Increased ROS Scavenging and Antioxidant Efficiency of Chlorogenic Acid Compound Delivered via a Chitosan Nanoparticulate System for Efficient In Vitro Visualization and Accumulation in Human Renal Adenocarcinoma Cells

 , ,
, ,
Abstract
:
1. Introduction
2. Results
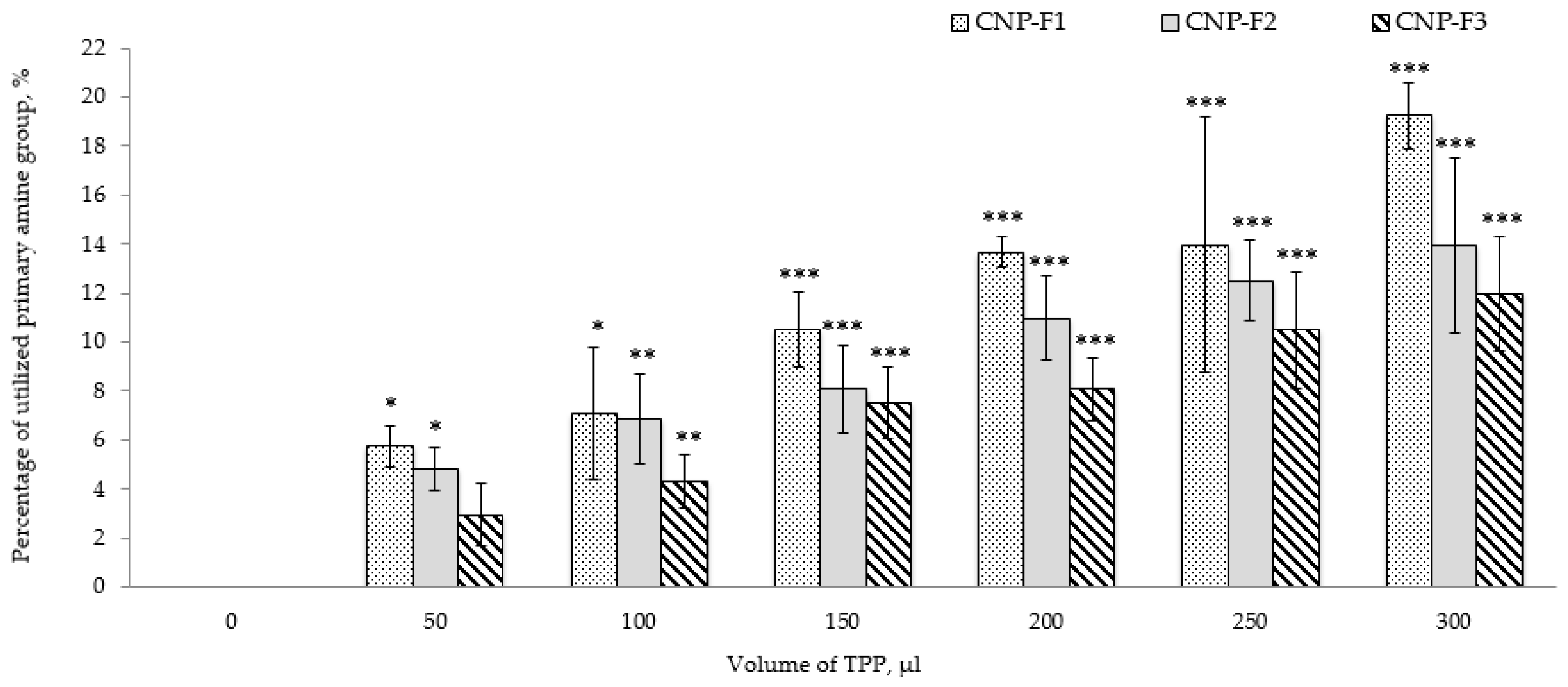
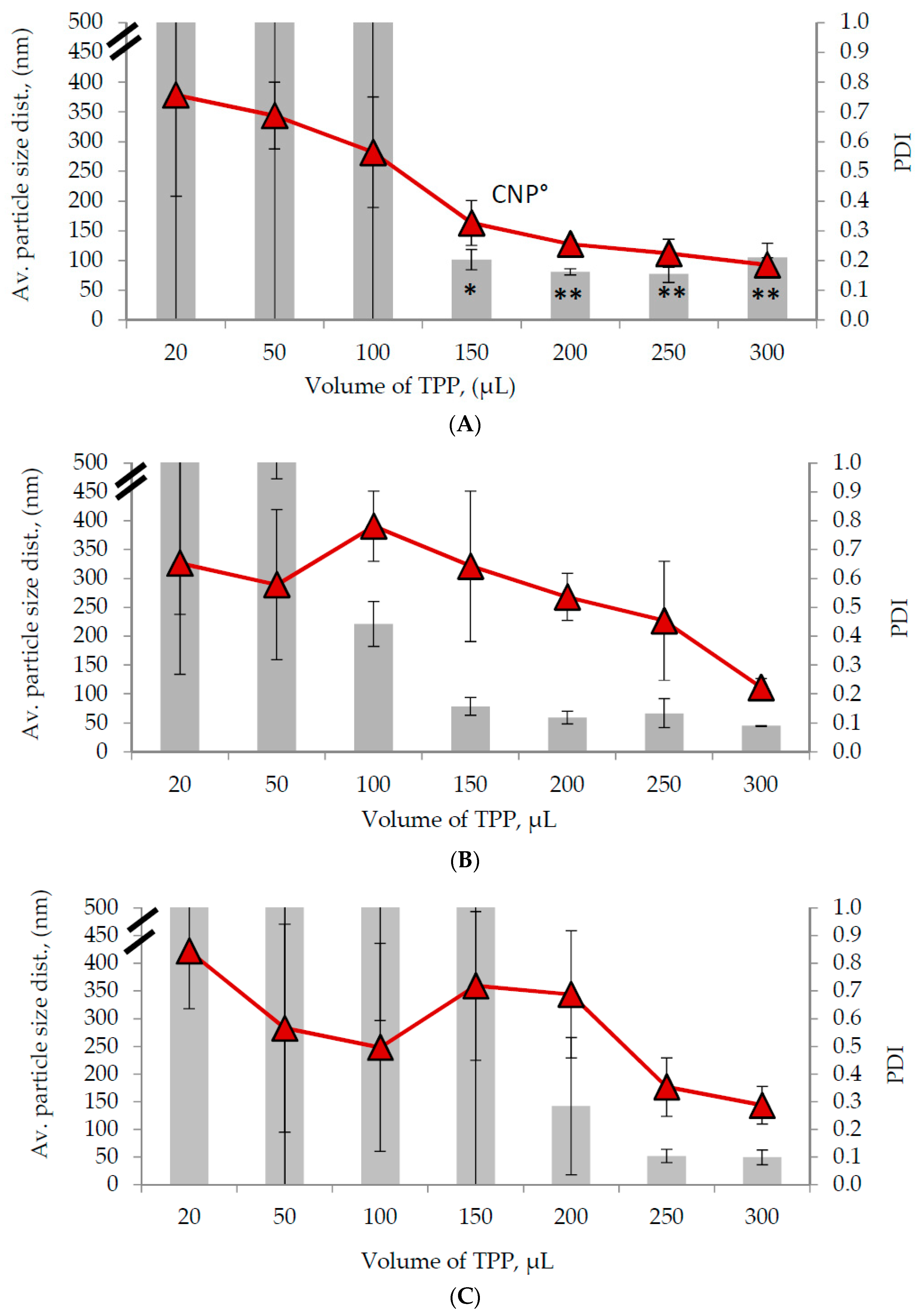
2.1. Characterization of Synthesized CNP and Determination of Optimal Parameters
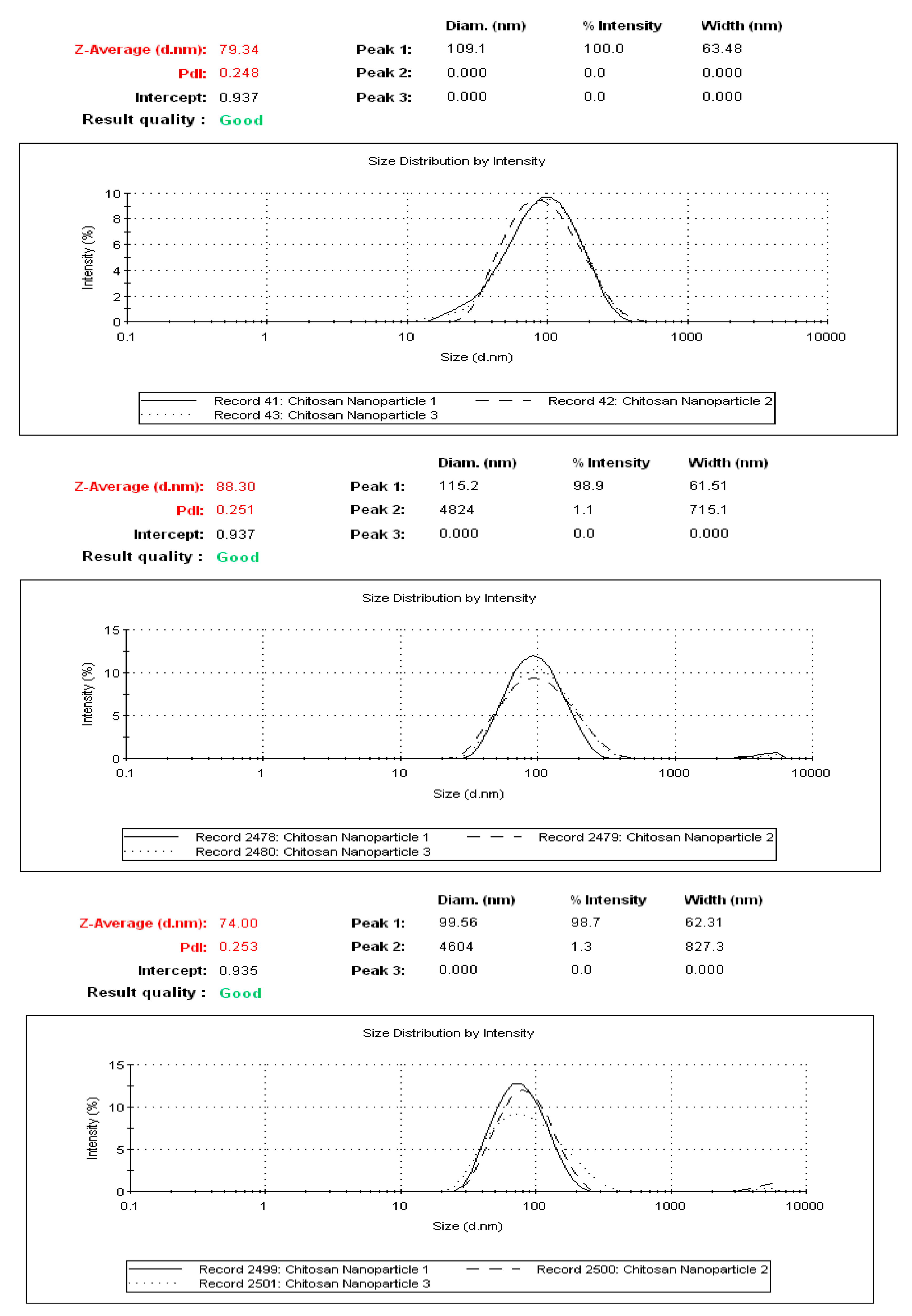
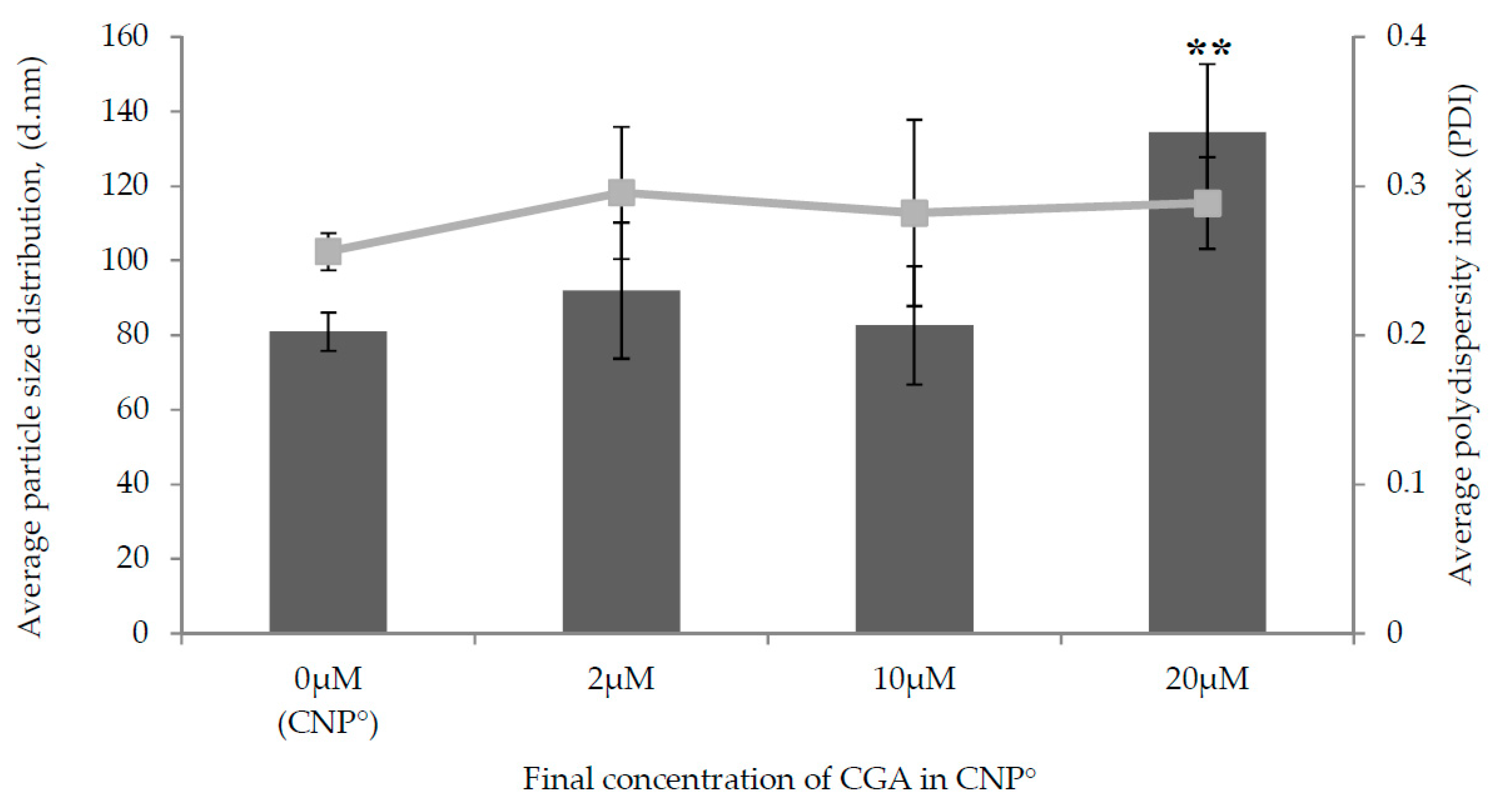
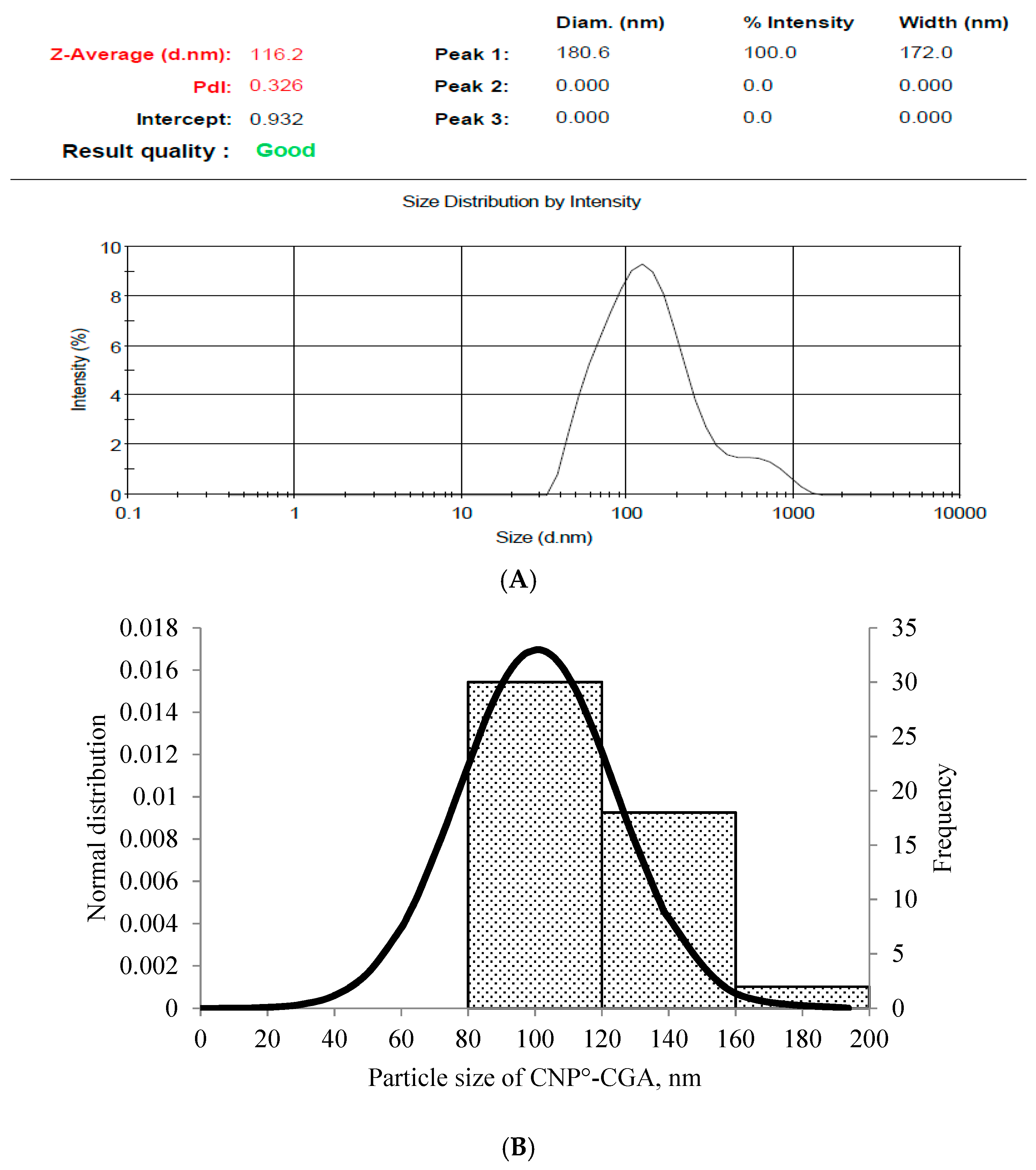
2.2. Physicochemical Characterization of CNP°-CGA Nanoparticles
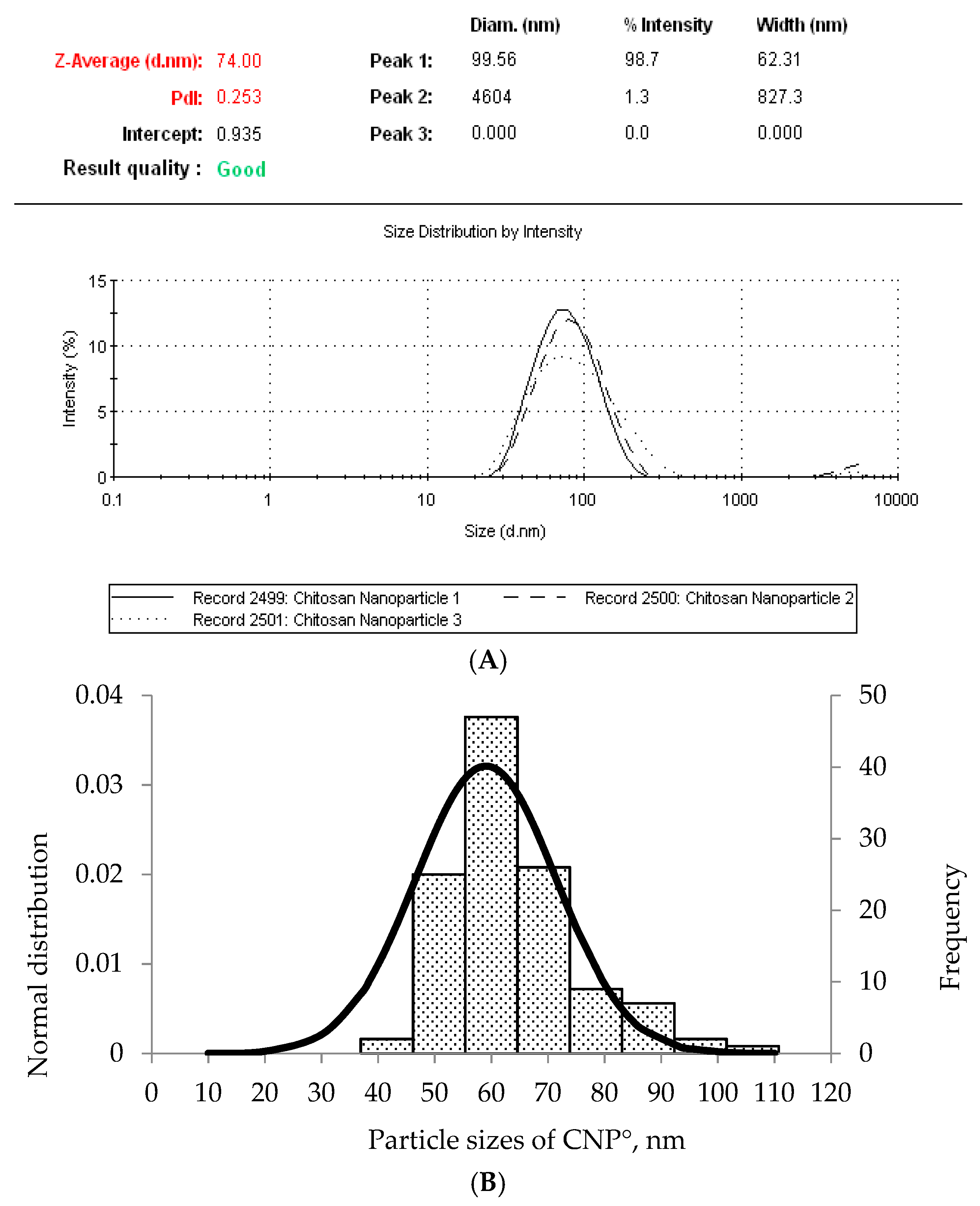
2.3. Morphological Characterization of CNP° and CNP°-CGA Samples
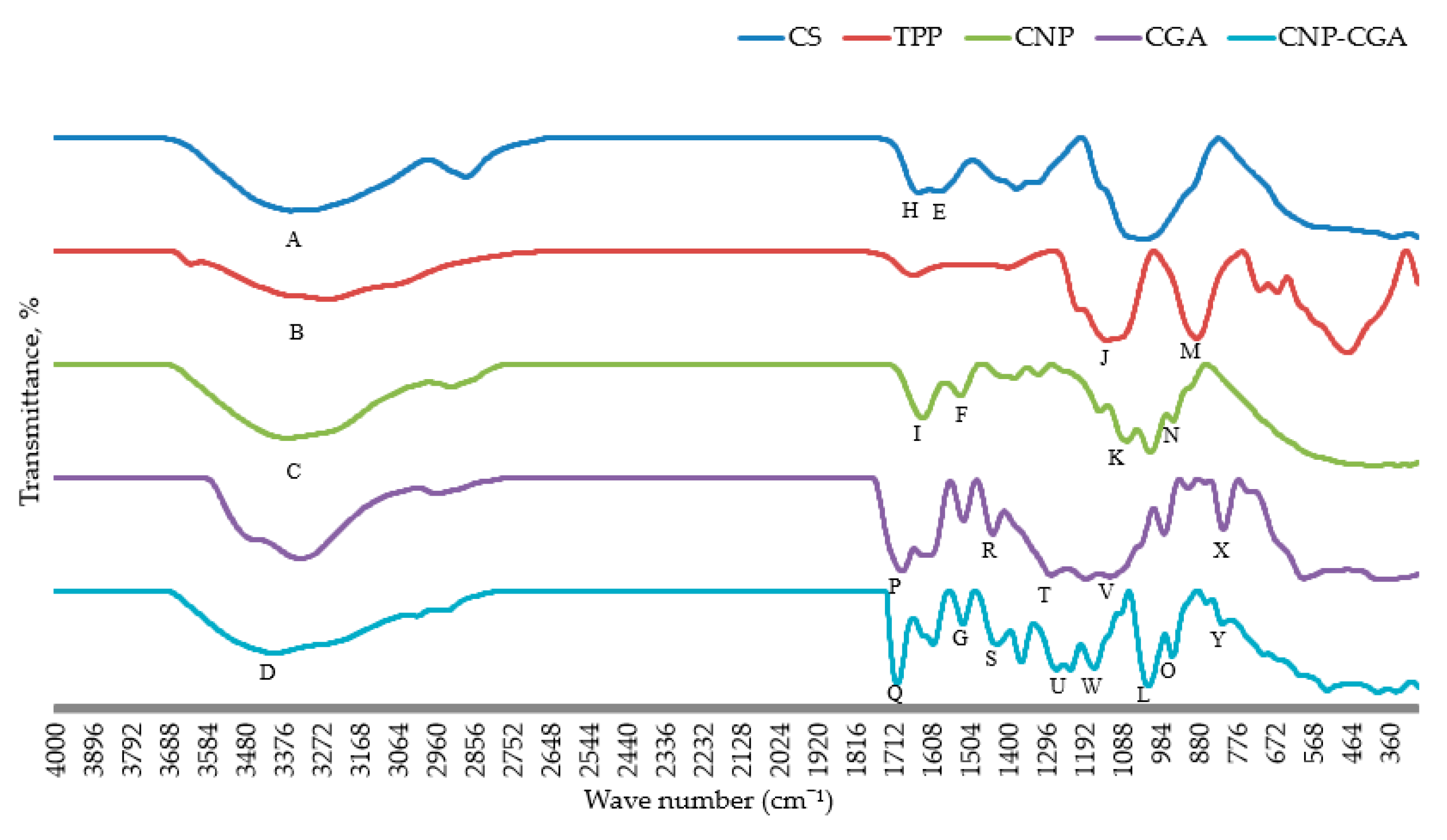
2.4. Functional Group Analysis of CNP° and CNP°-CGA Samples
2.5. Assessment of Antioxidant Potentials of CGA and CNP°-CGA
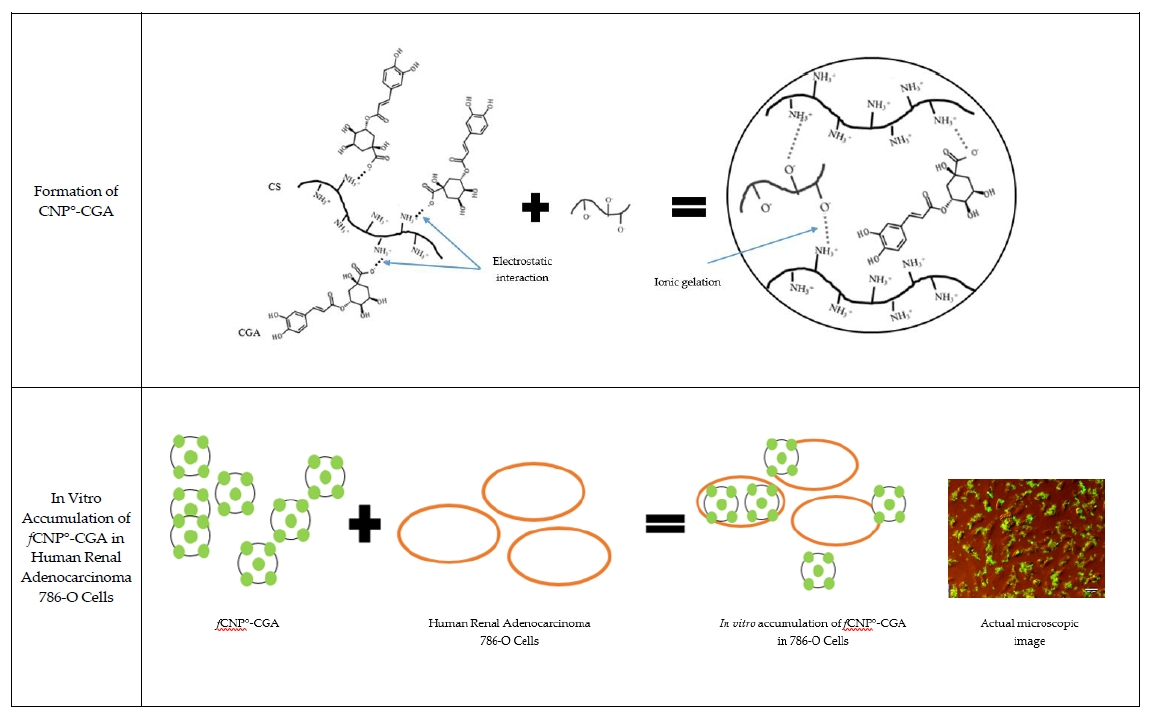
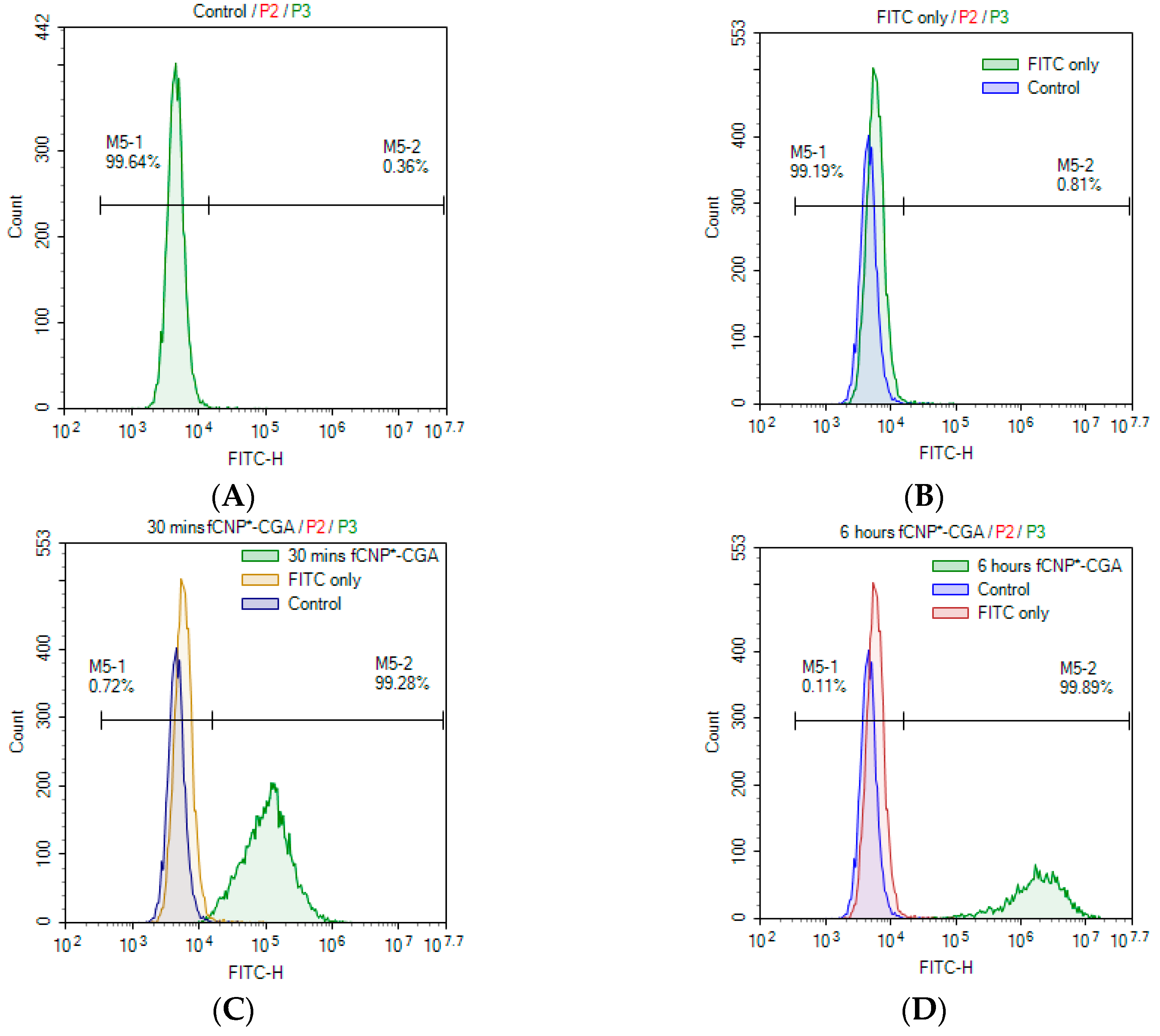
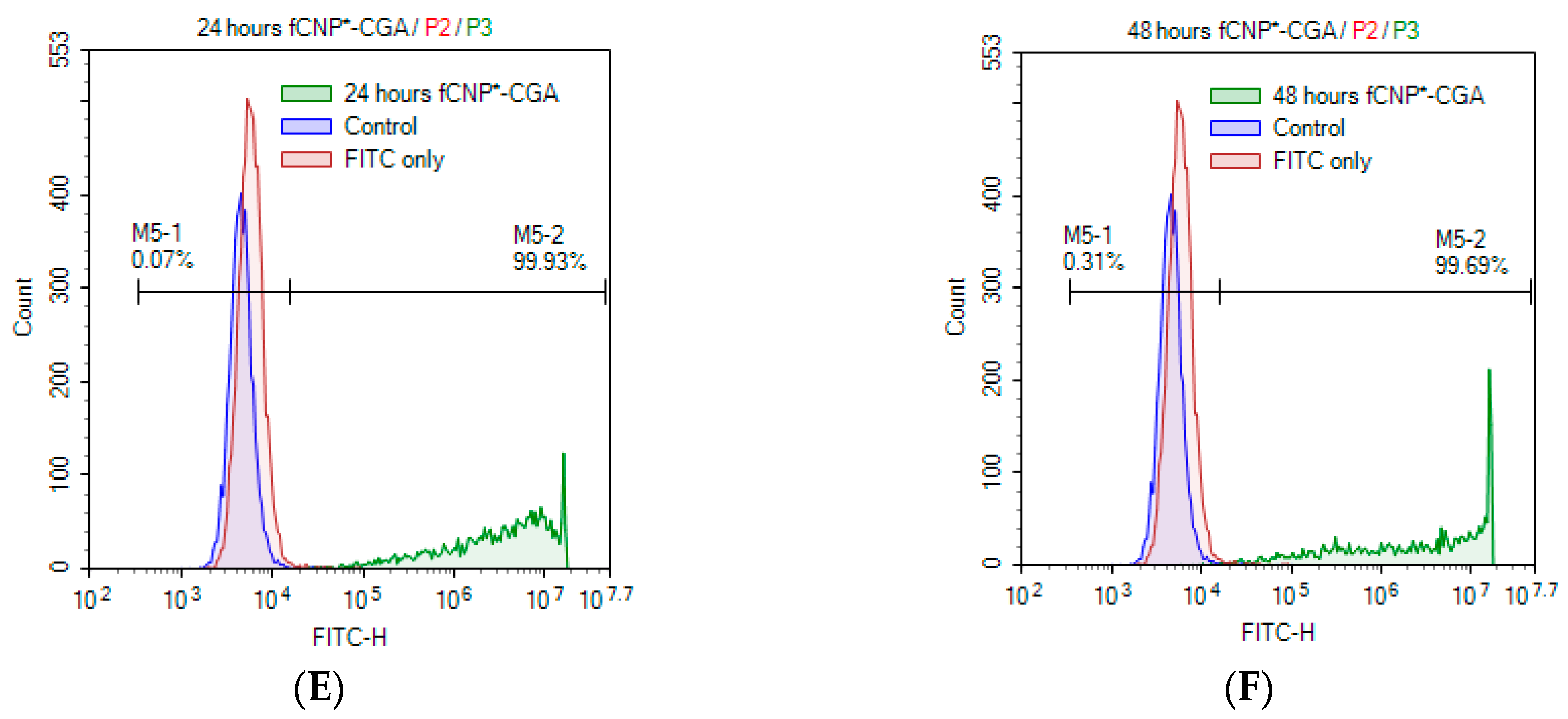
2.6. In Vitro fCNP°-CGA Uptake in Human Renal Adenocarcinoma 786-O Cells
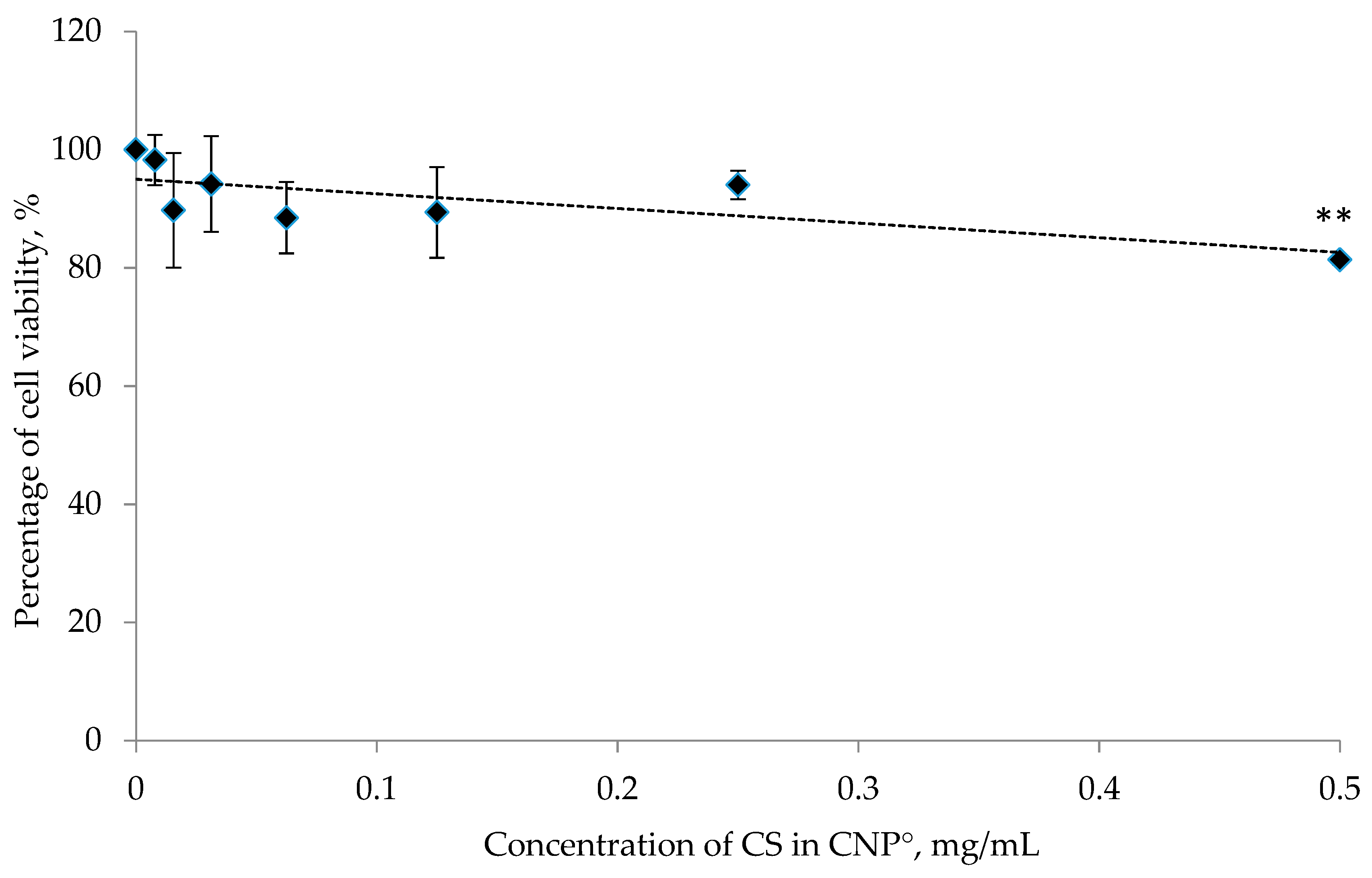
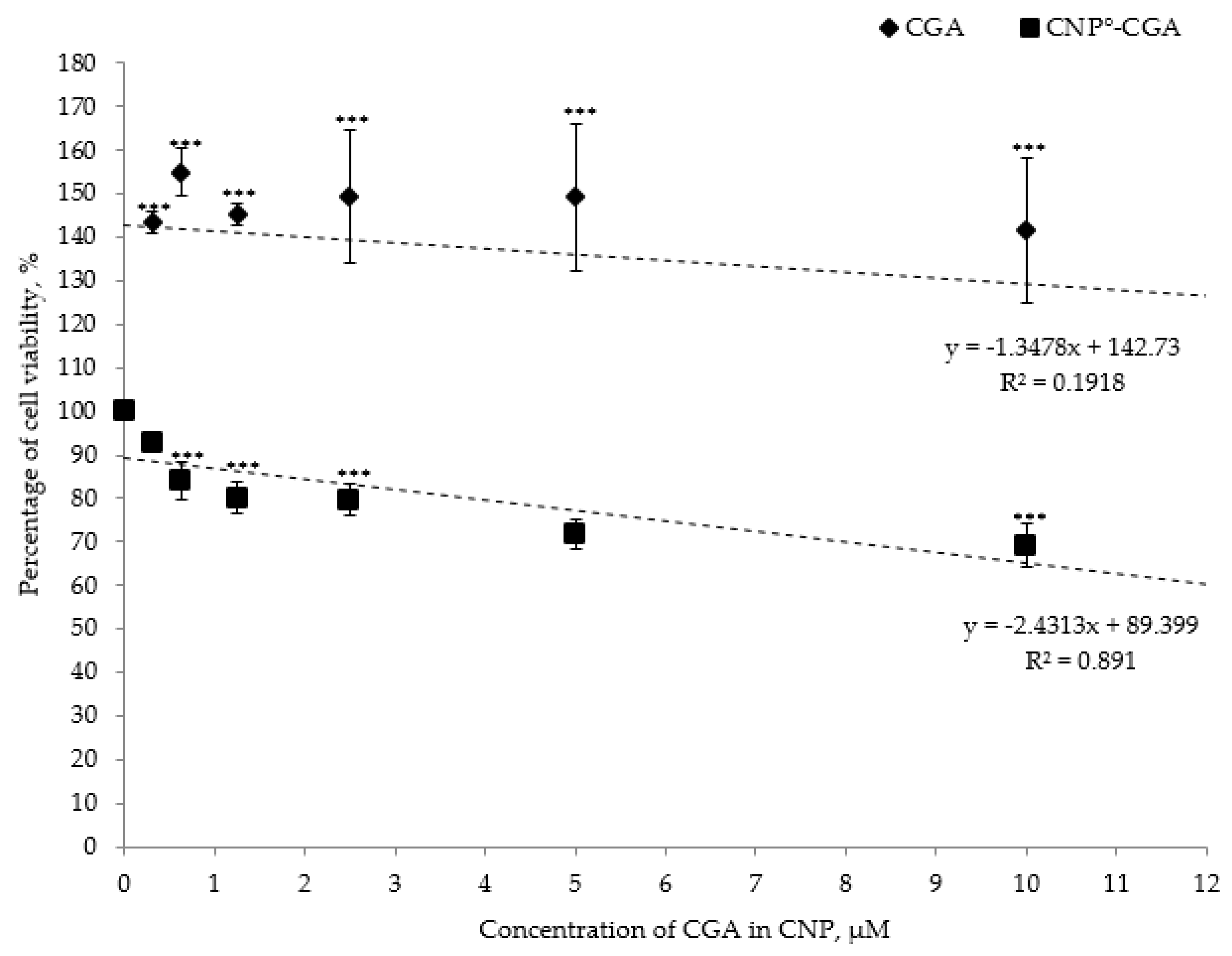
2.7. Cytotoxicity Analysis of CNP°, CGA, and CNP°-CGA
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Chitosan Nanoparticles, CNP
3.3. Quantification of Free Amines in CNP Formulations
3.4. Measurement of Particle Size Distribution, and Polydispersity (PDI) of Nanoparticle Samples
3.5. Synthesis of Chitosan Nanoparticles-Encapsulated Chlorogenic Acid (CNP°-CGA) and Determination of Optimum Parameters for CNP°-CGA Formation Using DLS and Encapsulation Efficiency Analyses
3.6. Morphological Assessment of CNP°, and CNP°-CGA via Electron Microscopy
3.7. Characterization of Functional Groups via Fourier-Transform-Infrared (FTIR) Spectroscopy
3.8. Evaluation of Free Radical Scavenging Activities of CGA Alone and CNP°-CGA
3.9. Propagation and Maintenance of Human Renal Adenocarcinoma Cells, 786–O Cells
3.10. MTT-Based Cytotoxicity Analyses of CGA, CNP°, and CNP°-CGA
3.11. Fluorescent Labeling of Chitosan Nanoparticles-Encapsulated Chlorogenic Acid, fCNP°-CGA
3.12. In Vitro Visualization and Quantification of fCNP°-CGA in 786–O Cancer Cells
3.13. Statistical Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| µL | Microliter |
| μM | Micromolar |
| 5-CQA | 5-O-caffeoylquinic acid |
| 786-O | Human renal cancer cell line |
| ANOVA | One Way Analysis of Variance |
| ARE | Antioxidant response element |
| °C | Degree Celsius |
| CGA | Chlorogenic acid |
| CGA-EE% | CGA encapsulation efficacy |
| CGA-L | CGA loading |
| CGM | Complete growth media |
| cm | Centimeter |
| CNP | Chitosan nanoparticles |
| CNP° | Chitosan nanoparticles at optimum parameter |
| CNP°-CGA | Chitosan nanoparticles-encapsulated chlorogenic acid at optimal parameter |
| CNP-F1 | Chitosan nanoparticle-Formulation 1 |
| CNP-F2 | Chitosan nanoparticle-Formulation 2 |
| CNP-F3 | Chitosan nanoparticle-Formulation 3 |
| CO2 | Carbon dioxide |
| CS | Chitosan |
| DDS | Drug delivery systems |
| DLS | Dynamic light scattering |
| DMSO | Dimethyl sulfoxide |
| DNA | Deoxyribonucleic acid |
| DPPH | 1,1-Diphenyl-2-picrylhydrazine |
| EDTA | Ethylenediaminetetraacetic acid |
| EE% | Encapsulation efficiency percentage |
| EPR | Enhanced permeability and retention effect |
| FBS | Fetal Bovine Serum solution |
| fCNP° | FITC labeled chitosan nanoparticles |
| fCNP°-CGA | FITC labeled chitosan nanoparticles-encapsulated chlorogenic acid |
| FESEM | Field emission scanning electron microscope |
| FITC | Fluorescein 5(6)-isothiocyanate |
| FTIR | Fourier-transform infrared spectroscopy |
| GST | Glutathione S-transferases |
| HCl | Hydrochloric acid |
| HeLa | Human epithelial cell line |
| IC50 | Concentration of a sample required to reduce the biological target by 50% |
| MCF-7 | Breast cancer cell line |
| MDA-MB-435 | Breast cancer cell line |
| Mg | Milligram |
| mL | Milliliter |
| mM | Millimolar |
| MTT | 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide |
| NaCO3 | Sodium carbonate |
| NaOH | Sodium hydroxide |
| NF-κB | Nuclear factor κB |
| NH2 | Primary amine functional groups |
| -NH3+ | Protonated primary amine groups of CS |
| NIH 3T3 | Mouse fibroblast NIH 3T3 cell line |
| NQO1 | NAD(P)H:quinone oxidoreductase 1 |
| nm | Nanometer |
| OH | Hydroxyl |
| OH- | Hydroxide ions |
| PBS | Phosphate buffered saline |
| PDI | Polydispersity index |
| pH | potential of Hydrogen |
