotransferase in mammalian liver and excretion of beta-aminoisobutyrate by man. J. Biol. Chem. 1969, 244, 335–340. [Google Scholar]
- Pollitt, R.J.; Green, A.; Smith, R. Excessive excretion of beta-alanine and of 3-hydroxypropionic, R- and S-3-aminoisobutyric, R- and S-3-hydroxyisobutyric and S-2-(hydroxymethyl)butyric acids probably due to a defect in the metabolism of the corresponding malonic semialdehydes. J. Inherit. Metab. Dis. 1985, 8, 75–79. [Google Scholar] [CrossRef]
- Fink, K.; Cline, R.E.; Henderson, R.B.; Fink, R.M. Metabolism of thymine (methyl-C14 or -2-C14) by rat liver in vitro. J. Biol. Chem. 1956, 221, 425–433. [Google Scholar]
- Kontani, Y.; Kaneko, M.; Kikugawa, M.; Fujimoto, S.; Tamaki, N. Identity of D-3-aminoisobutyrate-pyruvate aminotransferase with alanine-glyoxylate aminotransferase 2. Biochim. Biophys. Acta 1993, 1156, 161–166. [Google Scholar] [CrossRef]
- Rhee, E.P.; Ho, J.E.; Chen, M.H.; Shen, D.; Cheng, S.; Larson, M.G.; Ghorbani, A.; Shi, X.; Helenius, I.T.; O’Donnell, C.J.; et al. A Genome-wide Association Study of the Human Metabolome in a Community-Based Cohort. Cell Metab. 2013, 18, 130–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, L.D.; Boström, P.; O’Sullivan, J.F.; Schinzel, R.T.; Lewis, G.D.; Dejam, A.; Lee, Y.K.; Palma, M.J.; Calhoun, S.; Georgiadi, A.; et al. Beta-Aminoisobutyric acid induces browning of white fat and hepatic beta-oxidation and is inversely correlated with cardiometabolic risk factors. Cell Metab. 2014, 19, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.C.; Tallon, M.J.; Dunnett, M.; Boobis, L.; Coakley, J.; Kim, H.J.; Fallowfield, J.L.; Hill, C.A.; Sale, C.; Wise, J.A. The absorption of orally supplied beta-alanine and its effect on muscle carnosine synthesis in human vastus lateralis. Amino Acids 2006, 30, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Kleber, M.E.; Seppälä, I.; Pilz, S.; Hoffmann, M.M.; Tomaschitz, A.; Oksala, N.; Raitoharju, E.; Lyytikäinen, L.P.; Mäkelä, K.M.; Laaksonen, R.; et al. Genome-wide association study identifies 3 genomic loci significantly associated with serum levels of homoarginine: The AtheroRemo Consortium. Circ. Cardiovasc. Genet. 2013, 6, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Pilz, S.; Meinitzer, A.; Gaksch, M.; Grübler, M.; Verheyen, N.; Drechsler, C.; Hartaigh, B.Ó.; Lang, F.; Alesutan, I.; Voelkl, J.; et al. Homoarginine in the renal and cardiovascular systems. Amino Acids 2015, 47, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Atzler, D.; Schwedhelm, E.; Choe, C.U. L-homoarginine and cardiovascular disease. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, R.N.; Oppici, E.; Martens-Lobenhoffer, J.; Jarzebska, N.; Brilloff, S.; Burdin, D.; Demyanov, A.; Kolouschek, A.; Leiper, J.; Maas, R.; et al. A Novel Pathway for Metabolism of the Cardiovascular Risk Factor Homoarginine by alanine: Glyoxylate aminotransferase 2. Sci. Rep. 2016, 6, 35277. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, R.N.; Begmatov, H.; Jarzebska, N.; Patel, K.; Mills, M.T.; Ghani, Z.; Khakshour, D.; Tamboli, P.; Patel, M.N.; Abdalla, M.; et al. Homoarginine Supplementation Prevents Left Ventricular Dilatation and Preserves Systolic Function in a Model of Coronary Artery Disease. J. Am. Heart Assoc. 2019, 8, e012486. [Google Scholar] [CrossRef] [Green Version]
- Baylis, C. Nitric oxide deficiency in chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2008, 294, F1–F9. [Google Scholar] [CrossRef] [Green Version]
