Osteoclast-Like Cells in Aneurysmal Disease Exhibit an Enhanced Proteolytic Phenotype

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
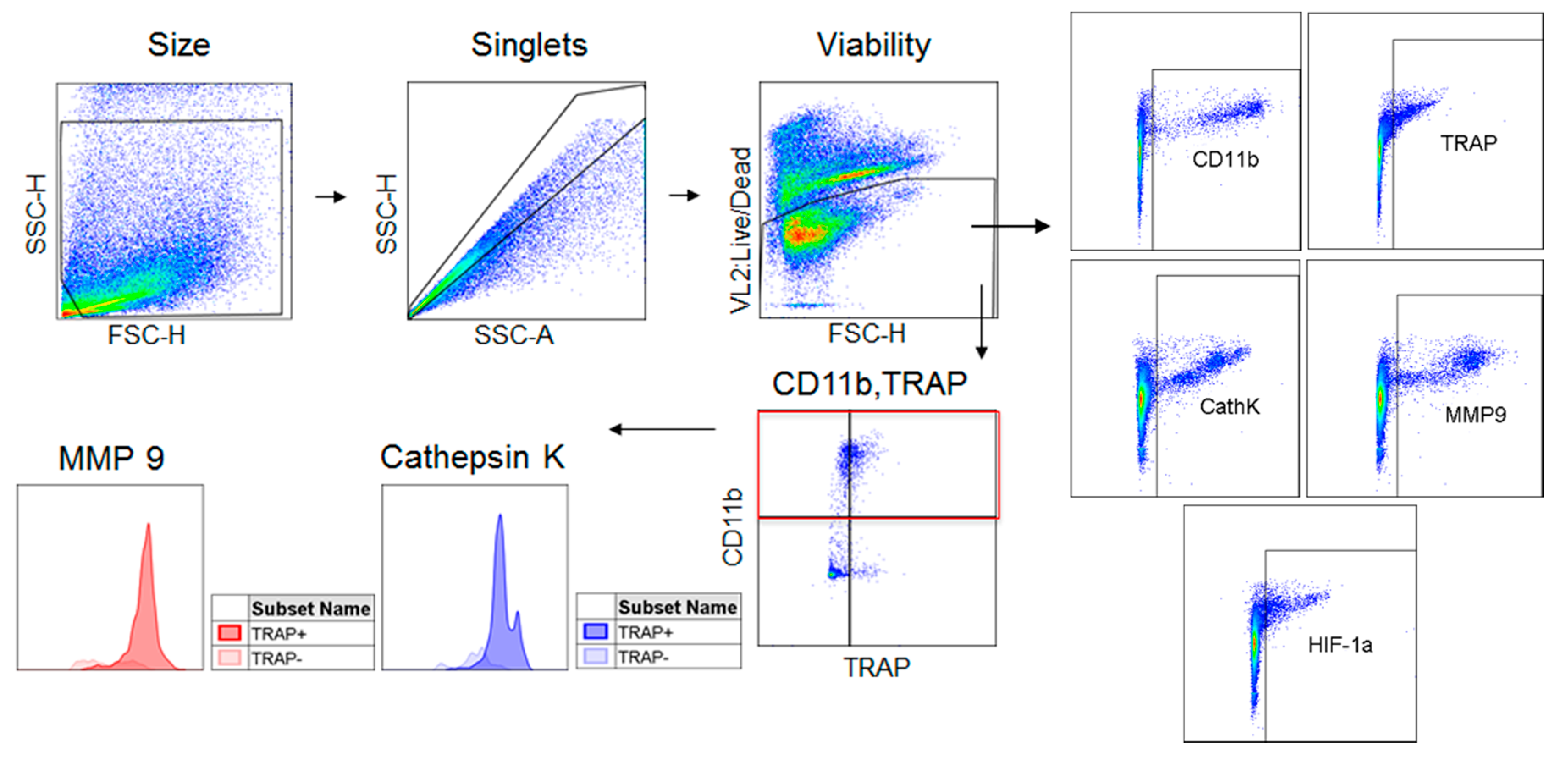
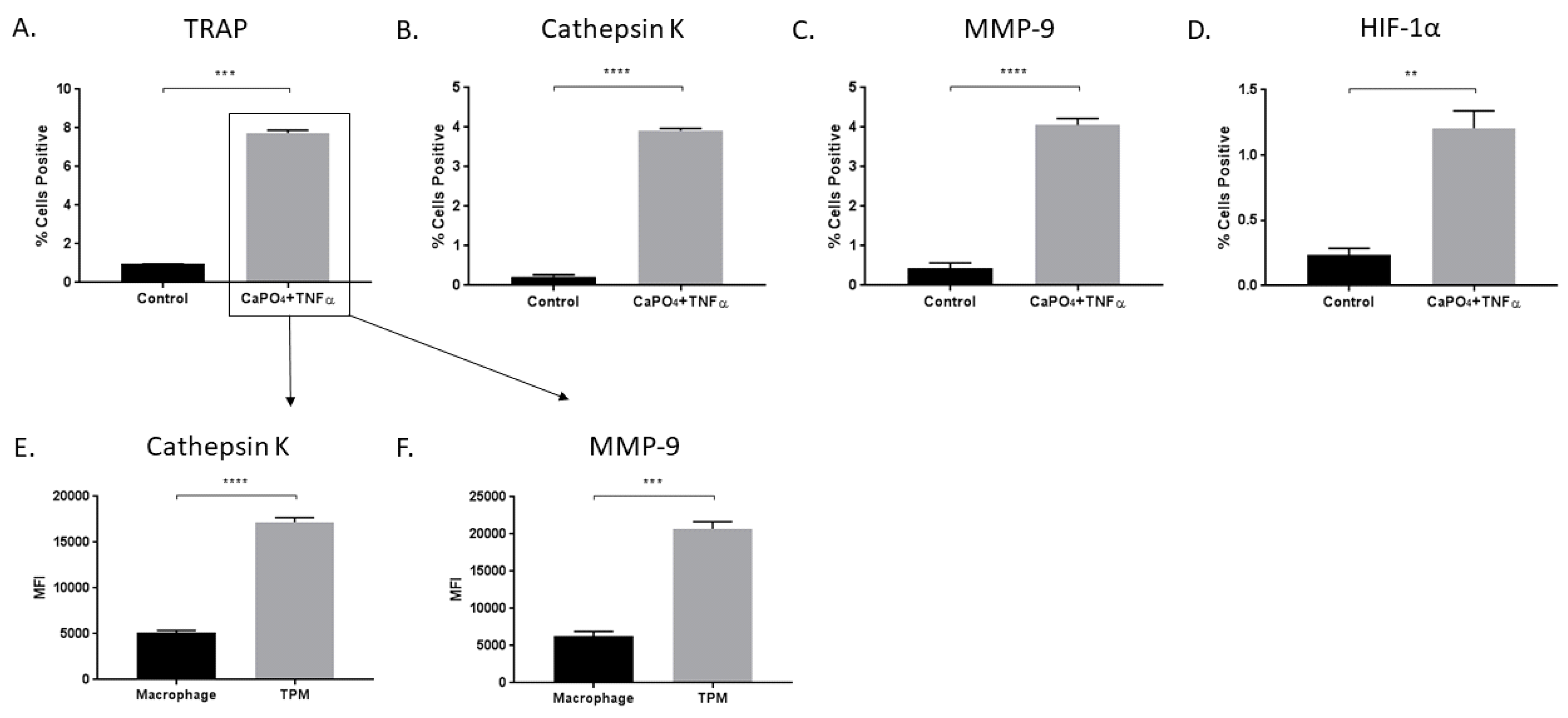
2.1. TRAP-Positive Macrophages Produce More Cathepsin K and MMP-9 than TRAP-Negative Macrophages
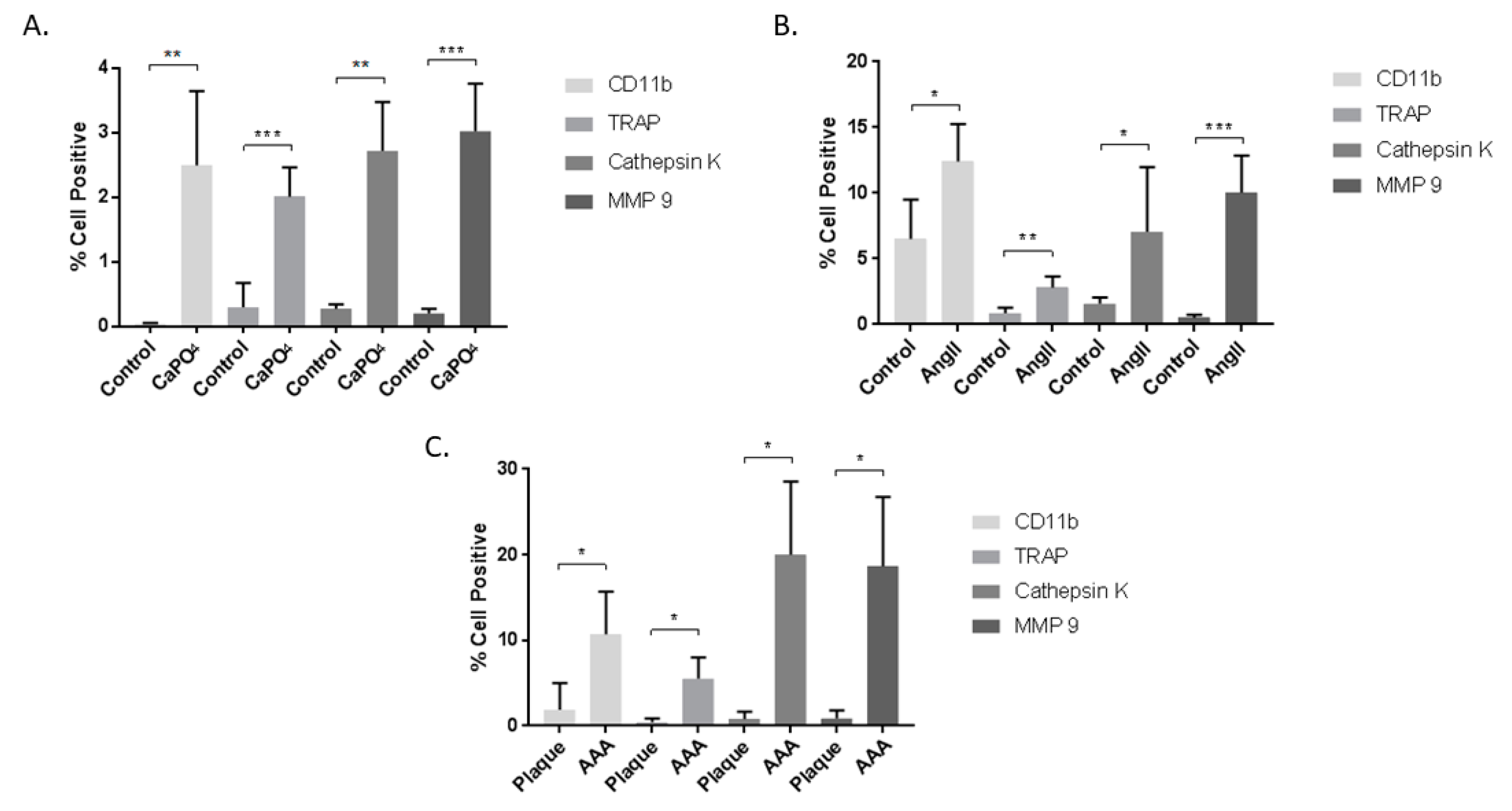
2.2. Aneurysmal Tissues Exhibit Increased Expression of Osteoclastogenic Markers
2.3. HIF-1α Expression is Enhanced in Aneurysmal Tissues and TPMs
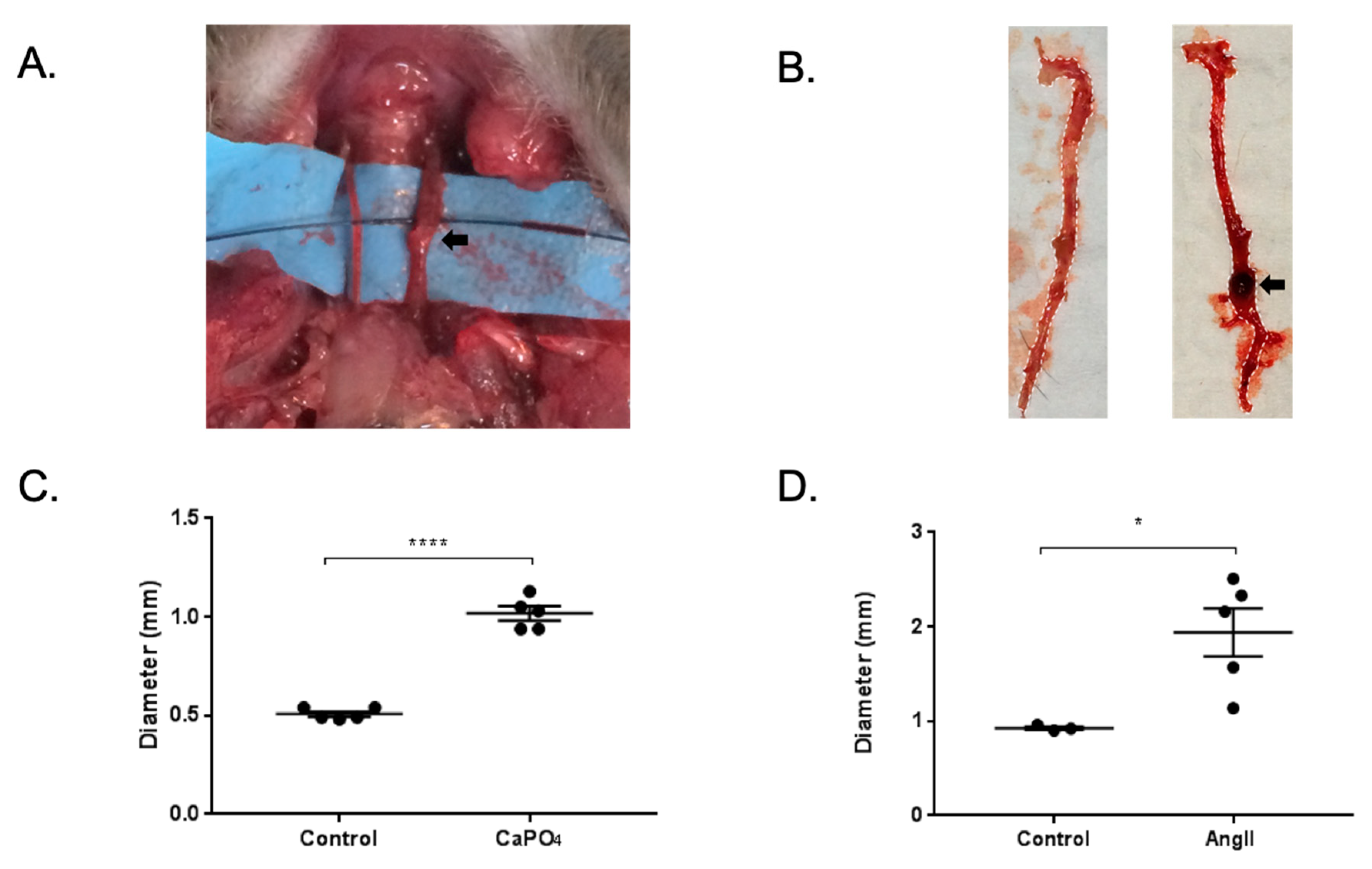
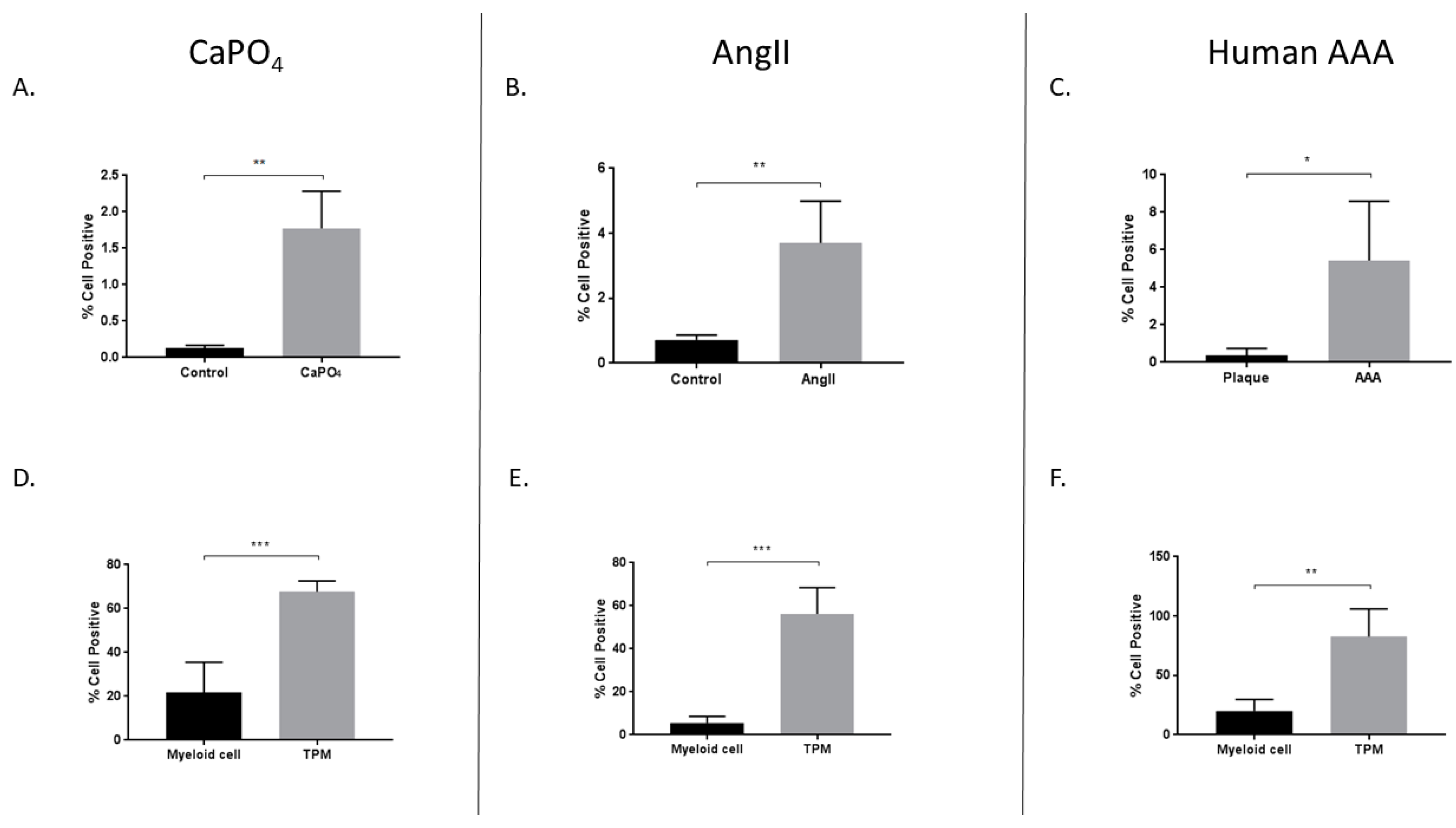
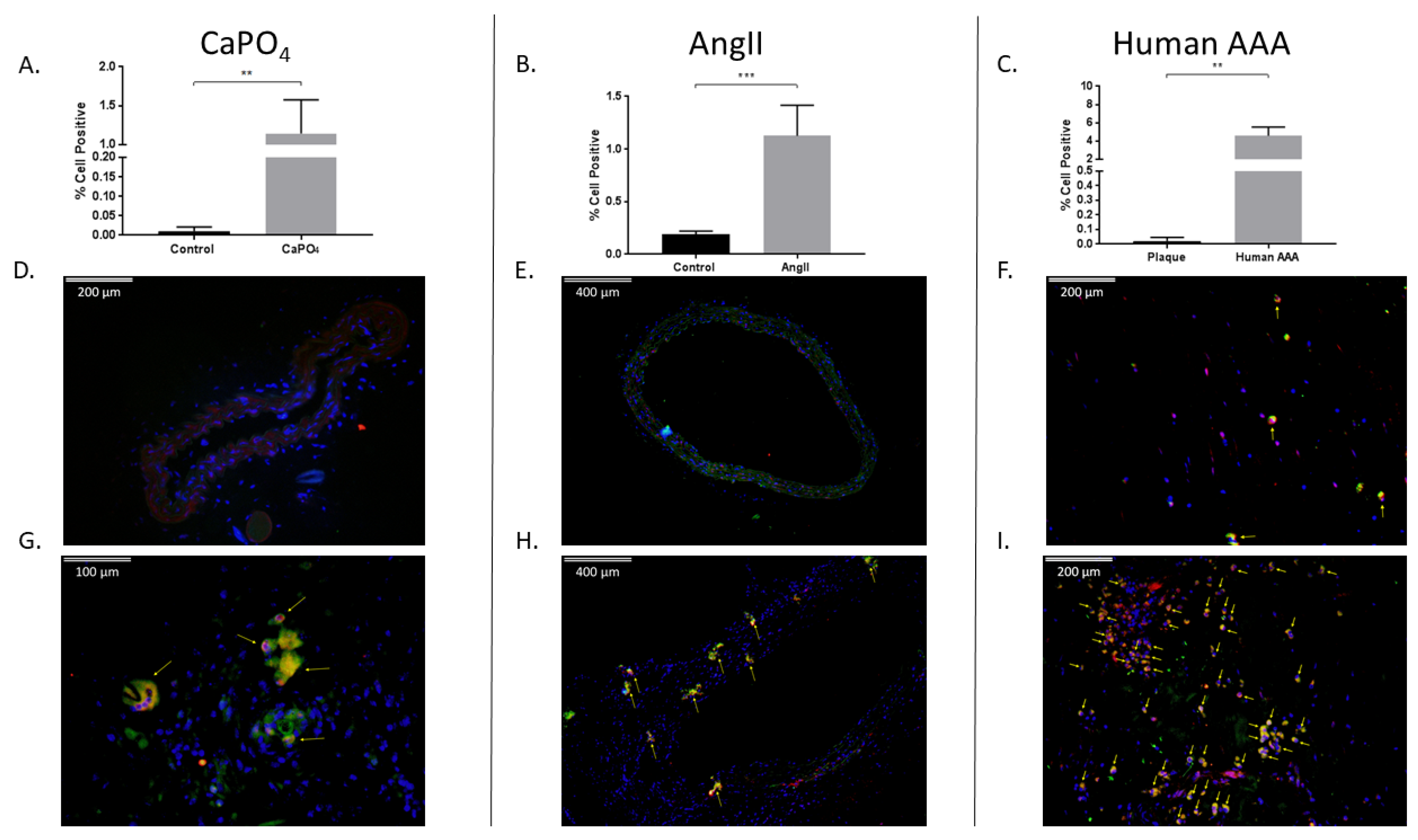
2.4. Aneurysmal Tissues Exhibit Increased Infiltration of TPMs
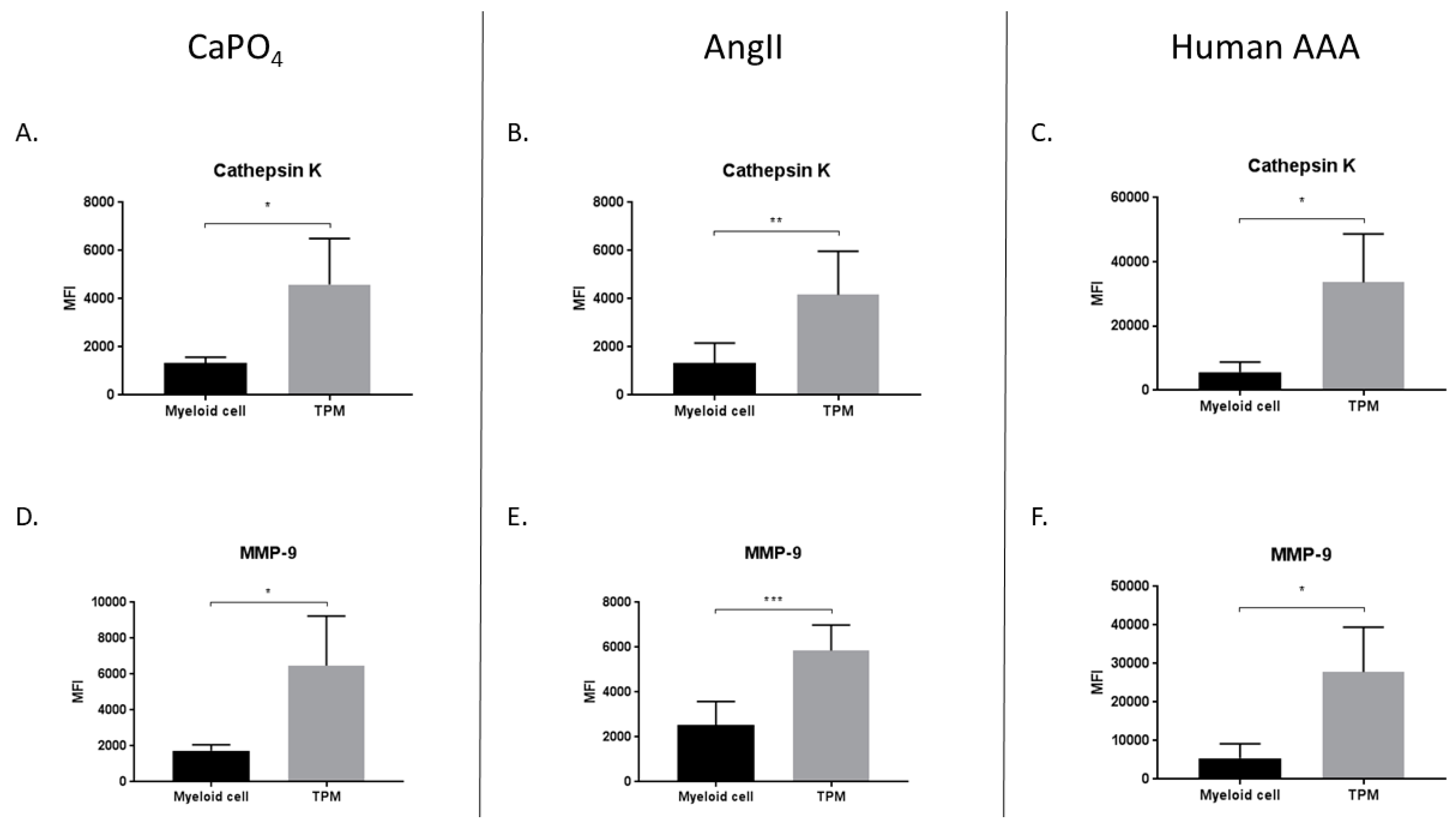
2.5. TPMs From Aneurysmal Tissues Express Elevated Levels of Cathepsin K and MMP-9
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Human Tissue
4.3. Mouse Arterial Aneurysm Models and Treatments
4.4. Flow Cytometry
4.5. Immunofluorescence Staining
4.6. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAA | Abdominal aortic aneurysm |
| OLC | Osteoclast-like cell |
| TRAP | Tartrate-resistant acid phosphatase |
| NF-κB | Nuclear factor kappa-B |
| RANKL | Receptor activator of nuclear factor kappa-B ligand |
| MMP-9 | Matrix metalloproteinase-9 |
| TRAF | TNF receptor-associated factor |
| MAPK | mitogen-activated protein kinase |
| NFATc1 | Nuclear factor of activated T cells cytoplasmic 1 |
| TNFα | Tumor necrosis factor α |
| TPM | TRAP-positive macrophage |
| Ang II | Angiotensin II |
| apoE−/− | Apolipoprotein E-deficient |
| HIF-1α | Hypoxia-inducible factor-1α |
| MFI | Median fluorescence intensity |
| KO | Knockout |
| DMEM | Dulbecco’s modified Eagle’s medium |
| FBS | Fetal bovine serum |
| MEM-α | Modified Eagle’s medium-α |
| PBS | Phosphate-buffered saline |
| TBST | Tris-buffered saline-Tween 20 |
| DAPI | 4,6-diamidino-2-phenylindole dihydrochloride |
| SEM | Standard error of the means |
References
- Kent, K.C.; Zwolak, R.M.; Egorova, N.N.; Riles, T.S.; Manganaro, A.; Moskowitz, A.J.; Gelijns, A.C.; Greco, G. Analysis of risk factors for abdominal aortic aneurysm in a cohort of more than 3 million individuals. J. Vasc. Surg. 2010, 52, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart disease and stroke statistics-2018 update: A report from the american heart association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef] [PubMed]