| -PO43- | Anionic phosphate groups |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| rpm | Revolutions per minute |
| RPMI | Roswell Park Memorial Institute |
| SD | Standard deviation |
| sdH2O | Sterile distilled water |
| SDS | Sodium dodecyl sulfate |
| siRNA | Small interfering RNA |
| TEM | Transmission electron microscopy |
| TNBS | 2,4,6-trinitrobenzene sulfonic acid |
| TPP | Sodium tripolyphosphate |
| UV | Ultraviolet |
References
- Farah, A.; Monteiro, M.; Donangelo, C.M.; Lafay, S. Chlorogenic acids from green coffee extract are highly bioavailable in humans. J. Nutr. 2008, 138, 2309–2315. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Itagaki, S.; Kurokawa, T.; Ogura, J.; Kobayashi, M.; Hirano, T.; Sugawara, M.; Iseki, K. In vitro and in vivo antioxidant properties of chlorogenic acid and caffeic acid. Int. J. Pharm. 2011, 403, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Cui, M.; Zhao, S.; Chen, X.; Tang, X. Salinity stress is beneficial to the accumulation of chlorogenic acids in honeysuckle (Lonicera japonica Thunb.). Front. Plant. Sci. 2016, 7, 1563. [Google Scholar] [CrossRef] [PubMed]
- Clifford, M.N. Chlorogenic acids and other cinnamates–nature, occurrence and dietary burden. J. Sci. Food Agric. 1999, 79, 362–372. [Google Scholar] [CrossRef]
- Clifford, M.N. Chlorogenic acids and other cinnamates–nature, occurrence, dietary burden, absorption and metabolism. J. Sci. Food Agric. 2000, 80, 1033–1043. [Google Scholar] [CrossRef]
- Niggeweg, R.; Michael, A.J.; Martin, C. Engineering plants with increased levels of the antioxidant chlorogenic acid. Nat. Biotechnol. 2004, 22, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Farah, A.; Donangelo, C.M. Phenolic compounds in coffee. Braz. J. Plant. Physiol. 2006, 18, 23–36. [Google Scholar] [CrossRef]
- Nallamuthu, I.; Devi, A.; Khanum, F. Chlorogenic acid loaded chitosan nanoparticles with sustained release property, retained antioxidant activity and enhanced bioavailability. Asian J. Pharm. Sci. 2015, 10, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic. Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Kono, Y.; Kobayashi, K.; Tagawa, S.; Adachi, A.; Ueda, A.; Sawa, Y.; Shibata, H. Antioxidant activity of polyphenolics in diets: Rate constants of reactions of chlorogenic acid and caffeic acid with reactive species of oxygen and nitrogen. Biochim. Et Biophys. Acta 1997, 1335, 335–342. [Google Scholar] [CrossRef]
- Kono, Y.; Shibata, H.; Kodama, Y.; Sawa, Y. The suppression of the N-nitrosating reaction by chlorogenic acid. Biochem. J. 1995, 312, 947–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.Y.; Cosma, G.; Gardner, H.; Vallyathan, V.; Castranova, V. Effect of chlorogenic acid on hydroxyl radical. Mol. Cell. Biochem. 2003, 247, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Santana-Gálvez, J.; Cisneros-Zevallos, L.; Jacobo-Velázquez, D.A. Chlorogenic acid: Recent advances on its dual role as a food additive and a nutraceutical against metabolic syndrome. Molecules 2017, 22, 358. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Foo, L.Y. Antioxidant and radical scavenging activities of polyphenols from apple pomace. Food Chem. 2000, 68, 81–85. [Google Scholar] [CrossRef]
- Wang, G.F.; Shi, L.P.; Ren, Y.D.; Liu, Q.F.; Liu, H.F.; Zhang, R.J.; Tao, P.Z. Anti-hepatitis B virus activity of chlorogenic acid, quinic acid and caffeic acid in vivo and in vitro. Antivir. Res. 2009, 83, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.S.; Lee, D.G. Antifungal action of chlorogenic acid against pathogenic fungi, mediated by membrane disruption. Pure Appl. Chem. 2010, 82, 219–226. [Google Scholar] [CrossRef]
- Lou, Z.; Wang, H.; Zhu, S.; Ma, C.; Wang, Z. Antibacterial activity and mechanism of action of chlorogenic acid. J. Food Sci. 2011, 76, M398–M403. [Google Scholar] [CrossRef]
- Cho, A.S.; Jeon, S.M.; Kim, M.J.; Yeo, J.; Seo, K.; Choi, M.S.; Lee, M.K. Chlorogenic acid exhibits anti-obesity property and improves lipid metabolism in high-fat diet-induced-obese mice. Food Chem. Toxicol. 2010, 48, 937–943. [Google Scholar] [CrossRef]
- Kasai, H.; Fukada, S.; Yamaizumi, Z.; Sugie, S.; Mori, H. Action of chlorogenic acid in vegetables and fruits as an inhibitor of 8-hydroxydeoxyguanosine formation in vitro and in a rat carcinogenesis model. Food Chem. Toxicol. 2000, 38, 467–471. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, J.; Ballevre, O.; Luo, H.; Zhang, W. Antihypertensive effects and mechanisms of chlorogenic acids. Hypertens. Res. 2012, 35, 370–374. [Google Scholar] [CrossRef]
