Ovarian Follicle Depletion Induced by Chemotherapy and the Investigational Stages of Potential Fertility-Protective Treatments—A Review

Abstract
:1. Introduction
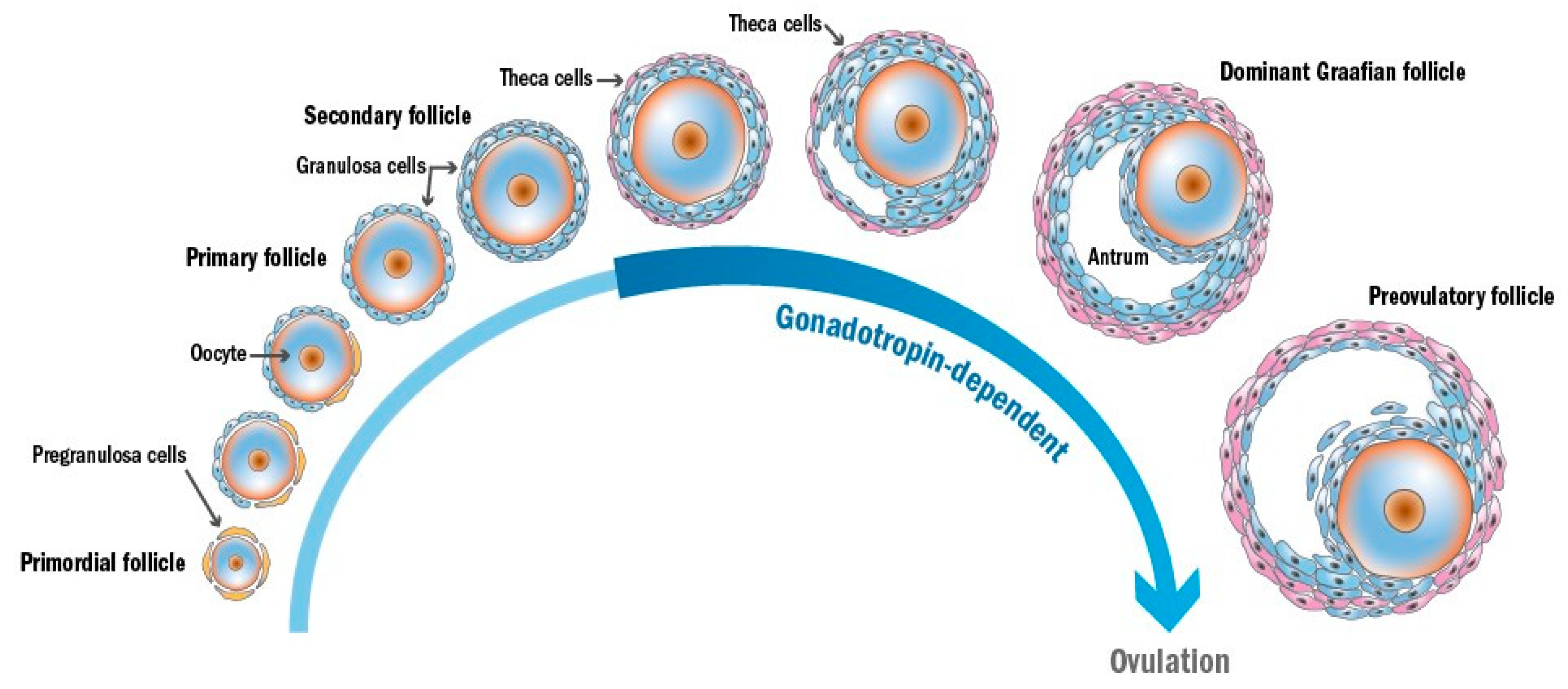
2. Gametogenesis and Folliculogenesis
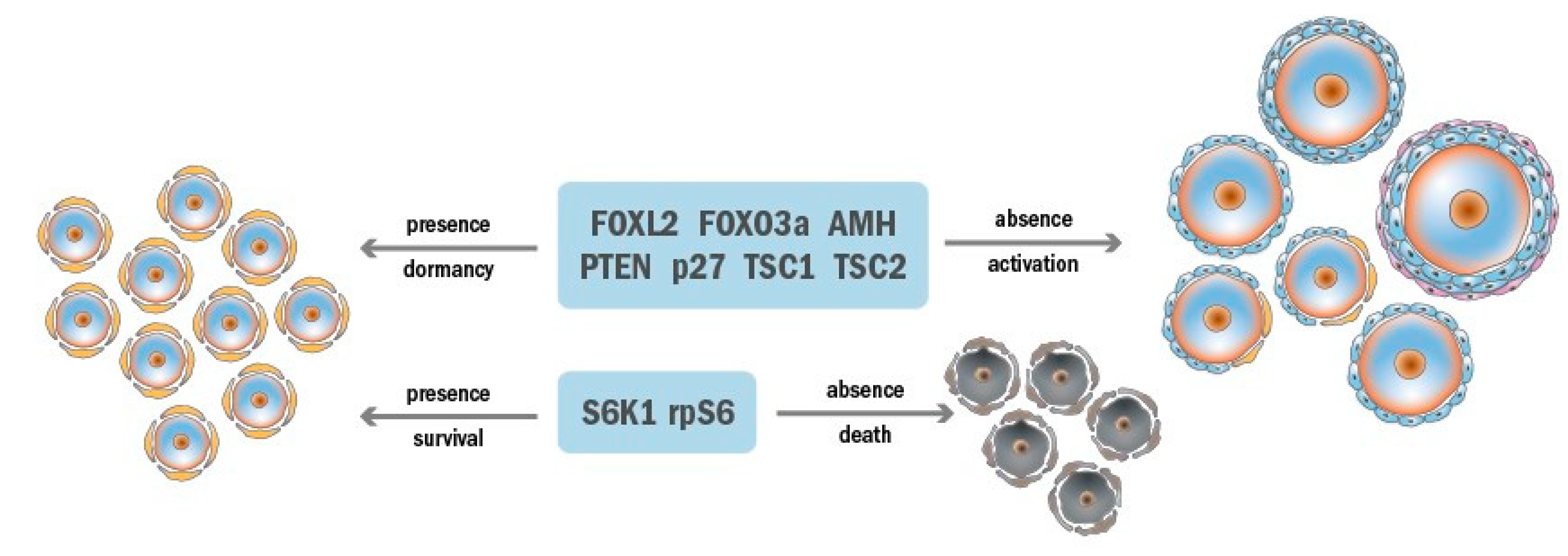
2.1. Ovarian Reserve Dormancy
2.2. Ovarian Reserve Maintenance
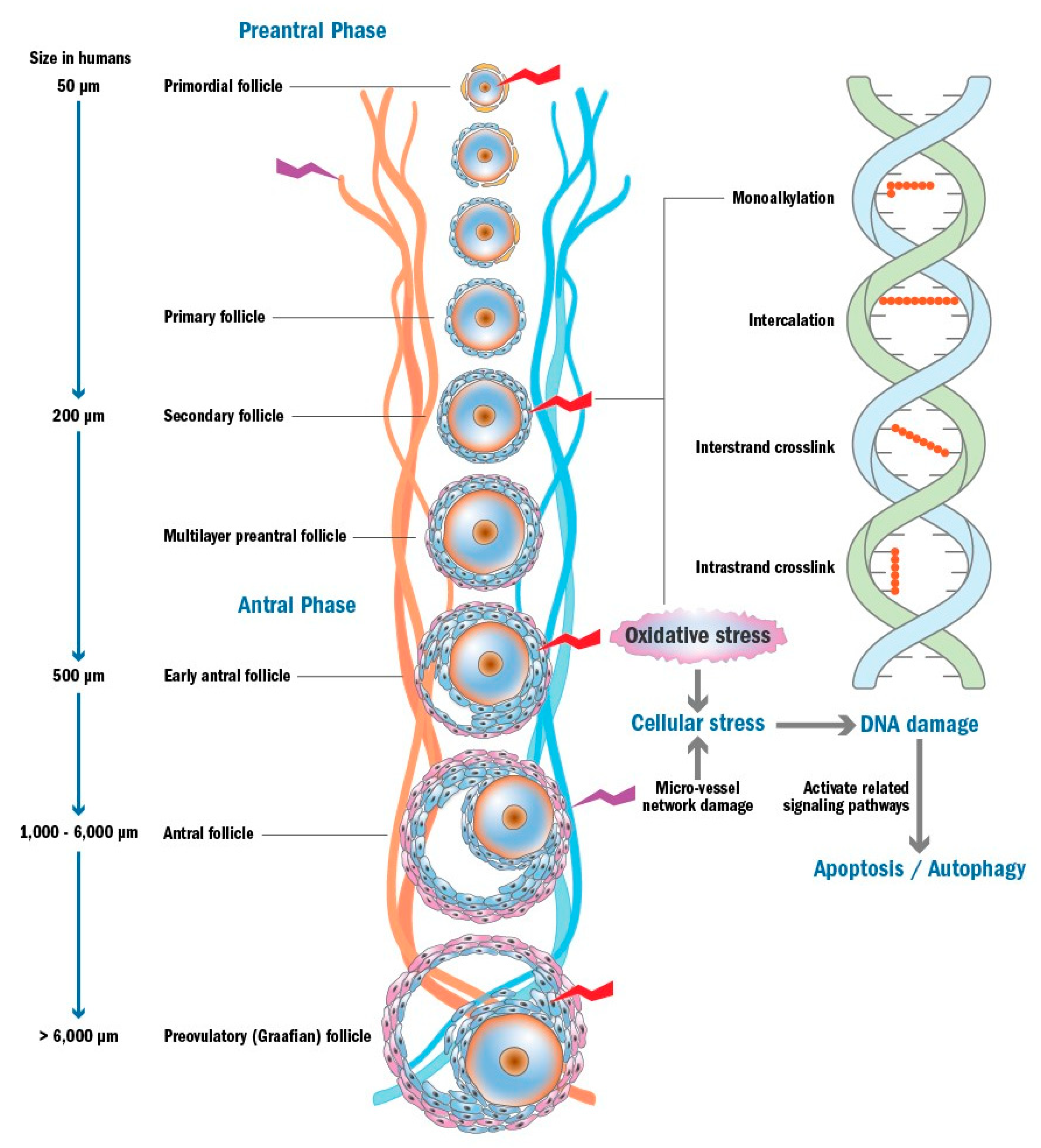
3. Chemotherapy-Induced Ovarian Damage
3.1. Apoptosis Triggered by DNA Damage and/or Oxidative Stress
3.2. Activation of Primordial Follicles and Ovarian Reserve Burnout
3.3. Inducing Autophagy
3.4. Micro-Vessel Network Damage
4. Potential Protective Treatments to Reduce/Recover Chemotherapy-Induced Ovarian Damage
4.1. Proposed Treatments to Protect Against Apoptosis in Ovarian Follicles
4.1.1. Antioxidants
4.1.2. Improving DNA Repair
4.1.3. Mediating the Nuclear Accumulation of Chemotherapeutic Drugs
4.1.4. Blocking the Apoptosis Pathway
4.2. Proposed Treatments to Reduce Overactivation of the Primordial Follicle Pool
4.3. Decreased Micro-Vessel Loss in the Ovary
4.4. Others
4.4.1. Caloric Restriction
4.4.2. Gene Therapy
4.4.3. Pharmaceutical Development and Improvement of Chemotherapeutic Treatment
4.5. Repair or Replace Damaged Cells in the Ovary After Chemotherapy
4.5.1. Human Amniotic Fluid Cells (hAFCs) and Human Amniotic Epithelial Cells (hAECs)
4.5.2. Bone Marrow-Derived Mesenchymal Stem Cells (BMMSCs)
4.6. Other Possible Protective Substance Resources
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-HC | 4-hydroperoxycyclophosphamide |
| 4-OH-CPA | 4-hydroxycyclophosphamide |
| AMH | Anti-Müllerian hormone |
| AS101 | Ammonium-trichloro (0,0-dioxyethylene) tellurate |
| ATM | Ataxia telangiectasia mutated |
| BMMSC | Bone marrow-derived mesenchymal stem cells |
| CPA | Cyclophosphamide |
| DSBs | DNA double strand breaks |
| FGSCs | Female germline stem cells |
| FOX | Forkhead box |
| FSH | Follicle-stimulating hormone |
| G-CSF | Granulocyte colony-stimulating factor |
| GnRHa | Gonadotropin-releasing hormone analogues |
| GSH | Glutathione |
| GST | Glutathione-S-transferase |
| hAECs | Human amniotic epithelial cells |
| hAFCs | Human amniotic fluid cells |
| LH | Luteinizing hormone |
| MDR1 | Multidrug resistance gene |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| p27Kip1 (p27) | Cyclin-dependent kinase inhibitor |
| PD | Postnatal day |
| PDK1 | 3-phosphoinositide-dependent kinase-1 |
| PGCs | Primordial germ cells |
| PI3K | Phosphatidylinositol 3-kinase |
| PM | Phosphoramide mustard |
| POF | Premature ovarian failure |
| PTEN | Phosphatase and tensin homolog |
| ROS | Reactive oxygen species |
| SCF | Stem cell factor |
| rpS6 | Ribosomal protein S6 |
| S1P | Sphingosine-1-phosphate |
| S6K1 | S6 kinase 1 |
| TSC | Tuberous sclerosis complex |
References
- Turan, V.; Oktay, K. Sexual and fertility adverse effects associated with chemotherapy treatment in women. Expert Opin. Drug Saf. 2014, 13, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Letourneau, J.; Chan, S.W.; Rosen, M.P. Accelerating ovarian age: Cancer treatment in the premenopausal woman. Semin. Reprod Med. 2013, 31, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Kasum, M.; Beketic-Oreskovic, L.; Peddi, P.F.; Oreskovic, S.; Johnson, R.H. Fertility after breast cancer treatment. Eur. J. Obstet Gynecol. Reprod. Biol. 2014, 173, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Wallberg, K.A.; Oktay, K. Fertility preservation during cancer treatment: Clinical guidelines. Cancer Manag. Res. 2014, 6, 105–117. [Google Scholar] [PubMed]
- Salama, M.; Woodruff, T.K. Anticancer treatments and female fertility: Clinical concerns and role of oncologists in oncofertility practice. Expert. Rev. Anticancer Ther. 2017, 17, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Van der Ven, H.; Liebenthron, J.; Beckmann, M.; Toth, B.; Korell, M.; Krussel, J.; Frambach, T.; Kupka, M.; Hohl, M.K.; Winkler-Crepaz, K.; et al. Ninety-five orthotopic transplantations in 74 women of ovarian tissue after cytotoxic treatment in a fertility preservation network: Tissue activity, pregnancy and delivery rates. Hum. Reprod. 2016, 31, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Wallberg, K.A.; Gemzell-Danielsson, K. Twenty years of development in fertility preservation of women and girls and the challenges that remain. Acta Obstet. Et Gynecol. Scand. 2019, 98, 543–544. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Wallberg, K.A.; Marklund, A.; Lundberg, F.; Wikander, I.; Milenkovic, M.; Anastacio, A.; Sergouniotis, F.; Wanggren, K.; Ekengren, J.; Lind, T.; et al. A prospective study of women and girls undergoing fertility preservation due to oncologic and non-oncologic indications in Sweden-Trends in patients’ choices and benefit of the chosen methods after long-term follow up. Acta Obstet. Gynecol. Scand. 2019, 98, 604–615. [Google Scholar] [CrossRef]
- Donnez, J.; Dolmans, M.M.; Pellicer, A.; Diaz-Garcia, C.; Sanchez Serrano, M.; Schmidt, K.T.; Ernst, E.; Luyckx, V.; Andersen, C.Y. Restoration of ovarian activity and pregnancy after transplantation of cryopreserved ovarian tissue: A review of 60 cases of reimplantation. Fertil. Steril. 2013, 99, 1503–1513. [Google Scholar] [CrossRef]
- Rodriguez-Wallberg, K.A.; Tanbo, T.; Tinkanen, H.; Thurin-Kjellberg, A.; Nedstrand, E.; Kitlinski, M.L.; Macklon, K.T.; Ernst, E.; Fedder, J.; Tiitinen, A.; et al. Ovarian tissue cryopreservation and transplantation among alternatives for fertility preservation in the Nordic countries—Compilation of 20 years of multicenter experience. Acta Obstet. Gynecol. Scand. 2016, 95, 1015–1026. [Google Scholar] [CrossRef]