- MacSweeney, S.T.; Ellis, M.; Worrell, P.C.; Greenhalgh, R.M.; Powell, J.T. Smoking and growth rate of small abdominal aortic aneurysms. Lancet 1994, 344, 651–652. [Google Scholar] [CrossRef]
- Lederle, F.A.; Johnson, G.R.; Wilson, S.E.; Chute, E.P.; Hye, R.J.; Makaroun, M.S.; Barone, G.W.; Bandyk, D.; Moneta, G.L.; Makhoul, R.G. The aneurysm detection and management study screening program: Validation cohort and final results. Aneurysm detection and management veterans affairs cooperative study investigators. Arch. Intern. Med. 2000, 160, 1425–1430. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.M.; Faggioli, G.L.; Fiedler, R.; Curl, G.R.; Ricotta, J.J. Ruptured abdominal aortic aneurysms: Factors affecting mortality rates. J. Vasc. Surg. 1991, 14, 812–818; disscussion 819–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, A.T.; Haskal, Z.J.; Hertzer, N.R.; Bakal, C.W.; Creager, M.A.; Halperin, J.L.; Hiratzka, L.F.; Murphy, W.R.; Olin, J.W.; Puschett, J.B.; et al. Acc/aha guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): A collaborative report from the american associations for vascular surgery/society for vascular surgery, society for cardiovascular angiography and interventions, society for vascular medicine and biology, society of interventional radiology, and the acc/aha task force on practice guidelines (writing committee to develop guidelines for the management of patients with peripheral arterial disease)—Summary of recommendations. J. Vasc. Interv. Radiol. 2006, 17, 1383–1397. [Google Scholar] [PubMed]
- Sweeting, M.J.; Thompson, S.G.; Brown, L.C.; Powell, J.T.; collaborators, R. Meta-analysis of individual patient data to examine factors affecting growth and rupture of small abdominal aortic aneurysms. Br. J. Surg. 2012, 99, 655–665. [Google Scholar] [CrossRef]
- Thompson, S.G.; Brown, L.C.; Sweeting, M.J.; Bown, M.J.; Kim, L.G.; Glover, M.J.; Buxton, M.J.; Powell, J.T. Systematic review and meta-analysis of the growth and rupture rates of small abdominal aortic aneurysms: Implications for surveillance intervals and their cost-effectiveness. Health Technol. Assess 2013, 17, 1–118. [Google Scholar] [CrossRef]
- Kurosawa, K.; Matsumura, J.S.; Yamanouchi, D. Current status of medical treatment for abdominal aortic aneurysm. Circ. J. 2013, 77, 2860–2866. [Google Scholar] [CrossRef]
- Takayama, T.; Yamanouchi, D. Aneurysmal disease: The abdominal aorta. Surg. Clin. N. Am. 2013, 93, 877–891. [Google Scholar] [CrossRef]