- Meng, S.; Cao, J.; Feng, Q.; Peng, J.; Hu, Y. Roles of chlorogenic acid on regulating glucose and lipids metabolism: A review. Evid. Based Complement. Alternat. Med. 2013, 2013, 801457. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shen, D.; Tang, X.; Li, X.; Wo, D.; Yan, H.; Li, J. Chlorogenic acid prevents isoproterenol-induced hypertrophy in neonatal rat myocytes. Toxicol. Lett. 2014, 226, 257–263. [Google Scholar] [CrossRef] [PubMed]
- De Maria, C.A.B.; Trugo, L.C.; e Miranda, L.D.M.; Salvador, E. Stability of 5-caffeoylquinic acid under different conditions of heating. Food Res. Int. 1998, 31, 475–477. [Google Scholar] [CrossRef]
- Friedman, M.; Jürgens, H.S. Effect of pH on the stability of plant phenolic compounds. J. Agric. Food Chem. 2000, 48, 2101–2110. [Google Scholar] [CrossRef] [PubMed]
- Zanoelo, E.F.; Beninca, C. Chemical kinetics of 5-o-caffeoylquinic acid in superheated steam: Effect of isomerization on mate (Ilex paraguariensis) manufacturing. J. Agric. Food Chem. 2009, 57, 11564–11569. [Google Scholar] [CrossRef] [PubMed]
- Dawidowicz, A.L.; Wianowska, D.; Olszowy, M. On practical problems in estimation of antioxidant activity of compounds by DPPH method (Problems in estimation of antioxidant activity). Food Chem. 2012, 131, 1037–1043. [Google Scholar] [CrossRef]
- Ma, Z.; Lim, L.Y. Uptake of chitosan and associated insulin in Caco-2 cell monolayers: A comparison between chitosan molecules and chitosan nanoparticles. Pharm. Res. 2003, 20, 1812–1819. [Google Scholar] [CrossRef]
- Olthof, M.R.; Hollman, P.C.H.; Katan, M.B. Chlorogenic acid and caffeic acid are absorbed in humans. J. Nutr. 2001, 131, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Gonthier, M.P.; Verny, M.A.; Besson, C.; Rémésy, C.; Scalbert, A. Chlorogenic acid bioavailability largely depends on its metabolism by the gut microflora in rats. J. Nutr. 2003, 133, 1853–1859. [Google Scholar] [CrossRef]
- Dupas, C.; Marsset Baglieri, A.; Ordonaud, C.; Tomé, D.; Maillard, M.N. Chlorogenic acid is poorly absorbed, independently of the food matrix: A Caco-2 cells and rat chronic absorption study. Mol. Nutr. Food Res. 2006, 50, 1053–1060. [Google Scholar] [CrossRef]
- Barahuie, F.; Hussein, M.Z.; Arulselvan, P.; Fakurazi, S.; Zainal, Z. Drug delivery system for an anticancer agent, chlorogenate-Zn/Al-layered double hydroxide nanohybrid synthesized using direct co-precipitation and ion exchange methods. J. Solid State Chem. 2014, 217, 31–41. [Google Scholar] [CrossRef]
- Burgos-Morón, E.; Calderón-Montaño, J.M.; Orta, M.L.; Pastor, N.; Pérez-Guerrero, C.; Austin, C.; López-Lázaro, M. The coffee constituent chlorogenic acid induces cellular DNA damage and formation of topoisomerase I–and II–DNA complexes in cells. J. Agric. Food Chem. 2012, 60, 7384–7391. [Google Scholar] [CrossRef] [PubMed]
- Olthof, M.R.; Hollman, P.C.; Zock, P.L.; Katan, M.B. Consumption of high doses of chlorogenic acid, present in coffee, or of black tea increases plasma total homocysteine concentrations in humans. Am. J. Clin. Nutr. 2001, 73, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Du, W.Y.; Chang, C.; Zhang, Y.; Liu, Y.Y.; Sun, K.; Wang, C.S.; Wang, M.X.; Liu, Y.; Wang, F.; Fan, J.Y.; et al. High-dose chlorogenic acid induces inflammation reactions and oxidative stress injury in rats without implication of mast cell degranulation. J. Ethnopharmacol. 2013, 147, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Barahuie, F.; Hussein, M.Z.; Arulselvan, P.; Fakurazi, S.; Zainal, Z. Controlled in vitro release of the anticancer drug chlorogenic acid using magnesium/aluminium-layered double hydroxide as a nanomatrix. Sci. Adv. Mater. 2016, 8, 501–513. [Google Scholar] [CrossRef]
- Gouthamchandra, K.; Sudeep, H.V.; Venkatesh, B.J.; Prasad, K.S. Chlorogenic acid complex (CGA7), standardized extract from green coffee beans exerts anticancer effects against cultured human colon cancer HCT-116 cells. Food Sci. Hum. Wellness 2017, 6, 147–153. [Google Scholar] [CrossRef]
- Shi, G.; Rao, L.; Yu, H.; Xiang, H.; Pen, G.; Long, S.; Yang, C. Yeast-cell-based microencapsulation of chlorogenic acid as a water-soluble antioxidant. J. Food Eng. 2007, 80, 1060–1067. [Google Scholar] [CrossRef]
- Naso, L.G.; Valcarcel, M.; Roura-Ferrer, M.; Kortazar, D.; Salado, C.; Lezama, L.; Ferrer, E.G. Promising antioxidant and anticancer (human breast cancer) oxidovanadium (IV) complex of chlorogenic acid. Synthesis, characterization and spectroscopic examination on the transport mechanism with bovine serum albumin. J. Inorg. Biochem. 2014, 135, 86–99. [Google Scholar] [CrossRef]