- Vu, J.V.; Llarena, N.C.; Estevez, S.L.; Moravek, M.B.; Jeruss, J.S. Oncofertility program implementation increases access to fertility preservation options and assisted reproductive procedures for breast cancer patients. J. Surg. Oncol. 2017, 115, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Poirot, C.; Brugieres, L.; Yakouben, K.; Prades-Borio, M.; Marzouk, F.; de Lambert, G.; Pacquement, H.; Bernaudin, F.; Neven, B.; Paye-Jaouen, A.; et al. Ovarian tissue cryopreservation for fertility preservation in 418 girls and adolescents up to 15 years of age facing highly gonadotoxic treatment. Twenty years of experience at a single center. Acta Obstet. Gynecol. Scand. 2019, 98, 630–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapira, M.; Raanani, H.; Cohen, Y.; Meirow, D. Fertility preservation in young females with hematological malignancies. Acta Haematol. 2014, 132, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.; Donnez, J.; Ginsburg, E.; Meirow, D. Emergency IVF versus ovarian tissue cryopreservation: Decision making in fertility preservation for female cancer patients. Fertil. Steril. 2013, 99, 1534–1542. [Google Scholar] [CrossRef] [PubMed]
- Dolmans, M.M.; Luyckx, V.; Donnez, J.; Andersen, C.Y.; Greve, T. Risk of transferring malignant cells with transplanted frozen–thawed ovarian tissue. Fertil. Steril. 2013, 99, 1514–1522. [Google Scholar] [CrossRef] [PubMed]
- Quinn, G.; Vadaparampil, S.; Gwede, C.; Miree, C.; King, L.; Clayton, H.; Wilson, C.; Munster, P. Discussion of fertility preservation with newly diagnosed patients: oncologists’ views. J. Cancer Surviv. 2007, 1, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Schover, L.R.; Brey, K.; Lichtin, A.; Lipshultz, L.I.; Jeha, S. Oncologists’ attitudes and practices regarding banking sperm before cancer treatment. J. Clin. Oncol. 2002, 20, 1890–1897. [Google Scholar] [CrossRef]
- Armuand, G.M.; Rodriguez-Wallberg, K.A.; Wettergren, L.; Ahlgren, J.; Enblad, G.; Höglund, M.; Lampic, C. Sex differences in fertility-related information received by young adult cancer survivors. J. Clin. Oncol. 2012, 30, 2147. [Google Scholar] [CrossRef]
- Li, J.; Eriksson, M.; Czene, K.; Hall, P.; Rodriguez-Wallberg, K.A. Common diseases as determinants of menopausal age. Hum. Reprod. 2016, 31, 2856–2864. [Google Scholar] [CrossRef]
- Bedoschi, G.; Navarro, P.A.; Oktay, K. Chemotherapy-induced damage to ovary: Mechanisms and clinical impact. Future Oncol. 2016, 12, 2333–2344. [Google Scholar] [CrossRef]
- Gonfloni, S.; Di Tella, L.; Caldarola, S.; Cannata, S.M.; Klinger, F.G.; Di Bartolomeo, C.; Mattei, M.; Candi, E.; De Felici, M.; Melino, G.; et al. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat. Med. 2009, 15, 1179–1185. [Google Scholar] [CrossRef]
- Ganesan, S.; Keating, A.F. The ovarian DNA damage repair response is induced prior to phosphoramide mustard-induced follicle depletion, and ataxia telangiectasia mutated inhibition prevents PM-induced follicle depletion. Toxicol. Appl. Pharm. 2016, 292, 65–74. [Google Scholar] [CrossRef]
- Petrillo, S.K.; Desmeules, P.; Truong, T.Q.; Devine, P.J. Detection of DNA damage in oocytes of small ovarian follicles following phosphoramide mustard exposures of cultured rodent ovaries in vitro. Toxicol. Appl. Pharm. 2011, 253, 94–102. [Google Scholar] [CrossRef]
- Tsai-Turton, M.; Luong, B.T.; Tan, Y.; Luderer, U. Cyclophosphamide-induced apoptosis in COV434 human granulosa cells involves oxidative stress and glutathione depletion. Toxicol. Sci. 2007, 98, 216–230. [Google Scholar] [CrossRef]
- Chang, E.M.; Lim, E.; Yoon, S.; Jeong, K.; Bae, S.; Lee, D.R.; Yoon, T.K.; Choi, Y.; Lee, W.S. Cisplatin Induces Overactivation of the dormant primordial follicle through PTEN/AKT/FOXO3a Pathway which leads to loss of ovarian reserve in mice. PLoS ONE 2015, 10, e0144245. [Google Scholar] [CrossRef]
- Kalich-Philosoph, L.; Roness, H.; Carmely, A.; Fishel-Bartal, M.; Ligumsky, H.; Paglin, S.; Wolf, I.; Kanety, H.; Sredni, B.; Meirow, D. Cyclophosphamide triggers follicle activation and ″burnout″; AS101 prevents follicle loss and preserves fertility. Sci. Transl. Med. 2013, 5, 185ra62. [Google Scholar] [CrossRef]
- Madden, J.A.; Thomas, P.Q.; Keating, A.F. Phosphoramide mustard induces autophagy markers and mTOR inhibition prevents follicle loss due to phosphoramide mustard exposure. Reprod. Toxicol. 2017, 67, 65–78. [Google Scholar] [CrossRef]
- Meirow, D.; Dor, J.; Kaufman, B.; Shrim, A.; Rabinovici, J.; Schiff, E.; Raanani, H.; Levron, J.; Fridman, E. Cortical fibrosis and blood-vessels damage in human ovaries exposed to chemotherapy. Potential mechanisms of ovarian injury. Hum. Reprod. 2007, 22, 1626–1633. [Google Scholar] [CrossRef] [Green Version]
- Bar-Joseph, H.; Ben-Aharon, I.; Tzabari, M.; Tsarfaty, G.; Stemmer, S.M.; Shalgi, R. In vivo bioimaging as a novel strategy to detect doxorubicin-induced damage to gonadal blood vessels. PLoS ONE 2011, 6, e23492. [Google Scholar] [CrossRef]
- Hoffman, B.L.; Schorge, J.O.; Bradshaw, K.D.; Halvorson, L.M.; Schaffer, J.I.; Corton, M.M. Williams Gynecology, 3rd ed.; McGraw Hill Professional: New York, NY, USA, 2016. [Google Scholar]
- Brevini, T.A.L. Gametogenesis, Early Embryo Development and Stem Cell Derivation; Springer: New York, NY, USA, 2013. [Google Scholar]
- Piprek, R.P. Molecular Mechanisms of Cell Differentiation in Gonad Development; Springer: New York, NY, USA, 2016; Volume 58. [Google Scholar]
- Boron, W.F.; Boulpaep, E.L. The female reproductive system. In Medical Physiology: A Cellular and Molecular Approach, 2nd ed.; Elsevier Health Sciences: New York, NY, USA, 2012. [Google Scholar]
- McGee, E.A.; Hsueh, A.J. Initial and cyclic recruitment of ovarian follicles. Endocr. Rev. 2000, 21, 200–214. [Google Scholar] [CrossRef]