- Lindholt, J.S. Aneurysmal wall calcification predicts natural history of small abdominal aortic aneurysms. Atherosclerosis 2008, 197, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.L.; Kratzberg, J.; Castro de Tolosa, E.M.; Hanaoka, M.M.; Walker, P.; da Silva, E.S. Regional distribution of wall thickness and failure properties of human abdominal aortic aneurysm. J. Biomech. 2006, 39, 3010–3016. [Google Scholar] [CrossRef] [PubMed]
- Yamanouchi, D.; Takei, Y.; Komori, K. Balanced mineralization in the arterial system: Possible role of osteoclastogenesis/osteoblastogenesis in abdominal aortic aneurysm and stenotic disease. Circ. J. 2012, 76, 2732–2737. [Google Scholar] [CrossRef] [PubMed]
- Liberman, M.; Pesaro, A.E.; Carmo, L.S.; Serrano, C.V., Jr. Vascular calcification: Pathophysiology and clinical implications. Einstein (Sao Paulo) 2013, 11, 376–382. [Google Scholar] [CrossRef]
- Minkin, C. Bone acid phosphatase: Tartrate-resistant acid phosphatase as a marker of osteoclast function. Calcif. Tissue Int. 1982, 34, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. Rank is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, O.; Niwa, S.; Kadoyama, K.; Inui, T. Mmp-9 antisense oligodeoxynucleotide exerts an inhibitory effect on osteoclastic bone resorption by suppressing cell migration. Life Sci. 2006, 79, 1657–1660. [Google Scholar] [CrossRef] [PubMed]
- Takei, Y.; Tanaka, T.; Kent, K.C.; Yamanouchi, D. Osteoclastogenic differentiation of macrophages in the development of abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1962–1971. [Google Scholar] [CrossRef]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor nfatc1 (nfat2) integrate rankl signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Wong, B.R.; Josien, R.; Lee, S.Y.; Vologodskaia, M.; Steinman, R.M.; Choi, Y. The traf family of signal transducers mediates nf-kappab activation by the trance receptor. J. Biol. Chem. 1998, 273, 28355–28359. [Google Scholar] [CrossRef]
- Kobayashi, N.; Kadono, Y.; Naito, A.; Matsumoto, K.; Yamamoto, T.; Tanaka, S.; Inoue, J. Segregation of traf6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J. 2001, 20, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kelly, M.; Takei, Y.; Yamanouchi, D. Rankl-mediated osteoclastogenic differentiation of macrophages in the abdominal aorta of angiotensin ii-infused apolipoprotein e knockout mice. J. Vasc. Surg. 2018, 68, 48S–59S. [Google Scholar] [CrossRef] [PubMed]