- Barahuie, F.; Saifullah, B.; Dorniani, D.; Fakurazi, S.; Karthivashan, G.; Hussein, M.Z.; Elfghi, F.M. Graphene oxide as a nanocarrier for controlled release and targeted delivery of an anticancer active agent, chlorogenic acid. Mater. Sci. Eng. Cmaterials Biol. Appl. 2017, 74, 177–185. [Google Scholar] [CrossRef]
- Agnihotri, S.A.; Mallikarjuna, N.N.; Aminabhavi, T.M. Recent advances on chitosan-based micro-and nanoparticles in drug delivery. J. Control. Release 2004, 100, 5–28. [Google Scholar] [CrossRef]
- Shapi¿i, R.A.; Othman, S.H.; Nazli Naim, M.; Kadir Basha, R. Effect of ball milling and ultrasonication time on particle size of chitosan for potential nanofiller in food packaging film. Acta Hortic 2017, 1152, 125–130. [Google Scholar] [CrossRef]
- Mohanraj, V.J.; Chen, Y.; Suresh, B. Chitosan-based nanoparticles for delivery of proteins and peptides. Indian J. Pharm. Educ. Res. 2006, 40, 106. [Google Scholar]
- Szymańska, E.; Winnicka, K. Stability of chitosan—A challenge for pharmaceutical and biomedical applications. Mar. Drugs 2015, 13, 1819–1846. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Ahmad, M.; Swami, B.L.; Ikram, S. A review on plants extract mediated synthesis of silver nanoparticles for antimicrobial applications: A green expertise. J. Adv. Res. 2016, 7, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.K.; Escargueil, A.E.; Skladanowski, A. Resistance mechanisms associated with altered intracellular distribution of anticancer agents. Pharmacol. Ther. 2000, 85, 217–229. [Google Scholar] [CrossRef]
- Panyam, J.; Dali, M.M.; Sahoo, S.K.; Ma, W.; Chakravarthi, S.S.; Amidon, G.L.; Levy, R.J.; Labhasetwar, V. Polymer degradation and in vitro release of a model protein from poly(d,l-lactide-co-glycolide) nano-and microparticles. J. Control. Release 2003, 92, 173–187. [Google Scholar] [CrossRef]
- Cho, K.; Wang, X.U.; Nie, S.; Shin, D.M. Therapeutic nanoparticles for drug delivery in cancer. Clin. Cancer Res. 2008, 14, 1310–1316. [Google Scholar] [CrossRef]
- Zhang, L.; Gu, F.X.; Chan, J.M.; Wang, A.Z.; Langer, R.S.; Farokhzad, O.C. Nanoparticles in medicine: Therapeutic applications and developments. Clin. Pharmacol. Ther. 2008, 83, 761–769. [Google Scholar] [CrossRef]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef]
- Vila, A.; Sánchez, A.; Janes, K.; Behrens, I.; Kissel, T.; Jato, J.L.V.; Alonso, M.J. Low molecular weight chitosan nanoparticles as new carriers for nasal vaccine delivery in mice. Eur. J. Pharm. Biopharm. 2004, 57, 123–131. [Google Scholar] [CrossRef]
- Sannan, T.; Kurita, K.; Iwakura, Y. Studies on chitin, 2. Effect of deacetylation on solubility. Macromol. Chem. Phys. 1976, 177, 3589–3600. [Google Scholar] [CrossRef]
- Masarudin, M.J.; Cutts, S.M.; Evison, B.J.; Phillips, D.R.; Pigram, P.J. Factors determining the stability, size distribution, and cellular accumulation of small, monodisperse chitosan nanoparticles as candidate vectors for anticancer drug delivery: Application to the passive encapsulation of [14C]-doxorubicin. Nanotechnol. Sci. Appl. J. 2015, 8, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.P.; Labhasetwar, V.; Amidon, G.L.; Levy, R.J. Gastrointestinal uptake of biodegradable microparticles: Effect of particle size. Pharm. Res. 1996, 13, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- Barua, S.; Mitragotri, S. Challenges associated with penetration of nanoparticles across cell and tissue barriers: A review of current status and future prospects. Nano Today 2014, 9, 223–243. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Braun, G.B.; Friman, T.; Pang, H.B.; Pallaoro, A.; De Mendoza, T.H.; Willmore, A.M.A.; Reich, N.O. Etchable plasmonic nanoparticle probes to image and quantify cellular internalization. Nat. Mater. 2014, 13, 904. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Yan, W.; Xu, Z.; Ni, H. Formation mechanism of monodisperse, low molecular weight chitosan nanoparticles by ionic gelation technique. Colloids Surf. B Biointerfaces 2012, 90, 21–27. [Google Scholar] [CrossRef]
- Udaybhaskar, P.; Iyengar, L.; Rao, A.V.S. Hexavalent chromium interaction with chitosan. J. Appl. Polym. Sci. 1990, 39, 739–747. [Google Scholar] [CrossRef]
- Ramanery, F.P.; Mansur, A.A.; Mansur, H.S. One-step colloidal synthesis of biocompatible water-soluble ZnS quantum dot/chitosan nanoconjugates. Nanoscale Res. Lett. 2013, 8, 512. [Google Scholar] [CrossRef]