- Aittomäki, K.; Dieguez Lucena, J.; Pakarinen, P.; Sistonen, P.; Tapanainen, J.; Gromoll, J.; Kaskikari, R.; Sankila, E.-M.; Lehväslaiho, H.; Reyes Engel, A.; et al. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell 1995, 82, 959–968. [Google Scholar] [CrossRef] [Green Version]
- Cossigny, D.A.; Findlay, J.K.; Drummond, A.E. The effects of FSH and activin A on follicle development in vitro. Reproduction 2012, 143, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Gougeon, A. Dynamics of follicular growth in the human: A model from preliminary results. Hum. Reprod. 1986, 1, 7. [Google Scholar] [CrossRef]
- Fauser, B.C.; Van Heusden, A.M. Manipulation of human ovarian function: Physiological concepts and clinical consequences. Endocr. Rev. 1997, 18, 71–106. [Google Scholar]
- Faddy, M.J.; Gosden, R.G. A mathematical model of follicle dynamics in the human ovary. Hum. Reprod. 1995, 10, 770–775. [Google Scholar] [CrossRef]
- Practice Committee of the American Society for Reproductive Medicine. Smoking and infertility: A committee opinion. Fertil. Steril. 2012; 98, 1400–1406. [Google Scholar]
- Meeker, J.D.; Benedict, M.D. Infertility, pregnancy loss and adverse birth outcomes in relation to maternal secondhand tobacco smoke exposure. Curr. Womens Health Rev. 2013, 9, 41–49. [Google Scholar] [CrossRef]
- Meirow, D.; Biederman, H.; Anderson, R.A.; Wallace, W.H. Toxicity of chemotherapy and radiation on female reproduction. Clin. Obstet. Gynecol. 2010, 53, 727–739. [Google Scholar] [CrossRef]
- Keating, A.F.; Sen, N.; Sipes, I.G.; Hoyer, P.B. Dual protective role for glutathione S-transferase class pi against VCD-induced ovotoxicity in the rat ovary. Toxicol. Appl. Pharm. 2010, 247, 71–75. [Google Scholar] [CrossRef]
- Nam, E.Y.; Kim, S.A.; Kim, H.; Kim, S.H.; Han, J.H.; Lee, J.H.; Kim, D.I. Akt activation by evodiae fructus extract protects ovary against 4-vinylcyclohexene diepoxide-induced ovotoxicity. J. Ethnopharmacol. 2016, 194, 733–739. [Google Scholar] [CrossRef]
- Wandji, S.A.; Srsen, V.; Voss, A.K.; Eppig, J.J.; Fortune, J.E. Initiation in vitro of growth of bovine primordial follicles. Biol. Reprod. 1996, 55, 942–948. [Google Scholar] [CrossRef]
- Reddy, P.; Zheng, W.; Liu, K. Mechanisms maintaining the dormancy and survival of mammalian primordial follicles. Trends Endocrinol. Metab. 2010, 21, 96–103. [Google Scholar] [CrossRef]
- Kim, J.Y. Control of ovarian primordial follicle activation. Clin. Exp. Reprod. Med. 2012, 39, 10–14. [Google Scholar] [CrossRef] [Green Version]
- Uhlenhaut, N.H.; Treier, M. Forkhead transcription factors in ovarian function. Reproduction 2011, 142, 489–495. [Google Scholar] [CrossRef]
- Pisarska, M.D.; Barlow, G.; Kuo, F.T. Minireview: Roles of the forkhead transcription factor FOXL2 in granulosa cell biology and pathology. Endocrinology 2011, 152, 1199–1208. [Google Scholar] [CrossRef]
- Crisponi, L.; Deiana, M.; Loi, A.; Chiappe, F.; Uda, M.; Amati, P.; Bisceglia, L.; Zelante, L.; Nagaraja, R.; Porcu, S.; et al. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat. Genet. 2001, 27, 159–166. [Google Scholar] [CrossRef]
- Uda, M.; Ottolenghi, C.; Crisponi, L.; Garcia, J.E.; Deiana, M.; Kimber, W.; Forabosco, A.; Cao, A.; Schlessinger, D.; Pilia, G. Foxl2 disruption causes mouse ovarian failure by pervasive blockage of follicle development. Hum. Mol. Genet. 2004, 13, 1171–1181. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, D.; Ovitt, C.E.; Anlag, K.; Fehsenfeld, S.; Gredsted, L.; Treier, A.C.; Treier, M. The murine winged-helix transcription factor Foxl2 is required for granulosa cell differentiation and ovary maintenance. Development 2004, 131, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, N.; Ramaswamy, S.; Vazquez, F.; Signoretti, S.; Loda, M.; Sellers, W.R. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol. Cell. Biol. 2000, 20, 8969–8982. [Google Scholar] [CrossRef]
- Castrillon, D.H.; Miao, L.; Kollipara, R.; Horner, J.W.; DePinho, R.A. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science 2003, 301, 215–218. [Google Scholar] [CrossRef]
- Pelosi, E.; Omari, S.; Michel, M.; Ding, J.; Amano, T.; Forabosco, A.; Schlessinger, D.; Ottolenghi, C. Constitutively active Foxo3 in oocytes preserves ovarian reserve in mice. Nat. Commun. 2013, 4, 1843. [Google Scholar] [CrossRef]
- Durlinger, A.L.; Visser, J.A.; Themmen, A.P. Regulation of ovarian function: The role of anti-Mullerian hormone. Reproduction 2002, 124, 601–609. [Google Scholar] [CrossRef]
- Nilsson, E.; Rogers, N.; Skinner, M.K. Actions of anti-Mullerian hormone on the ovarian transcriptome to inhibit primordial to primary follicle transition. Reproduction 2007, 134, 209–221. [Google Scholar] [CrossRef]
- Visser, J.A.; Themmen, A.P. Anti-Mullerian hormone and folliculogenesis. Mol. Cell. Endocrinol. 2005, 234, 81–86. [Google Scholar] [CrossRef]
- Durlinger, A.L.; Kramer, P.; Karels, B.; de Jong, F.H.; Uilenbroek, J.T.; Grootegoed, J.A.; Themmen, A.P. Control of primordial follicle recruitment by anti-Mullerian hormone in the mouse ovary. Endocrinology 1999, 140, 5789–5796. [Google Scholar] [CrossRef]
- Carlsson, I.B.; Scott, J.E.; Visser, J.A.; Ritvos, O.; Themmen, A.P.; Hovatta, O. Anti-Mullerian hormone inhibits initiation of growth of human primordial ovarian follicles in vitro. Hum. Reprod. 2006, 21, 2223–2227. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef]
- Makker, A.; Goel, M.M.; Mahdi, A.A. PI3K/PTEN/Akt and TSC/mTOR signaling pathways, ovarian dysfunction, and infertility: An update. J. Mol. Endocrinol. 2014, 53, R103–R118. [Google Scholar] [CrossRef]