- Saraff, K.; Babamusta, F.; Cassis, L.A.; Daugherty, A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin ii-infused, apolipoprotein e-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1621–1626. [Google Scholar] [CrossRef] [PubMed]
- Sakalihasan, N.; Hustinx, R.; Limet, R. Contribution of pet scanning to the evaluation of abdominal aortic aneurysm. Semin. Vasc. Surg. 2004, 17, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Truijers, M.; Kurvers, H.A.; Bredie, S.J.; Oyen, W.J.; Blankensteijn, J.D. In vivo imaging of abdominal aortic aneurysms: Increased fdg uptake suggests inflammation in the aneurysm wall. J. Endovasc. Ther. 2008, 15, 462–467. [Google Scholar] [CrossRef]
- Wiernicki, I.; Parafiniuk, M.; Kolasa-Wołosiuk, A.; Gutowska, I.; Kazimierczak, A.; Clark, J.; Baranowska-Bosiacka, I.; Szumilowicz, P.; Gutowski, P. Relationship between aortic wall oxidative stress/proteolytic enzyme expression and intraluminal thrombus thickness indicates a novel pathomechanism in the progression of human abdominal aortic aneurysm. FASEB J. 2019, 33, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.W.; Holmes, D.R.; Mertens, R.A.; Liao, S.; Botney, M.D.; Mecham, R.P.; Welgus, H.G.; Parks, W.C. Production and localization of 92-kilodalton gelatinase in abdominal aortic aneurysms. An elastolytic metalloproteinase expressed by aneurysm-infiltrating macrophages. J. Clin. Investig. 1995, 96, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Longo, G.M.; Xiong, W.; Greiner, T.C.; Zhao, Y.; Fiotti, N.; Baxter, B.T. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J. Clin. Investig. 2002, 110, 625–632. [Google Scholar] [CrossRef]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (hif-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef]
- Hu, X.H.; Yang, J.; Liu, C.W.; Zhang, Z.S.; Zhang, Q. The expression and significance of hypoxia-inducible factor-1 alpha and related genes in abdominal aorta aneurysm. Zhonghua Wai Ke Za Zhi 2004, 42, 1509–1512. [Google Scholar]
- Van Vickle-Chavez, S.J.; Tung, W.S.; Absi, T.S.; Ennis, T.L.; Mao, D.; Cobb, J.P.; Thompson, R.W. Temporal changes in mouse aortic wall gene expression during the development of elastase-induced abdominal aortic aneurysms. J. Vasc. Surg. 2006, 43, 1010–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Shen, L.; Li, G.; Yuan, H.; Jin, X.; Wu, X. Silencing of hypoxia inducible factor-1α gene attenuated angiotensin ii-induced abdominal aortic aneurysm in apolipoprotein e-deficient mice. Atherosclerosis 2016, 252, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, B.; Xuan, H.; Ge, Y.; Wang, Y.; Wang, L.; Huang, J.; Fu, W.; Michie, S.A.; Dalman, R.L. Hypoxia-inducible factor 1 in clinical and experimental aortic aneurysm disease. J. Vasc. Surg. 2018, 68, 1538–1550. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.N.; Zhang, F.; Tang, P.; Qi, X.W.; Jiang, J. Hypoxia induces rank and rankl expression by activating hif-1α in breast cancer cells. Biochem. Biophys. Res. Commun. 2011, 408, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Igari, K.; Kelly, M.J.; Yamanouchi, D. Digoxin attenuates receptor activation of nf-κb ligand-induced osteoclastogenesis in macrophages. J. Vasc. Res. 2019, 56, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Arnett, T.R.; Gibbons, D.C.; Utting, J.C.; Orriss, I.R.; Hoebertz, A.; Rosendaal, M.; Meghji, S. Hypoxia is a major stimulator of osteoclast formation and bone resorption. J. Cell Physiol. 2003, 196, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.H.; Huang, P.H.; Hsu, Y.J.; Peng, Y.J.; Lee, C.H.; Wang, J.C.; Chen, J.W.; Lin, S.J. Inhibition of hypoxia inducible factor-1α attenuates abdominal aortic aneurysm progression through the down-regulation of matrix metalloproteinases. Sci. Rep. 2016, 6, 28612. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Hussien, H.; Soekhoe, R.G.; Weber, E.; von der Thüsen, J.H.; Kleemann, R.; Mulder, A.; van Bockel, J.H.; Hanemaaijer, R.; Lindeman, J.H. Collagen degradation in the abdominal aneurysm: A conspiracy of matrix metalloproteinase and cysteine collagenases. Am. J. Pathol. 2007, 170, 809–817. [Google Scholar] [CrossRef]
- Saftig, P.; Hunziker, E.; Wehmeyer, O.; Jones, S.; Boyde, A.; Rommerskirch, W.; Moritz, J.D.; Schu, P.; von Figura, K. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-k-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 13453–13458. [Google Scholar] [CrossRef]
- Matsumoto, M.; Kogawa, M.; Wada, S.; Takayanagi, H.; Tsujimoto, M.; Katayama, S.; Hisatake, K.; Nogi, Y. Essential role of p38 mitogen-activated protein kinase in cathepsin k gene expression during osteoclastogenesis through association of nfatc1 and pu.1. J. Biol. Chem. 2004, 279, 45969–45979. [Google Scholar] [CrossRef]
- Drake, F.H.; Dodds, R.A.; James, I.E.; Connor, J.R.; Debouck, C.; Richardson, S.; Lee-Rykaczewski, E.; Coleman, L.; Rieman, D.; Barthlow, R.; et al. Cathepsin k, but not cathepsins b, l, or s, is abundantly expressed in human osteoclasts. J. Biol. Chem. 1996, 271, 12511–12516. [Google Scholar] [CrossRef] [PubMed]