- Xiao, Z.; Tian, T.; Hu, J.; Wang, M.; Zhou, R. Preparation and characterization of chitosan nanoparticles as the delivery system for tuberose fragrance. Flavour Fragr. J. 2014, 29, 22–34. [Google Scholar] [CrossRef]
- Tsai, M.L.; Chen, R.H.; Bai, S.W.; Chen, W.Y. The storage stability of chitosan/tripolyphosphate nanoparticles in a phosphate buffer. Carbohydr. Polym. 2011, 84, 756–761. [Google Scholar] [CrossRef]
- Bhumkar, D.R.; Pokharkar, V.B. Studies on effect of pH on cross-linking of chitosan with sodium tripolyphosphate: A technical note. Aaps Pharmascitech 2006, 7, E138–E143. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.A.; Park, H.J.; Hwang, S.J.; Park, J.B.; Lee, J.S. Preparation and characterization of chitosan microparticles intended for controlled drug delivery. Int. J. Pharm. 2002, 249, 165–174. [Google Scholar] [CrossRef]
- Villegas, R.J.A.; Shimokawa, T.; Okuyama, H.; Kojima, M. Purification and characterization of chlorogenic acid: Chlorogenate caffeoyl transferase in sweet potato roots. Phytochemistry 1987, 26, 1577–1581. [Google Scholar] [CrossRef]
- Oliver, S.; Vittorio, O.; Cirillo, G.; Boyer, C. Enhancing the therapeutic effects of polyphenols with macromolecules. Polym. Chem. 2016, 7, 1529–1544. [Google Scholar] [CrossRef]
- Liu, D.; Mori, A.; Huang, L. Role of liposome size and RES blockade in controlling biodistribution and tumor uptake of GM1-containing liposomes. Biochim. Et Biophys. Acta (Bba)-Biomembr. 1992, 1104, 95–101. [Google Scholar] [CrossRef]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef] [PubMed]
- Ernsting, M.J.; Murakami, M.; Roy, A.; Li, S.D. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release 2013, 172, 782–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagda, C.D.; Chotai, N.P.; Patel, S.B.; Soni, T.J.; Patel, U.L. Preparation and in vitro evaluation of bioadhesive microparticulate system. Int. J. Pharm. Sci. Nanotechnol. 2008, 1, 257–266. [Google Scholar]
- Pavan Veena, C.; Kavitha, K.; Anil Kumar, S.N. Formulation and evaluation of Trimetazidine hydrochloride loaded chitosan microspheres. Int. J. Appl. Pharm. 2010, 2, 11–14. [Google Scholar]
- Raja, M.A.G.; Katas, H.; Wen, T.J. Stability, intracellular delivery, and release of siRNA from chitosan nanoparticles using different cross-linkers. PLoS ONE 2015, 10, e0128963. [Google Scholar]
- Pekarskaya, E.; Kim, C.P.; Johnson, W.L. In situ transmission electron microscopy studies of shear bands in a bulk metallic glass based composite. J. Mater. Res. 2001, 16, 2513–2518. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, B.; Cheng, W.H.; Wang, Q. Preparation, characterization and evaluation of selenite-loaded chitosan/TPP nanoparticles with or without zein coating. Carbohydr. Polym. 2010, 82, 942–951. [Google Scholar] [CrossRef]
- Chuah, L.H.; Billa, N.; Roberts, C.J.; Burley, J.C.; Manickam, S. Curcumin-containing chitosan nanoparticles as a potential mucoadhesive delivery system to the colon. Pharm. Dev. Technol. 2013, 18, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Ariff, S.A.Y.; Yusoff, K.; Masarudin, M.J. Encapsulation of miRNA in chitosan nanoparticles as a candidate for an anti-metastatic agent in cancer therapy. Malays. Appl. Biol. 2017, 46, 165–170. [Google Scholar]
- Xu, Y.; Du, Y. Effect of molecular structure of chitosan on protein delivery properties of chitosan nanoparticles. Int. J. Pharm. 2003, 250, 215–226. [Google Scholar] [CrossRef]
- Gierszewska-Drużyńska, M.; Ostrowska-Czubenko, J. The effect of ionic crosslinking on thermal properties of hydrogel chitosan membranes. Prog. Chem. Appl. Chitin Its Deriv. 2010, 16, 25–32. [Google Scholar]
- Rodrigues, S.; da Costa, A.M.R.; Grenha, A. Chitosan/carrageenan nanoparticles: Effect of cross-linking with tripolyphosphate and charge ratios. Carbohydr. Polym. 2012, 89, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Catauro, M.; Barrino, F.; Dal Poggetto, G.; Pacifico, F.; Piccolella, S.; Pacifico, S. Chlorogenic acid/PEG-based organic-inorganic hybrids: A versatile sol-gel synthesis route for new bioactive materials. Mater. Sci. Eng. 2019, 100, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Gierszewska-Drużyńska, M.; Ostrowska-Czubenko, J. Influence of crosslinking process conditions on molecular and supermolecular structure of chitosan hydrogel membrane. Prog. Chem. Appl. Chitin Its Deriv. 2011, 15, 22. [Google Scholar]