- Reddy, P.; Liu, L.; Adhikari, D.; Jagarlamudi, K.; Rajareddy, S.; Shen, Y.; Du, C.; Tang, W.; Hamalainen, T.; Peng, S.L.; et al. Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science 2008, 319, 611–613. [Google Scholar] [CrossRef]
- Adhikari, D.; Gorre, N.; Risal, S.; Zhao, Z.; Zhang, H.; Shen, Y.; Liu, K. The safe use of a PTEN inhibitor for the activation of dormant mouse primordial follicles and generation of fertilizable eggs. PLoS ONE 2012, 7, e39034. [Google Scholar] [CrossRef]
- Li, J.; Kawamura, K.; Cheng, Y.; Liu, S.; Klein, C.; Liu, S.; Duan, E.K.; Hsueh, A.J. Activation of dormant ovarian follicles to generate mature eggs. Proc. Natl. Acad. Sci. USA 2010, 107, 10280–10284. [Google Scholar] [CrossRef] [Green Version]
- Novella-Maestre, E.; Herraiz, S.; Rodriguez-Iglesias, B.; Diaz-Garcia, C.; Pellicer, A. Short-Term PTEN inhibition improves in vitro activation of primordial follicles, preserves follicular viability, and restores AMH levels in cryopreserved ovarian tissue from cancer patients. PLoS ONE 2015, 10, e0127786. [Google Scholar] [CrossRef]
- Rajareddy, S.; Reddy, P.; Du, C.; Liu, L.; Jagarlamudi, K.; Tang, W.; Shen, Y.; Berthet, C.; Peng, S.L.; Kaldis, P.; et al. p27kip1 (cyclin-dependent kinase inhibitor 1B) controls ovarian development by suppressing follicle endowment and activation and promoting follicle atresia in mice. Mol. Endocrinol. 2007, 21, 2189–2202. [Google Scholar] [CrossRef]
- Yang, Q.; Guan, K.L. Expanding mTOR signaling. Cell. Res. 2007, 17, 666–681. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, D.; Zheng, W.; Shen, Y.; Gorre, N.; Hamalainen, T.; Cooney, A.J.; Huhtaniemi, I.; Lan, Z.J.; Liu, K. Tsc/mTORC1 signaling in oocytes governs the quiescence and activation of primordial follicles. Hum. Mol. Genet. 2010, 19, 397–410. [Google Scholar] [CrossRef]
- Adhikari, D.; Flohr, G.; Gorre, N.; Shen, Y.; Yang, H.; Lundin, E.; Lan, Z.; Gambello, M.J.; Liu, K. Disruption of Tsc2 in oocytes leads to overactivation of the entire pool of primordial follicles. Mol. Hum. Reprod. 2009, 15, 765–770. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.; Adhikari, D.; Zheng, W.; Liang, S.; Hamalainen, T.; Tohonen, V.; Ogawa, W.; Noda, T.; Volarevic, S.; Huhtaniemi, I.; et al. PDK1 signaling in oocytes controls reproductive aging and lifespan by manipulating the survival of primordial follicles. Hum. Mol. Genet. 2009, 18, 2813–2824. [Google Scholar] [CrossRef] [Green Version]
- Gagliardi, P.A.; Puliafito, A.; Primo, L. PDK1: At the crossroad of cancer signaling pathways. Seminars in Cancer Biology 2018, 48, 27–35. [Google Scholar] [CrossRef]
- Zheng, W.; Nagaraju, G.; Liu, Z.; Liu, K. Functional roles of the phosphatidylinositol 3-kinases (PI3Ks) signaling in the mammalian ovary. Mol. Cell. Endocrinol. 2012, 356, 24–30. [Google Scholar] [CrossRef]
- Takimoto, C.H.; Calvo, E. Principles of oncologic pharmacotherapy. Cancer Manag. Multidiscipl. Approach 2008, 11, 1–9. [Google Scholar]
- Rodriguez-Wallberg, K.A.; Oktay, K. Fertility preservation and pregnancy in women with and without BRCA mutation-positive breast cancer. Oncologist 2012, 17, 1409–1417. [Google Scholar] [CrossRef]
- Morgan, S.; Anderson, R.A.; Gourley, C.; Wallace, W.H.; Spears, N. How do chemotherapeutic agents damage the ovary? Hum. Reprod. Update 2012, 18, 525–535. [Google Scholar] [CrossRef] [Green Version]
- Roness, H.; Kalich-Philosoph, L.; Meirow, D. Prevention of chemotherapy-induced ovarian damage: Possible roles for hormonal and non-hormonal attenuating agents. Hum. Reprod. Update 2014, 20, 759–774. [Google Scholar] [CrossRef]
- Kondo, N.; Takahashi, A.; Ono, K.; Ohnishi, T. DNA damage induced by alkylating agents and repair pathways. J. Nucleic Acids 2010, 2010, 543531. [Google Scholar] [CrossRef]
- Myers, M.; Hutt, K.J. Damage Control in the Female Germline: Protecting Primordial Follicles. In Oogenesis; Coticchio, G., Albertini, D.F., Eds.; Springer-Verlag: London, UK, 2013. [Google Scholar]
- Tingen, C.M.; Bristol-Gould, S.K.; Kiesewetter, S.E.; Wellington, J.T.; Shea, L.; Woodruff, T.K. Prepubertal primordial follicle loss in mice is not due to classical apoptotic pathways. Biol. Reprod. 2009, 81, 16–25. [Google Scholar] [CrossRef]
- Ludeman, S.M. The chemistry of the metabolites of cyclophosphamide. Curr. Pharm. Des. 1999, 5, 627–643. [Google Scholar]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell. Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Shimizu, S. Bcl-2 family: Life-or-death switch. FEBS Lett. 2000, 466, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.B.; Hutt, K.J.; Michalak, E.M.; Cook, M.; Vandenberg, C.J.; Liew, S.H.; Bouillet, P.; Mills, A.; Scott, C.L.; Findlay, J.K.; et al. DNA damage-induced primordial follicle oocyte apoptosis and loss of fertility require TAp63-mediated induction of Puma and Noxa. Mol. Cell. 2012, 48, 343–352. [Google Scholar] [CrossRef]
- Tuppi, M.; Kehrloesser, S.; Coutandin, D.W.; Rossi, V.; Luh, L.M.; Strubel, A.; Hotte, K.; Hoffmeister, M.; Schafer, B.; De Oliveira, T.; et al. Oocyte DNA damage quality control requires consecutive interplay of CHK2 and CK1 to activate p63. Nat. Struct. Mol. Biol. 2018, 25, 261. [Google Scholar] [CrossRef]
- Coutandin, D.; Osterburg, C.; Srivastav, R.K.; Sumyk, M.; Kehrloesser, S.; Gebel, J.; Tuppi, M.; Hannewald, J.; Schafer, B.; Salah, E.; et al. Quality control in oocytes by p63 is based on a spring-loaded activation mechanism on the molecular and cellular level. Elife 2016, 5, e13909. [Google Scholar] [CrossRef]
- Nguyen, Q.N.; Zerafa, N.; Liew, S.H.; Morgan, F.H.; Strasser, A.; Scott, C.L.; Findlay, J.K.; Hickey, M.; Hutt, K.J. Loss of PUMA protects the ovarian reserve during DNA-damaging chemotherapy and preserves fertility. Cell Death Dis. 2018, 9, 618. [Google Scholar] [CrossRef]