- Bossard, M.J.; Tomaszek, T.A.; Thompson, S.K.; Amegadzie, B.Y.; Hanning, C.R.; Jones, C.; Kurdyla, J.T.; McNulty, D.E.; Drake, F.H.; Gowen, M.; et al. Proteolytic activity of human osteoclast cathepsin k. Expression, purification, activation, and substrate identification. J. Biol. Chem. 1996, 271, 12517–12524. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Sukhova, G.K.; Zhang, J.; Chen, H.; Sjöberg, S.; Libby, P.; Xia, M.; Xiong, N.; Gelb, B.D.; Shi, G.P. Cathepsin k deficiency reduces elastase perfusion-induced abdominal aortic aneurysms in mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Beckers, L.; Wijnands, E.; Lutgens, S.P.; Herías, M.V.; Saftig, P.; Daemen, M.J.; Cleutjens, K.; Lutgens, E.; Biessen, E.A.; et al. Cathepsin k gene disruption does not affect murine aneurysm formation. Atherosclerosis 2010, 209, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Punturieri, A.; Filippov, S.; Allen, E.; Caras, I.; Murray, R.; Reddy, V.; Weiss, S.J. Regulation of elastinolytic cysteine proteinase activity in normal and cathepsin k-deficient human macrophages. J. Exp. Med. 2000, 192, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Kaakinen, R.; Lindstedt, K.A.; Sneck, M.; Kovanen, P.T.; Oörni, K. Angiotensin ii increases expression and secretion of cathepsin f in cultured human monocyte-derived macrophages: An angiotensin ii type 2 receptor-mediated effect. Atherosclerosis 2007, 192, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Knowles, H.J.; Athanasou, N.A. Acute hypoxia and osteoclast activity: A balance between enhanced resorption and increased apoptosis. J. Pathol. 2009, 218, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Hulley, P.A.; Bishop, T.; Vernet, A.; Schneider, J.E.; Edwards, J.R.; Athanasou, N.A.; Knowles, H.J. Hypoxia-inducible factor 1-alpha does not regulate osteoclastogenesis but enhances bone resorption activity via prolyl-4-hydroxylase 2. J. Pathol. 2017, 242, 322–333. [Google Scholar] [CrossRef]
- Indo, Y.; Takeshita, S.; Ishii, K.A.; Hoshii, T.; Aburatani, H.; Hirao, A.; Ikeda, K. Metabolic regulation of osteoclast differentiation and function. J. Bone Miner. Res. 2013, 28, 2392–2399. [Google Scholar] [CrossRef] [PubMed]
- Choke, E.; Cockerill, G.W.; Dawson, J.; Chung, Y.L.; Griffiths, J.; Wilson, R.W.; Loftus, I.M.; Thompson, M.M. Hypoxia at the site of abdominal aortic aneurysm rupture is not associated with increased lactate. Ann. N. Y. Acad. Sci. 2006, 1085, 306–310. [Google Scholar] [CrossRef]