- Martins, A.F.; de Oliveira, D.M.; Pereira, A.G.; Rubira, A.F.; Muniz, E.C. Chitosan/TPP microparticles obtained by microemulsion method applied in controlled release of heparin. Int. J. Biol. Macromol. 2012, 51, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, R.; Pulat, M. 5-Fluorouracil encapsulated chitosan nanoparticles for pH-stimulated drug delivery: Evaluation of controlled release kinetics. J. Nanomater. 2012, 2012, 42. [Google Scholar]
- Huang, Y.C.; Li, R.Y. Preparation and characterization of antioxidant nanoparticles composed of chitosan and fucoidan for antibiotics delivery. Mar. Drugs 2014, 12, 4379–4398. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, Y.; Ferrer, E.G.; Baró, A.G.; Williams, P.A.M. Oxovanadium (IV) complexes of quinic acid. Synthesis, characterization and potentiometric study. Polyhedron 2000, 19, 2613–2619. [Google Scholar] [CrossRef]
- Cornard, J.P.; Lapouge, C. Theoretical and spectroscopic investigations of a complex of Al (III) with caffeic acid. J. Phys. Chem. A 2004, 108, 4470–4478. [Google Scholar] [CrossRef]
- Biswas, N.; Kapoor, S.; Mahal, H.S.; Mukherjee, T. Adsorption of CGA on colloidal silver particles: DFT and SERS study. Chem. Phys. Lett. 2007, 444, 338–345. [Google Scholar] [CrossRef]
- El-Abassy, R.M.; Donfack, P.; Materny, A. Discrimination between Arabica and Robusta green coffee using visible micro Raman spectroscopy and chemometric analysis. Food Chem. 2011, 126, 1443–1448. [Google Scholar] [CrossRef]
- Barahuie, F.; Hussein, M.Z.; Arulselvan, P.; Fakurazi, S.; Zainal, Z. Development of the anticancer potential of a chlorogenate-zinc layered hydroxide nanohybrid with controlled release property against various cancer cells. Sci. Adv. Mater. 2013, 5, 1983–1993. [Google Scholar] [CrossRef]
- Leaves, L. Antioxidant activity by DPPH radical scavenging method of ageratum conyzoides. Am. J. Ethnomedicine. 2014, 1, 244–249. [Google Scholar]
- Pisoschi, A.M.; Cheregi, M.C.; Danet, A.F. Total antioxidant capacity of some commercial fruit juice: Electrochemical and spectrophotometrical approaches. Molecule 2009, 14, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Medhe, S.; Bansal, P.; Srivastava, M.M. Enhanced antioxidant activity of gold nanoparticle embedded 3, 6-dihydroxyflavone: A combinational study. Appl. Nanosci. 2014, 4, 153–161. [Google Scholar] [CrossRef]
- Valgimigli, L.; Amorati, R.; Fumo, M.G.; Dilabio, G.A.; Pedulli, G.F.; Ingold, K.U.; Pratt, D.A. The unusual reaction of semiquinone radicals with molecular oxygen. J. Org. Chem. 2008, 73, 1830–1841. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Houk, K.N. Substituent effects in the benzene dimer are due to direct interactions of the substituents with the unsubstituted benzene. J. Am. Chem. Soc. 2008, 130, 10854–10855. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Bloom, J.W. Towards a more complete understanding of noncovalent interactions involving aromatic rings. J. Phys. Chem. A 2014, 118, 6133–6147. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, P.; Subhapradha, N.; Shanmugam, V.; Shanmugam, A. Extraction, characterization and antioxidant property of chitosan from cuttlebone Sepia kobiensis (Hoyle 1885). Int. J. Biol. Macromol. 2014, 64, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, C.; Tan, W.; Gu, G.; Guo, Z. Novel amino-pyridine functionalized chitosan quaternary ammonium derivatives: Design, synthesis, and antioxidant activity. Molecules 2017, 22, 156. [Google Scholar] [CrossRef] [PubMed]
- Delie, F. Evaluation of nano-and microparticle uptake by the gastrointestinal tract. Adv. Drug Deliv. Rev. 1998, 34, 221–233. [Google Scholar] [CrossRef]
- Orienti, K.; Aiedeh, E.; Gianasi, V.; Bertasi, V.; Zecchi. Indomethacin loaded chitosan microspheres. Correlation between the erosion process and release kinetics. J. Microencapsul. 1996, 13, 463–472. [Google Scholar] [CrossRef]
- He, P.; Davis, S.S.; Illum, L. Chitosan microspheres prepared by spray drying. Int. J. Pharm. 1999, 187, 53–65. [Google Scholar] [CrossRef]
- Pan, Y.; Li, Y.J.; Zhao, H.Y.; Zheng, J.M.; Xu, H.; Wei, G.; Hao, J.S. Bioadhesive polysaccharide in protein delivery system: Chitosan nanoparticles improve the intestinal absorption of insulin in vivo. Int. J. Pharm. 2002, 249, 139–147. [Google Scholar] [CrossRef]