- Luan, Y.; Edmonds, M.E.; Woodruff, T.K.; Kim, S.-Y. Inhibitors of apoptosis protect the ovarian reserve from cyclophosphamide. J. Endocrinol. 2019, 240, 243. [Google Scholar] [CrossRef]
- Devine, P.J.; Perreault, S.D.; Luderer, U. Roles of reactive oxygen species and antioxidants in ovarian toxicity. Biol. Reprod. 2012, 86, 27. [Google Scholar] [CrossRef]
- Jeelani, R.; Khan, S.N.; Shaeib, F.; Kohan-Ghadr, H.R.; Aldhaheri, S.R.; Najafi, T.; Thakur, M.; Morris, R.; Abu-Soud, H.M. Cyclophosphamide and acrolein induced oxidative stress leading to deterioration of metaphase II mouse oocyte quality. Free Radic. Biol. Med. 2017, 110, 11–18. [Google Scholar] [CrossRef]
- Lande, Y.; Fisch, B.; Tsur, A.; Farhi, J.; Prag-Rosenberg, R.; Ben-Haroush, A.; Kessler-Icekson, G.; Zahalka, M.A.; Ludeman, S.M.; Abir, R. Short-term exposure of human ovarian follicles to cyclophosphamide metabolites seems to promote follicular activation in vitro. Reprod. Biomed. Online 2017, 34, 104–114. [Google Scholar] [CrossRef]
- Roness, H.; Gavish, Z.; Cohen, Y.; Meirow, D. Ovarian follicle burnout: A universal phenomenon? Cell Cycle 2013, 12, 3245–3246. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Xie, Y.; Li, S.; Liang, Y.; Qiu, Q.; Lin, H.; Zhang, Q. Rapamycin prevents cyclophosphamide-induced over-activation of primordial follicle pool through PI3K/Akt/mTOR signaling pathway in vivo. J. Ovarian Res. 2017, 10, 56. [Google Scholar] [CrossRef]
- Yadav, P.K.; Tiwari, M.; Gupta, A.; Sharma, A.; Prasad, S.; Pandey, A.N.; Chaube, S.K. Germ cell depletion from mammalian ovary: Possible involvement of apoptosis and autophagy. J. Biomed. Sci. 2018, 25, 36. [Google Scholar] [CrossRef]
- Ben-Aharon, I.; Meizner, I.; Granot, T.; Uri, S.; Hasky, N.; Rizel, S.; Yerushalmi, R.; Sulkes, A.; Stemmer, S.M. Chemotherapy-induced ovarian failure as a prototype for acute vascular toxicity. Oncologist 2012, 17, 1386–1393. [Google Scholar] [CrossRef]
- Soleimani, R.; Heytens, E.; Darzynkiewicz, Z.; Oktay, K. Mechanisms of chemotherapy-induced human ovarian aging: Double strand DNA breaks and microvascular compromise. Aging 2011, 3, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.M.; Sutcliffe, S.B. Protection of ovarian function by oral contraceptives in women receiving chemotherapy for Hodgkin’s disease. Blood 1981, 58, 849–851. [Google Scholar] [PubMed]
- Pandir, D.; Kara, O.; Kara, M. Protective effect of bilberry (Vaccinium myrtillus L.) on cisplatin induced ovarian damage in rat. Cytotechnology 2014, 66, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Khedr, N.F. Protective effect of mirtazapine and hesperidin on cyclophosphamide-induced oxidative damage and infertility in rat ovaries. Exp. Biol. Med. 2015, 240, 1682–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yang, S.; Lv, X.; Sun, H.; Weng, J.; Liang, Y.; Zhou, D. The mechanism of mesna in protection from cisplatin-induced ovarian damage in female rats. J. Gynecol. Oncol. 2013, 24, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Taskin, M.I.; Yay, A.; Adali, E.; Balcioglu, E.; Inceboz, U. Protective effects of sildenafil citrate administration on cisplatin-induced ovarian damage in rats. Gynecol. Endocrinol. 2015, 31, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Chen, H.; Wang, G.; Yu, Y.; Xie, K. Hydrogen-rich saline attenuates chemotherapy-induced ovarian injury via regulation of oxidative stress. Exp. Med. 2015, 10, 2277–2282. [Google Scholar] [CrossRef] [PubMed]
- Salih, S.M.; Ringelstetter, A.K.; Elsarrag, M.Z.; Abbott, D.H.; Roti, E.C. Dexrazoxane abrogates acute doxorubicin toxicity in marmoset ovary. Biol. Reprod. 2015, 92, 73. [Google Scholar] [CrossRef]
- Roti Roti, E.C.; Salih, S.M. Dexrazoxane ameliorates doxorubicin-induced injury in mouse ovarian cells. Biol. Reprod. 2012, 86, 96. [Google Scholar] [CrossRef]
- Kropp, J.; Roti Roti, E.C.; Ringelstetter, A.; Khatib, H.; Abbott, D.H.; Salih, S.M. Dexrazoxane Diminishes Doxorubicin-Induced Acute Ovarian Damage and Preserves Ovarian Function and Fecundity in Mice. PLoS ONE 2015, 10, e0142588. [Google Scholar] [CrossRef]
- Roti Roti, E.C.; Ringelstetter, A.K.; Kropp, J.; Abbott, D.H.; Salih, S.M. Bortezomib prevents acute doxorubicin ovarian insult and follicle demise, improving the fertility window and pup birth weight in mice. PLoS ONE 2014, 9, e108174. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Cordeiro, M.H.; Serna, V.A.; Ebbert, K.; Butler, L.M.; Sinha, S.; Mills, A.A.; Woodruff, T.K.; Kurita, T. Rescue of platinum-damaged oocytes from programmed cell death through inactivation of the p53 family signaling network. Cell Death Differ. 2013, 20, 987–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, S.; Lopes, F.; Gourley, C.; Anderson, R.A.; Spears, N. Cisplatin and doxorubicin induce distinct mechanisms of ovarian follicle loss; imatinib provides selective protection only against cisplatin. PLoS ONE 2013, 8, e70117. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.J.; Lee, L.J.; Tzeng, C.R.; Wang, C.W.; Hsu, M.I.; Chen, C.H. Targeted anti-apoptosis activity for ovarian protection against chemotherapy-induced ovarian gonadotoxicity. Reprod. Biomed. Online 2014, 29, 612–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Turan, V.; Lierman, S.; Cuvelier, C.; De Sutter, P.; Oktay, K. Sphingosine-1-phosphate prevents chemotherapy-induced human primordial follicle death. Hum. Reprod. 