- Vorp, D.A.; Lee, P.C.; Wang, D.H.; Makaroun, M.S.; Nemoto, E.M.; Ogawa, S.; Webster, M.W. Association of intraluminal thrombus in abdominal aortic aneurysm with local hypoxia and wall weakening. J. Vasc. Surg. 2001, 34, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Erdozain, O.J.; Pegrum, S.; Winrow, V.R.; Horrocks, M.; Stevens, C.R. Hypoxia in abdominal aortic aneurysm supports a role for hif-1α and ets-1 as drivers of matrix metalloproteinase upregulation in human aortic smooth muscle cells. J. Vasc. Res. 2011, 48, 163–170. [Google Scholar] [CrossRef]
- Koole, D.; Zandvoort, H.J.; Schoneveld, A.; Vink, A.; Vos, J.A.; van den Hoogen, L.L.; de Vries, J.P.; Pasterkamp, G.; Moll, F.L.; van Herwaarden, J.A. Intraluminal abdominal aortic aneurysm thrombus is associated with disruption of wall integrity. J. Vasc. Surg. 2013, 57, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Bønaa, K.H.; Jacobsen, B.K.; Bjørk, L.; Solberg, S. Prevalence of and risk factors for abdominal aortic aneurysms in a population-based study: The tromsø study. Am. J. Epidemiol. 2001, 154, 236–244. [Google Scholar] [CrossRef]
- Katz, D.J.; Stanley, J.C.; Zelenock, G.B. Gender differences in abdominal aortic aneurysm prevalence, treatment, and outcome. J. Vasc. Surg. 1997, 25, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Mukundan, H.; Kanagy, N.L.; Resta, T.C. 17-beta estradiol attenuates hypoxic induction of hif-1alpha and erythropoietin in hep3b cells. J. Cardiovasc. Pharmacol. 2004, 44, 93–100. [Google Scholar] [CrossRef]
- Miyauchi, Y.; Sato, Y.; Kobayashi, T.; Yoshida, S.; Mori, T.; Kanagawa, H.; Katsuyama, E.; Fujie, A.; Hao, W.; Miyamoto, K.; et al. Hif1α is required for osteoclast activation by estrogen deficiency in postmenopausal osteoporosis. Proc. Natl. Acad. Sci. USA 2013, 110, 16568–16573. [Google Scholar] [CrossRef]
- Tanaka, T.; Takei, Y.; Yamanouchi, D. Hyperglycemia suppresses calcium phosphate-induced aneurysm formation through inhibition of macrophage activation. J. Am. Heart Assoc. 2016, 5, e003062. [Google Scholar] [CrossRef]
- Kurihara, C.; Tanaka, T.; Yamanouchi, D. Hyperglycemia attenuates receptor activator of nf-κb ligand-induced macrophage activation by suppressing insulin signaling. J. Surg. Res. 2017, 214, 168–175. [Google Scholar] [CrossRef]
- Chen, C.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation of glut1 mrna by hypoxia-inducible factor-1. Interaction between h-ras and hypoxia. J. Biol. Chem. 2001, 276, 9519–9525. [Google Scholar] [CrossRef]