- Keawchaoon, L.; Yoksan, R. Preparation, characterization and in vitro release study of carvacrol-loaded chitosan nanoparticles. Colloids Surf. B Biointerfaces 2011, 84, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.F.; Zandi, M.; Rezaei, M.; Farahmandghavi, F. Two-step method for encapsulation of oregano essential oil in chitosan nanoparticles: Preparation, characterization and in vitro release study. Carbohydr. Polym. 2013, 95, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Liu, M.; Yang, X.; Zhai, G. The design of pH-sensitive chitosan-based formulations for gastrointestinal delivery. Drug Discov. Today 2015, 20, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Miladi, K.; Sfar, S.; Fessi, H.; Elaissari, A. Enhancement of alendronate encapsulation in chitosan nanoparticles. J. Drug Deliv. Sci. Technol. 2015, 30, 391–396. [Google Scholar] [CrossRef]
- Scheeren, L.E.; Nogueira, D.R.; Macedo, L.B.; Vinardell, M.P.; Mitjans, M.; Infante, M.R.; Rolim, C.M. PEGylated and poloxamer-modified chitosan nanoparticles incorporating a lysine-based surfactant for pH-triggered doxorubicin release. Colloids Surf. B Biointerfaces 2016, 138, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Deng, X.; Li, X. Investigation on a novel core-coated microspheres protein delivery system. J. Control. Release 2001, 75, 27–36. [Google Scholar] [CrossRef]
- Amidi, M.; Romeijn, S.G.; Borchard, G.; Junginger, H.E.; Hennink, W.E.; Jiskoot, W. Preparation and characterization of protein-loaded N-trimethyl chitosan nanoparticles as nasal delivery system. J. Control. Release 2006, 111, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Hu, J.; Park, H.; Lee, M. Chitosan-based nanoparticles as a sustained protein release carrier for tissue engineering applications. J. Biomed. Mater. Res. Part. A 2012, 100, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xu, M.; Li, S.; Shen, X.; Li, T.; Yan, J.; Zhang, C.; Wu, C.; Zeng, H.; Liu, Y. Chitosan hybrid nanoparticles as a theranostic platform for targeted doxorubicin/VEGF shRNA co-delivery and dual-modality fluorescence imaging. RSC Adv. 2016, 6, 29685–29696. [Google Scholar] [CrossRef]
- Jia, X.; Chen, X.; Xu, Y.; Han, X.; Xu, Z. Tracing transport of chitosan nanoparticles and molecules in Caco-2 cells by fluorescent labeling. Carbohydr. Polym. 2009, 78, 323–329. [Google Scholar] [CrossRef]
- Huang, M.; Ma, Z.; Khor, E.; Lim, L.Y. Uptake of FITC-chitosan nanoparticles by A549 cells. Pharm. Res. 2002, 19, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Morgenstern, R.; Nyström, A.M. Nanoparticle-directed sub-cellular localization of doxorubicin and the sensitization breast cancer cells by circumventing GST-mediated drug resistance. Biomaterials 2014, 35, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Liang, N.; Kitts, D.D. Role of chlorogenic acids in controlling oxidative and inflammatory stress conditions. Nutrients 2016, 8, 16. [Google Scholar] [CrossRef]
- Kono, Y.; Shibata, H.; Kodama, Y.; Ueda, A.; Sawa, Y. Chlorogenic acid as a natural scavenger for hypochlorous acid. Biochem. Biophys. Res. Commun. 1995, 217, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Lu, Y.; Bowman, L.L.; Qian, Y.; Castranova, V.; Ding, M. Inhibition of activator protein-1, NF-κB, and MAPKs and induction of phase 2 detoxifying enzyme activity by chlorogenic acid. J. Biol. Chem. 2005, 280, 27888–27895. [Google Scholar] [CrossRef]
- Lee, W.J.; Zhu, B.T. Inhibition of DNA methylation by caffeic acid and chlorogenic acid, two common catechol-containing coffee polyphenols. Carcinogenesis 2005, 27, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Boettler, U.; Sommerfeld, K.; Volz, N.; Pahlke, G.; Teller, N.; Somoza, V.; Lang, R.; Hofmann, T.; Marko, D. Coffee constituents as modulators of Nrf2 nuclear translocation and ARE (EpRE)-dependent gene expression. J. Nutr. Biochem. 2011, 22, 426–440. [Google Scholar] [CrossRef]
- Noratto, G.; Porter, W.; Byrne, D.; Cisneros-Zevallos, L. Identifying peach and plum polyphenols with chemopreventive potential against estrogen-independent breast cancer cells. J. Agric. Food Chem. 2009, 57, 5219–5226. [Google Scholar] [CrossRef]
- Jin, U.H.; Lee, J.Y.; Kang, S.K.; Kim, J.K.; Park, W.H.; Kim, J.G.; Moon, S.K.; Kim, C.H. A phenolic compound, 5-caffeoylquinic acid (chlorogenic acid), is a new type and strong matrix metalloproteinase-9 inhibitor: Isolation and identification from methanol extract of Euonymus alatus. Life Sci. 2005, 77, 2760–2769. [Google Scholar] [CrossRef]
- Dounighi, M.N.; Eskandari, R.; Avadi, M.R.; Zolfagharian, H.; Sadeghi, M.M.A.; Rezayat, M. Preparation and in vitro characterization of chitosan nanoparticles containing Mesobuthus eupeus scorpion venom as an antigen delivery system. J. Venom. Anim. Toxins Incl. Trop. Dis. 2012, 18, 44–52. [Google Scholar] [Green Version]