2014, 29, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Sosulski, A.E.; Zhang, L.; Saatcioglu, H.D.; Wang, D.; Nagykery, N.; Sabatini, M.E.; Gao, G.; Donahoe, P.K.; Pepin, D. AMH/MIS as a contraceptive that protects the ovarian reserve during chemotherapy. Proc. Natl. Acad. Sci. USA 2017, 114, E1688–E1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.; Lee, O.H.; Lee, Y.; Yoon, H.; Chang, E.M.; Park, M.; Lee, J.W.; Hong, K.; Kim, J.O.; Kim, N.K.; et al. Melatonin prevents cisplatin-induced primordial follicle loss via suppression of PTEN/AKT/FOXO3a pathway activation in the mouse ovary. J. Pineal Res. 2016, 60, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.J.; Yeh, Y.C.; Shang, W.J.; Wu, G.J.; Liu, J.Y.; Chen, C.H. Protective effect of a gonadotropin-releasing hormone analogue on chemotherapeutic agent-induced ovarian gonadotoxicity: A mouse model. Eur. J. Obstet. Gynecol. Reprod. Biol. 2010, 149, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Gao, H.; Yuan, Z. Effect of leuprolide acetate on ovarian function after cyclophosphamide-doxorubicin-based chemotherapy in premenopausal patients with breast cancer: Results from a phase II randomized trial. Med. Oncol. 2013, 30, 667. [Google Scholar] [CrossRef] [PubMed]
- Moore, H.C.; Unger, J.M.; Phillips, K.A.; Boyle, F.; Hitre, E.; Porter, D.; Francis, P.A.; Goldstein, L.J.; Gomez, H.L.; Vallejos, C.S.; et al. Goserelin for ovarian protection during breast-cancer adjuvant chemotherapy. N. Engl. J. Med. 2015, 372, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Skaznik-Wikiel, M.E.; McGuire, M.M.; Sukhwani, M.; Donohue, J.; Chu, T.; Krivak, T.C.; Rajkovic, A.; Orwig, K.E. Granulocyte colony-stimulating factor with or without stem cell factor extends time to premature ovarian insufficiency in female mice treated with alkylating chemotherapy. Fertil. Steril. 2013, 99, 2045–2054.e2043. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, A.H. NRF2 much of a good thing. Sci. Transl. Med. 2017, 9, eaar4435. [Google Scholar] [CrossRef]
- Herman, E.H.; Ferrans, V.J. Examination of the potential long-lasting protective effect of ICRF-187 against anthracycline-induced chronic cardiomyopathy. Cancer Treat. Rev. 1990, 17, 155–160. [Google Scholar] [CrossRef]
- Pouillart, P. Evaluating the role of dexrazoxane as a cardioprotectant in cancer patients receiving anthracyclines. Cancer Treat. Rev. 2004, 30, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, M.; Jendiroba, D.; Pagliaro, L.; Keyhani, A.; Liu, B.; Freireich, E.J. Dexrazoxane in combination with anthracyclines lead to a synergistic cytotoxic response in acute myelogenous leukemia cell lines. Leuk. Res. 2003, 27, 617–626. [Google Scholar] [CrossRef]
- Styczynski, J.; Wysocki, M.; Balwierz, W.; Kowalczyk, J.R. Dexrazoxane has no impact on sensitivity of childhood leukemic blasts to daunorubicin. Leukemia 2002, 16, 820–825. [Google Scholar] [CrossRef]
- Barry, E.V.; Vrooman, L.M.; Dahlberg, S.E.; Neuberg, D.S.; Asselin, B.L.; Athale, U.H.; Clavell, L.A.; Larsen, E.C.; Moghrabi, A.; Samson, Y.; et al. Absence of secondary malignant neoplasms in children with high-risk acute lymphoblastic leukemia treated with dexrazoxane. J. Clin. Oncol. 2008, 26, 1106–1111. [Google Scholar] [CrossRef]
- Vrooman, L.M.; Neuberg, D.S.; Stevenson, K.E.; Asselin, B.L.; Athale, U.H.; Clavell, L.; Cole, P.D.; Kelly, K.M.; Larsen, E.C.; Laverdiere, C.; et al. The low incidence of secondary acute myelogenous leukaemia in children and adolescents treated with dexrazoxane for acute lymphoblastic leukaemia: A report from the Dana-Farber Cancer Institute ALL Consortium. Eur. J. Cancer 2011, 47, 1373–1379. [Google Scholar] [CrossRef] [Green Version]
- Tebbi, C.K.; London, W.B.; Friedman, D.; Villaluna, D.; De Alarcon, P.A.; Constine, L.S.; Mendenhall, N.P.; Sposto, R.; Chauvenet, A.; Schwartz, C.L. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin’s disease. J. Clin. Oncol. 2007, 25, 493–500. [Google Scholar] [CrossRef]
- Marty, M.; Espie, M.; Llombart, A.; Monnier, A.; Rapoport, B.L.; Stahalova, V.; Dexrazoxane Study, G. Multicenter randomized phase III study of the cardioprotective effect of dexrazoxane (Cardioxane) in advanced/metastatic breast cancer patients treated with anthracycline-based chemotherapy. Ann. Oncol. 2006, 17, 614–622. [Google Scholar] [CrossRef]
- Testore, F.; Milanese, S.; Ceste, M.; de Conciliis, E.; Parello, G.; Lanfranco, C.; Manfredi, R.; Ferrero, G.; Simoni, C.; Miglietta, L.; et al. Cardioprotective effect of dexrazoxane in patients with breast cancer treated with anthracyclines in adjuvant setting: A 10-year single institution experience. Am. J. Cardiovasc. Drugs 2008, 8, 257–263. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends in Molecular Medicine 2006, 12, 440–450. [Google Scholar] [CrossRef]
- Van Brocklyn, J.R.; Williams, J.B. The control of the balance between ceramide and sphingosine-1-phosphate by sphingosine kinase: Oxidative stress and the seesaw of cell survival and death. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2012, 163, 26–36. [Google Scholar] [CrossRef]
- Slater, C.A.; Liang, M.H.; McCune, J.W.; Christman, G.M.; Laufer, M.R. Preserving ovarian function in patients receiving cyclophosphamide. Lupus 1999, 8, 3–10. [Google Scholar] [CrossRef]
- Kishk, E.A.; Mohammed Ali, M.H. Effect of a gonadotropin-releasing hormone analogue on cyclophosphamide-induced ovarian toxicity in adult mice. Arch. Gynecol. Obstet. 2013, 287, 1023–1029. [Google Scholar] [CrossRef]
- Meirow, D.; Assad, G.; Dor, J.; Rabinovici, J. The GnRH antagonist cetrorelix reduces cyclophosphamide-induced ovarian follicular destruction in mice. Hum. Reprod. 2004, 19, 1294–1299. [Google Scholar] [CrossRef]
- Meistrich, M.L. Failure to demonstrate hormonal protection of chemotherapy-induced fertility reduction in female rats. Reprod. Toxicol. 1994, 8, 277. [Google Scholar] [CrossRef]