- Watanabe, A.; Ichiki, T.; Sankoda, C.; Takahara, Y.; Ikeda, J.; Inoue, E.; Tokunou, T.; Kitamoto, S.; Sunagawa, K. Suppression of abdominal aortic aneurysm formation by inhibition of prolyl hydroxylase domain protein through attenuation of inflammation and extracellular matrix disruption. Clin. Sci. (Lond.) 2014, 126, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Takahara, Y.; Tokunou, T.; Kojima, H.; Hirooka, Y.; Ichiki, T. Deletion of hypoxia-inducible factor-1α in myeloid lineage exaggerates angiotensin ii-induced formation of abdominal aortic aneurysm. Clin. Sci. (Lond.) 2017, 131, 609–620. [Google Scholar] [CrossRef]
- Imanishi, M.; Chiba, Y.; Tomita, N.; Matsunaga, S.; Nakagawa, T.; Ueno, M.; Yamamoto, K.; Tamaki, T.; Tomita, S. Hypoxia-inducible factor-1α in smooth muscle cells protects against aortic aneurysms-brief report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2158–2162. [Google Scholar] [CrossRef]
- Yamanouchi, D.; Morgan, S.; Stair, C.; Seedial, S.; Lengfeld, J.; Kent, K.C.; Liu, B. Accelerated aneurysmal dilation associated with apoptosis and inflammation in a newly developed calcium phosphate rodent abdominal aortic aneurysm model. J. Vasc. Surg. 2012, 56, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Galkina, E.; Kadl, A.; Sanders, J.; Varughese, D.; Sarembock, I.J.; Ley, K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially l-selectin dependent. J. Exp. Med. 2006, 203, 1273–1282. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kelly, M.J.; Igari, K.; Yamanouchi, D. Osteoclast-Like Cells in Aneurysmal Disease Exhibit an Enhanced Proteolytic Phenotype. Int. J. Mol. Sci. 2019, 20, 4689. https://doi.org/10.3390/ijms20194689
Kelly MJ, Igari K, Yamanouchi D. Osteoclast-Like Cells in Aneurysmal Disease Exhibit an Enhanced Proteolytic Phenotype. International Journal of Molecular Sciences. 2019; 20(19):4689. https://doi.org/10.3390/ijms20194689
Chicago/Turabian StyleKelly, Matthew J., Kimihiro Igari, and Dai Yamanouchi. 2019. "Osteoclast-Like Cells in Aneurysmal Disease Exhibit an Enhanced Proteolytic Phenotype" International Journal of Molecular Sciences 20, no. 19: 4689. https://doi.org/10.3390/ijms20194689
APA StyleKelly, M. J., Igari, K., & Yamanouchi, D. (2019). Osteoclast-Like Cells in Aneurysmal Disease Exhibit an Enhanced Proteolytic Phenotype. International Journal of Molecular Sciences, 20(19), 4689. https://doi.org/10.3390/ijms20194689