- Saravanabhavan, S.S.; Bose, R.; Skylab, S.; Dharmalingam, S. Fabrication of chitosan/tpp nano particles as a carrier towards the treatment of cancer. Int. J. Drug Deliv. 2013, 5, 35. [Google Scholar]
- Calvo, P.; Remuñan-Lopez, C.; Vila-Jato, J.L.; Alonso, M.J. Novel hydrophilic chitosanpolyethylene oxide nanoparticles as protein carriers. J. Appl. Polym. Sci. 1997, 63, 125–132. [Google Scholar] [CrossRef]
- Blois, M.S. Antioxidant determinations by the use of a stable free radical. Nature 1958, 181, 1199–1200. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | Solution | Concentration (mg/mL) | pH | Volume (µL) |
|---|---|---|---|---|
| CNP-F1 | CS | 0.50 | 5 | 600 |
| TPP | 0.70 | 2 | 20, 50, 100, 150, 200, 250, 300 | |
| CNP-F2 | CS | 0.25 | 5 | 600 |
| TPP | 0.35 | 2 | 20, 50, 100, 150, 200, 250, 300 | |
| CNP-F3 | CS | 0.10 | 5 | 600 |
| TPP | 0.20 | 2 | 20, 50, 100, 150, 200, 250, 300 |
| Final Concentration of CGA in CNP | Average Particle Size Distribution, (nm) | Polydispersity Index, (PDI) | Encapsulation Efficiency, (EE %) | CGA Loading, (CGA-L, µM) |
|---|---|---|---|---|
| 0 µM (CNP°) | 80.89 ± 5.16 | 0.26 ± 0.01 | - | - |
| 2 µM | 91.99 ± 18.28 | 0.30 ± 0.04 | 74.43 ± 0.31 | 1.49 |
| 10 µM | 82.60 ± 15.81 | 0.28 ± 0.06 | 62.30 ± 0.05 | 6.23 |
| 20 µM | 134.44 ± 18.29 | 0.29 ± 0.03 | 60.21 ± 0.03 | 12.04 |
| Functional Group | Samples | Wavenumber (cm−1) | Transmittance Percentage (%) | Figure 9 Label |
|---|---|---|---|---|
| -OH/-NH2 stretching vibration | CS | 3353.91 | 35.26 | A |
| TPP | 3253.92 | 57.22 | B | |
| CNP° | 3364.97 | 34.74 | C | |
| CNP°-CGA | 3398.61 | 44.97 | D | |
| N-H bending, amide | CS | 1587.00 | 52.24 | E |
| CNP° | 1527.82 | 72.08 | F | |
| CNP°-CGA | 1520.64 | 70.60 | G | |
| Stretching of C=O, amide | CS | 1641.06 | 51.11 | H |
| CNP° | 1630.94 | 52.27 | I | |
| Stretching of P=O | TPP | 1126.89 | 20.92 | J |
| CNP° | 1073.91 | 32.03 | K | |
| CNP°-CGA | 1014.92 | 15.61 | L | |
| Asymmetrical stretching vibration of P-O-P | TPP | 883.70 | 22.73 | M |
| CNP° | 948.77 | 49.86 | N | |
| CNP°-CGA | 949.14 | 41.68 | O | |
| C=O stretching vibration of carboxyl and ester group | CGA | 1687.62 | 17.02 | P |
| CNP°-CGA | 1700.27 | 16.99 | Q | |
| Aromatic ring C=C stretching vibration | CGA | 1439.88 | 49.94 | R |
| CNP°-CGA | 1428.52 | 52.90 | S | |
| Stretching vibration of C-O-C and C-O of carboxyl and ester group | CGA | 1279.45, 1185.17 | 13.86, 10.39 | T |
| CNP°-CGA | 1265.04, 1228.71 | 30.34, 30.00 | U | |
| Bending of C-H and COH | CGA | 1119.50 | 12.37 | V |
| CNP°-CGA | 1163.43 | 31.83 | W | |
| CH aromatic bending | CGA | 810.46 | 53.71 | X |
| CNP°-CGA | 812.13 | 70.81 | Y |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kavi Rajan, R.; Hussein, M.Z.; Fakurazi, S.; Yusoff, K.; Masarudin, M.J. Increased ROS Scavenging and Antioxidant Efficiency of Chlorogenic Acid Compound Delivered via a Chitosan Nanoparticulate System for Efficient In Vitro Visualization and Accumulation in Human Renal Adenocarcinoma Cells. Int. J. Mol. Sci. 2019, 20, 4667. https://doi.org/10.3390/ijms20194667
Kavi Rajan R, Hussein MZ, Fakurazi S, Yusoff K, Masarudin MJ. Increased ROS Scavenging and Antioxidant Efficiency of Chlorogenic Acid Compound Delivered via a Chitosan Nanoparticulate System for Efficient In Vitro Visualization and Accumulation in Human Renal Adenocarcinoma Cells. International Journal of Molecular Sciences. 2019; 20(19):4667. https://doi.org/10.3390/ijms20194667
Chicago/Turabian StyleKavi Rajan, Revathi, Mohd Zobir Hussein, Sharida Fakurazi, Khatijah Yusoff, and Mas Jaffri Masarudin. 2019. "Increased ROS Scavenging and Antioxidant Efficiency of Chlorogenic Acid Compound Delivered via a Chitosan Nanoparticulate System for Efficient In Vitro Visualization and Accumulation in Human Renal Adenocarcinoma Cells" International Journal of Molecular Sciences 20, no. 19: 4667. https://doi.org/10.3390/ijms20194667
APA StyleKavi Rajan, R., Hussein, M. Z., Fakurazi, S., Yusoff, K., & Masarudin, M. J. (2019). Increased ROS Scavenging and Antioxidant Efficiency of Chlorogenic Acid Compound Delivered via a Chitosan Nanoparticulate System for Efficient In Vitro Visualization and Accumulation in Human Renal Adenocarcinoma Cells. International Journal of Molecular Sciences, 20(19), 4667. https://doi.org/10.3390/ijms20194667