- Horicks, F.; Van Den Steen, G.; Houben, S.; Englert, Y.; Demeestere, I. Folliculogenesis Is Not Fully Inhibited during GnRH Analogues Treatment in Mice Challenging Their Efficiency to Preserve the Ovarian Reserve during Chemotherapy in This Model.(Gonadotropin-releasing hormone)(Report). PLoS ONE 2015, 10, e0137164. [Google Scholar] [CrossRef]
- Yuce, M.A.; Balkanli Kaplan, P.; Gucer, F.; Doganay, L.; Altaner, S.; Canda, T.; Yardim, T. Prevention of cyclophosphamide-induced ovarian damage by concomitant administration of GnRHa in mice: A dose-dependent relationship? Eur. J. Gynaecol. Oncol. 2004, 25, 628–631. [Google Scholar]
- Horicks, F.; Van Den Steen, G.; Gervy, C.; Clarke, H.J.; Demeestere, I. Both in vivo FSH depletion and follicular exposure to Gonadotrophin-releasing hormone analogues in vitro are not effective to prevent follicular depletion during chemotherapy in mice. Mol. Hum. Reprod. 2018, 24, 221–232. [Google Scholar] [CrossRef]
- Bildik, G.; Akin, N.; Senbabaoglu, F.; Sahin, G.N.; Karahuseyinoglu, S.; Ince, U.; Taskiran, C.; Selek, U.; Yakin, K.; Guzel, Y.; et al. GnRH agonist leuprolide acetate does not confer any protection against ovarian damage induced by chemotherapy and radiation in vitro. Hum. Reprod. 2015, 30, 2912–2925. [Google Scholar]
- Kilic, S.; Pinarli, F.; Ozogul, C.; Tasdemir, N.; Naz Sarac, G.; Delibasi, T. Protection from cyclophosphamide-induced ovarian damage with bone marrow-derived mesenchymal stem cells during puberty. Gynecol. Endocrinol. 2014, 30, 135–140. [Google Scholar] [CrossRef]
- Lambertini, M.; Moore, H.C.F.; Leonard, R.C.F.; Loibl, S.; Munster, P.; Bruzzone, M.; Boni, L.; Unger, J.M.; Anderson, R.A.; Mehta, K.; et al. Gonadotropin-Releasing Hormone Agonists During Chemotherapy for Preservation of Ovarian Function and Fertility in Premenopausal Patients With Early Breast Cancer: A Systematic Review and Meta-Analysis of Individual Patient-Level Data. J. Clin. Oncol. 2018, 36, 1981–1990. [Google Scholar] [CrossRef]
- Oktay, K.; Rodriguez-Wallberg, K.; Munster, P. Ovarian protection during adjuvant chemotherapy. N. Engl. J. Med. 2015, 372, 2268–2269. [Google Scholar]
- Bedaiwy, M.A.; Abou-Setta, A.M.; Desai, N.; Hurd, W.; Starks, D.; El-Nashar, S.A.; Al-Inany, H.G.; Falcone, T. Gonadotropin-releasing hormone analog cotreatment for preservation of ovarian function during gonadotoxic chemotherapy: A systematic review and meta-analysis. Fertil. Steril. 2011, 95, 906–914.e904. [Google Scholar] [CrossRef]
- Chen, H.; Xiao, L.; Li, J.; Cui, L.; Huang, W. Adjuvant gonadotropin-releasing hormone analogues for the prevention of chemotherapy-induced premature ovarian failure in premenopausal women. Cochrane Database Syst. Rev. 2019, 3, CD008018. [Google Scholar] [CrossRef]
- Oktay, K.; Harvey, B.E.; Partridge, A.H.; Quinn, G.P.; Reinecke, J.; Taylor, H.S.; Wallace, W.H.; Wang, E.T.; Loren, A.W. Fertility Preservation in Patients With Cancer: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2018, 36, 1994–2001. [Google Scholar] [CrossRef]
- Ting, A.Y.; Petroff, B.K. Tamoxifen decreases ovarian follicular loss from experimental toxicant DMBA and chemotherapy agents cyclophosphamide and doxorubicin in the rat. J. Assist. Reprod. Genet. 2010, 27, 591–597. [Google Scholar] [CrossRef] [Green Version]
- Muhonen, T.; Jantunen, I.; Pertovaara, H.; Voutilainen, L.; Maiche, A.; Blomqvist, C.; Pyrhonen, S.; Kellokumpu-Lehtinen, P. Prophylactic filgrastim (G-CSF) during mitomycin-C, mitoxantrone, and methotrexate (MMM) treatment for metastatic breast cancer. A randomized study. Am. J. Clin. Oncol. 1996, 19, 232–234. [Google Scholar] [CrossRef]
- Ganesan, S.; Nteeba, J.; Madden, J.A.; Keating, A.F. Obesity alters phosphoramide mustard-induced ovarian DNA repair in mice. Biol. Reprod. 2017, 96, 491–501. [Google Scholar] [CrossRef]
- Nteeba, J.; Ganesan, S.; Madden, J.A.; Dickson, M.J.; Keating, A.F. Progressive obesity alters ovarian insulin, phosphatidylinositol-3 kinase, and chemical metabolism signaling pathways and potentiates ovotoxicity induced by phosphoramide mustard in mice. Biol. Reprod. 2017, 96, 478–490. [Google Scholar] [CrossRef]
- Xiang, Y.; Xu, J.; Li, L.; Lin, X.; Chen, X.; Zhang, X.; Fu, Y.; Luo, L. Calorie restriction increases primordial follicle reserve in mature female chemotherapy-treated rats. Gene 2012, 493, 77–82. [Google Scholar] [CrossRef]
- Matsushita, N.; Takami, Y.; Kimura, M.; Tachiiri, S.; Ishiai, M.; Nakayama, T.; Takata, M. Role of NAD-dependent deacetylases SIRT1 and SIRT2 in radiation and cisplatin-induced cell death in vertebrate cells. Genes Cells 2005, 10, 321–332. [Google Scholar] [CrossRef]
- Salih, S.M. Retrovirus-mediated multidrug resistance gene (MDR1) overexpression inhibits chemotherapy-induced toxicity of granulosa cells. Fertil. Steril. 2011, 95, 1390–1396.e6. [Google Scholar] [CrossRef] [Green Version]
- Ahn, R.W.; Barrett, S.L.; Raja, M.R.; Jozefik, J.K.; Spaho, L.; Chen, H.; Bally, M.B.; Mazar, A.P.; Avram, M.J.; Winter, J.N.; et al. Nano-encapsulation of arsenic trioxide enhances efficacy against murine lymphoma model while minimizing its impact on ovarian reserve in vitro and in vivo. PLoS ONE 2013, 8, e58491. [Google Scholar] [CrossRef]
- Lai, D.M.; Wang, F.Y.; Chen, Y.F.; Wang, L.; Wang, Y.L.; Cheng, W.W. Human amniotic fluid stem cells have a potential to recover ovarian function in mice with chemotherapy-induced sterility. BMC Dev. Biol. 2013, 13, 34. [Google Scholar] [CrossRef]
- Wang, F.; Wang, L.; Yao, X.; Lai, D.; Guo, L. Human amniotic epithelial cells can differentiate into granulosa cells and restore folliculogenesis in a mouse model of chemotherapy-induced premature ovarian failure. Stem Cell Res. Ther. 2013, 4, 124. [Google Scholar] [CrossRef]