- Martens, C.R.; Edwards, D.G. Peripheral vascular dysfunction in chronic kidney disease. Cardiol. Res. Pract. 2011, 2011, 267257. [Google Scholar] [CrossRef]
- Baylis, C. Nitric oxide synthase derangements and hypertension in kidney disease. Curr. Opin. Nephrol. Hypertens. 2012, 21, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Fliser, D.; Kronenberg, F.; Kielstein, J.T.; Morath, C.; Bode-Böger, S.M.; Haller, H.; Ritz, E. Asymmetric dimethylarginine and progression of chronic kidney disease: The mild to moderate kidney disease study. J. Am. Soc. Nephrol. 2005, 16, 2456–2461. [Google Scholar] [CrossRef]
- Schwedhelm, E.; Boger, R.H. The role of asymmetric and symmetric dimethylarginines in renal disease. Nat. Rev. Nephrol. 2011, 7, 275–285. [Google Scholar] [CrossRef]
- Caplin, B.; Leiper, J. Endogenous nitric oxide synthase inhibitors in the biology of disease: Markers, mediators, and regulators? Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1343–1353. [Google Scholar] [CrossRef]
- Tomlinson, J.A.; Caplin, B.; Boruc, O.; Bruce-Cobbold, C.; Cutillas, P.; Dormann, D.; Faull, P.; Grossman, R.C.; Khadayate, S.; Mas, V.R.; et al. Reduced Renal Methylarginine Metabolism Protects against Progressive Kidney Damage. J. Am. Soc. Nephrol. 2015, 26, 3045–3059. [Google Scholar] [CrossRef]
- Martens-Lobenhoffer, J.; Emrich, I.E.; Zawada, A.M.; Fliser, D.; Wagenpfeil, S.; Heine, G.H.; Bode-Böger, S.M. L-Homoarginine and its AGXT2-metabolite GOCA in chronic kidney disease as markers for clinical status and prognosis. Amino Acids 2018, 50, 1347–1356. [Google Scholar] [CrossRef]
- Hu, X.L.; Zeng, W.J.; Li, M.P.; Yang, Y.L.; Kuang, D.B.; Li, H.; Zhang, Y.J.; Jiang, C.; Peng, L.M.; Qi, H.; et al. AGXT2 rs37369 polymorphism predicts the renal function in patients with chronic heart failure. Gene 2017, 637, 145–151. [Google Scholar] [CrossRef]
- Sawada, M.; Yamamoto, H.; Ogasahara, A.; Tanaka, Y.; Kihara, S. Beta-aminoisobutyric acid protects against vascular inflammation through PGC-1beta-induced antioxidative properties. Biochem. Biophys. Res. Commun. 2019, 516, 963–968. [Google Scholar] [CrossRef]
- Albrecht, T.; Schilperoort, M.; Zhang, S.; Braun, J.D.; Qiu, J.; Rodriguez, A.; Pastene, D.O.; Krämer, B.K.; Köppel, H.; Baelde, H.; et al. Carnosine Attenuates the Development of both Type 2 Diabetes and Diabetic Nephropathy in BTBR ob/ob Mice. Sci. Rep. 2017, 7, 44492. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarzebska, N.; Mangoni, A.A.; Martens-Lobenhoffer, J.; Bode-Böger, S.M.; Rodionov, R.N. The Second Life of Methylarginines as Cardiovascular Targets. Int. J. Mol. Sci. 2019, 20, 4592. https://doi.org/10.3390/ijms20184592
Jarzebska N, Mangoni AA, Martens-Lobenhoffer J, Bode-Böger SM, Rodionov RN. The Second Life of Methylarginines as Cardiovascular Targets. International Journal of Molecular Sciences. 2019; 20(18):4592. https://doi.org/10.3390/ijms20184592
Chicago/Turabian StyleJarzebska, Natalia, Arduino A. Mangoni, Jens Martens-Lobenhoffer, Stefanie M. Bode-Böger, and Roman N. Rodionov. 2019. "The Second Life of Methylarginines as Cardiovascular Targets" International Journal of Molecular Sciences 20, no. 18: 4592. https://doi.org/10.3390/ijms20184592
APA StyleJarzebska, N., Mangoni, A. A., Martens-Lobenhoffer, J., Bode-Böger, S. M., & Rodionov, R. N. (2019). The Second Life of Methylarginines as Cardiovascular Targets. International Journal of Molecular Sciences, 20(18), 4592. https://doi.org/10.3390/ijms20184592