- Leibowitz, B.J.; Yang, L.; Wei, L.; Buchanan, M.E.; Rachid, M.; Parise, R.A.; Beumer, J.H.; Eiseman, J.L.; Schoen, R.E.; Zhang, L.; et al. Targeting p53-dependent stem cell loss for intestinal chemoprotection. Sci. Transl. Med. 2018, 10, eaam7610. [Google Scholar] [CrossRef] [Green Version]
- Oronsky, B.; Scribner, C.; Aggarwal, R.; Cabrales, P. RRx-001 protects normal tissues but not tumors via Nrf2 induction and Bcl-2 inhibition. J. Cancer Res. Clin. Oncol. 2019, 145, 2045–2050. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Agents | Representatives | Anti-Tumor Mechanisms | Targeting Cell Cycle |
|---|---|---|---|
| Alkylating agents | Cyclophosphamide, iphosphamide, melphalan, busulfan, nitrogen mustard, nitrosoureas, procarbazine, chlorambucil | Produce highly reactive intermediates that form covalent bonds with nucleophilic substances; cause DNA inter- and intra-chain cross links; interfere with DNA transcription and replication | Non-specific |
| Platinum analogs | Cisplatin, carboplatin | Crosslink with purine bases; cause DNA damage; and interfere with DNA repair | Non-specific |
| Taxanes and Plant alkaloids | Paclitaxel, docetaxel, vincristine | Bind tubulin to inhibit its polymerization into microtubules; prevent spindle formation; and cause metaphase arrest | M-phase specific |
| Antitumor Antibiotics | Mitomycin, bleomycin, doxorubicin, valrubicin | Intercalate into the minor groove of double-stranded DNA between guanine-cytosine base pairs; interfere with RNA polymerase movement along the DNA; prevent transcription; cause DNA double-strand breaks; and stabilize DNA-topoisomerase II complexes | Specific and non-specific |
| Topoisomerase inhibitors | Epipodophyllotoxins etoposide, teniposide | Inhibit topoisomerase; suppress microtubule aggregation; and inhibit spindle formation | S- and G2-phase specific |
| Antimetabolites | Methotrexate, 5-fluorouracil, 6-mercaptopurine, hydroxyurea, | Inhibit the synthesis of or compete with purine or pyrimidine nucleotide precursors during DNA or RNA synthesis | S-phase specific |
| Enzymes | l-asparaginase | Inhibit the enzyme ribonucleotide diphosphate reductase; limit ribonucleotide conversion thus block DNA synthesis; deprive exogenously supplied asparagine thus limits protein synthesis | S-phase specific |
| Group | Mechanism Proposed | Substance | Chemotherapy | Experimental Model | Reference |
|---|---|---|---|---|---|
| Anti-oxidants | Alleviate free radical damage | Bilberry | Cisplatin | In vivo; rat | [99] |
| Mirtazapine or Hesperidin | CPA | In vivo; rat | [100] | ||
| Mesna | Cisplatin | In vivo; rat | [101] | ||
| Sildenafil citrate | Cisplatin | In vivo; rat | [102] | ||
| Hydrogen-rich saline | Cisplatin | In vivo; rat | [103] | ||
| Iron chelate drugs | Reduce the number of metal ions complexed with anthracycline decreasing the formation of superoxide radicals | Dexrazoxane | Doxorubicin | In vitro; marmoset ovarian tissue | [104] |
| In vitro; mouse cell line and mouse ovary | [105] | ||||
| In vivo; mouse | [106] | ||||
| Proteasome inhibitors | Inhibit chemotherapeutic drugs’ nuclear accumulation | Bortezomib | Doxorubicin | In vivo; mouse | [107] |
| c -Abl kinase inhibitors | Block apoptosis pathway | Imatinib | Cisplatin | In vitro, mouse ovary; in vivo, mouse ovary sub-renal graft | [21,108,109] |
| ATM inhibitors | KU55933 | CPA | In vitro; rat ovary | [22] | |
| Ceramide-induced death pathway inhibitors | S1P | Busulfan, | In vivo; mouse | [110] | |
| CPA, doxorubicin | In vivo; human ovarian xenograft to mouse | [111] | |||
| CHK2 inhibitors | CK2II | 4-HC | In vitro; mouse | [89] | |
| ATR inhibitors | ETP46464 | 4-HC | In vitro; mouse | [89] | |
| Glycoprotein hormones | Inhibit primordial follicle overactivation | AMH | Carboplatin, doxorubicin, CPA | In vivo; mouse | [112] |
| Immunomodulators | AS101 | CPA | In vivo; mouse | [26] | |
| mTOR inhibitors | Rapamycin | CPA | In vitro; mouse ovary | [27] | |
| Hormones & Free radical scavengers | Melatonin | Cisplatin | In vivo; mouse | [113] | |
| GnRHa | Triptorelin | Busulfan | In vivo; mouse | [114] | |
| Leuprolide acetate | CPA, doxorubicin | Breast cancer patients | [115] | ||
| Goserelin | CPA, anthracycline | Breast cancer patients | [116] | ||
| Colony and stem cell factors | Decrease ovarian micro-vessel loss | G-CSF ± SCF | CPA, busulfan | In vivo; mouse | [117] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, X.; Anastácio, A.; Liu, K.; Rodriguez-Wallberg, K.A. Ovarian Follicle Depletion Induced by Chemotherapy and the Investigational Stages of Potential Fertility-Protective Treatments—A Review. Int. J. Mol. Sci. 2019, 20, 4720. https://doi.org/10.3390/ijms20194720
Hao X, Anastácio A, Liu K, Rodriguez-Wallberg KA. Ovarian Follicle Depletion Induced by Chemotherapy and the Investigational Stages of Potential Fertility-Protective Treatments—A Review. International Journal of Molecular Sciences. 2019; 20(19):4720. https://doi.org/10.3390/ijms20194720
Chicago/Turabian StyleHao, Xia, Amandine Anastácio, Kui Liu, and Kenny A. Rodriguez-Wallberg. 2019. "Ovarian Follicle Depletion Induced by Chemotherapy and the Investigational Stages of Potential Fertility-Protective Treatments—A Review" International Journal of Molecular Sciences 20, no. 19: 4720. https://doi.org/10.3390/ijms20194720
APA StyleHao, X., Anastácio, A., Liu, K., & Rodriguez-Wallberg, K. A. (2019). Ovarian Follicle Depletion Induced by Chemotherapy and the Investigational Stages of Potential Fertility-Protective Treatments—A Review. International Journal of Molecular Sciences, 20(19), 4720. https://doi.org/10.3390/ijms20194720