1. Introduction
Glioblastoma multiforme (GBM) is a central nervous system tumor classified as grade IV of high-grade malignant gliomas (HGG), according to the World Health Organization (WHO) guidelines [
1]. GBM belongs to the group of diffuse astrocytic and oligodendroglial tumor, joining oligodendrocytomas, ependymomas, and mixed gliomas, under the glioma classification [
2]. According to the ASTRO guidelines statements, the current standard care for GBM is surgical resection to the feasible extent, followed by conventional radiotherapy (RT) of 60 Gy delivered by fractions of 2 Gy, up to seven weeks. Moreover, chemotherapy is concurrent to RT with daily temozolomide (TMZ) administration [
3,
4,
5]. These treatment modalities are not currently curative and the resistance to both chemotherapy and RT plans is the main cause of GBM care failures (the median survival time is 14.6 months) [
6]. Moreover, the percentage of relapses and side effects post TMZ and RT treatments is more than 90% [
7]. More precisely, even if the application of TMZ has significantly improved clinical GBM outcomes, cases of drug resistance related to the activity of the enzyme methyl guanine methyl transferase (MGMT) have been observed [
8]. The hypermethylation of its promoter, is indeed associated with a better survival rate in patients receiving TMZ with or without RT [
9]. In addition, the dose release onto healthy brain tissue or surrounding organs at risk during irradiation may, substantially, contribute to late tissue toxicities, such as radionecrosis and neurocognitive dysfunction, because of their limited dose tolerance.
In recent years, different dose fractionation schedules have been improved to have a better prognosis, avoiding the large side effect even in case of focal re-irradiation of recurrences. In this scenario, proton therapy (PT) could be used as a successful strategy for GBM treatment, being able to regulate the balance between tumor control and the normal tissue tolerance [
10,
11,
12,
13,
14]. In particular, when heavy particles cross the tissues, they deposit a minimal radiation dose on their track to the tumor. The depth-dose distribution, described by the Bragg peak trend, gradually increases as a function of the depth. So, the so-called spread-out Bragg peak (SOBP) lead to a complete irradiation of the target volume and a more conformal dose distribution, sparing the surrounding healthy tissues from damage [
15,
16]. This specific dose distribution curve represents a key topic for GBM tumor treatments in which the sparing of healthy tissue is a key factor for the patient’s quality of life. Therefore, there is a robust scientific rationale motivating the need to enlarge studies that guide towards new clinical trials for PT combined with targeted therapy rather than conventional RT with photons or electrons [
17,
18].
Today, in the context of personalized medicine, prognostic and predictive molecular biomarkers are useful to select cancer therapeutic planning [
19,
20]. A critical point in RT success is the prediction of cancer radiosensitivity. At the molecular level, the idea that genes may behave as biomarkers of a disease response represents the base for the development of gene signatures, to predict response to cancer radiation treatments [
21]. Several genes have been shown to be responsive to radiation exposure and thanks to the use of high-throughput technologies, such as gene expression profiling (GEP) by microarray, radiosensitivity assays have been developed with gene signatures predicting radioresponse in many cancer types, including GBM [
22]. However, the response to radiation is highly cell-line dependent and some specific genes and pathways may be linked both to tumor subtypes and dose delivered [
23,
24,
25].
Actually, few published studies have evaluated the effectiveness of radiosensitizing agents combined with PT in GBM and none of them consider genes and response pathways induced by RT. Most studies have demonstrated that different genetic pathways and molecular features can provide reliable prognostic biomarkers, overlooking the treatment responses and predictive outcomes. However, according to WHO guidelines, IDH1/IDH2 gene status distinguishes a more radioresistant tumor type (primary GBM, IDH-wild type) from a more sensitive one (secondary GBM,
IDH-mutant). IDH mutation is correlated with epigenetic modifications of the MGMT gene and assumes a prognostic value together with other biomarkers such as, the presence of LOH 10q, epidermal growth factor receptor (EGFR) amplification, p16
INK4a deletion, TP53 mutation, PTEN mutation, and the co-deletion of 1p/19q [
26,
27,
28].
Based on this evidence, a large group of molecularly targeted agents have been designed, but none of them seem to overcome tumor radioresistance [
29]. Previous studies support an involvement of the SRC-family protein kinases in the irradiation induction of radioresistance mechanisms. SRC protein is a non-receptor tyrosine kinase that interacts with many intracellular proteins involved in GBM carcinogenesis and progression. In addition, in vitro and in vivo studies confirmed the correlation between SRC activity and GBM carcinogenesis. [
30].
In this work we analyzed the GEP on the U87 MG human glioblastoma cell line after treatment with PT alone or in combination with a new targeted compound, named Si306 (Lead Discovery Siena, Siena, Italy), inhibiting SRC proteins. The Si306 molecule is a new TKI, chosen among the family of pyrazolo[3,4-d] pyrimidines, that exhibited the ability to specifically bind the ATP site of SRC protein, making it inactive. Furthermore, previous in vitro and in vivo studies have shown that the Si306 determines a significant reduction of the β-PDGFR active phosphorylated form and a greater loss of the migratory ability in GBM cells stimulated by Epidermal Growth Factor (EGF). In addition, the antiproliferative effect of Si306 has been tested in association with conventional RT treatments both in vitro and in vivo [
31].
Here, in order to clarify the Si306 activity in GBM cells exposed to PT, we firstly evaluated radiosensitive effects of different amounts of the Si306 compound on the U87 cell line in combination with PT exposed at the doses of 1, 2, 3, 4, 10, and 21 Gy. Clonogenic assay and dose modifying factor (DMF) calculations were performed. We also analyzed the U87 cell radiosensitivity by applying the radiobiological linear-quadratic (LQ) model and calculated the α, β, and
ratio, commonly used to predict radiosensitivity of normal and tumor cells [
32].
In addition, at molecular level we selected 2 and 10 Gy of proton radiation doses combined with the Si306 to evaluate GEP induced responses, by using whole genome cDNA microarray. We described networks and specific gene signatures of response to both treatments, highlighting for the first time, the cell pathways induced by Si306.
3. Discussion
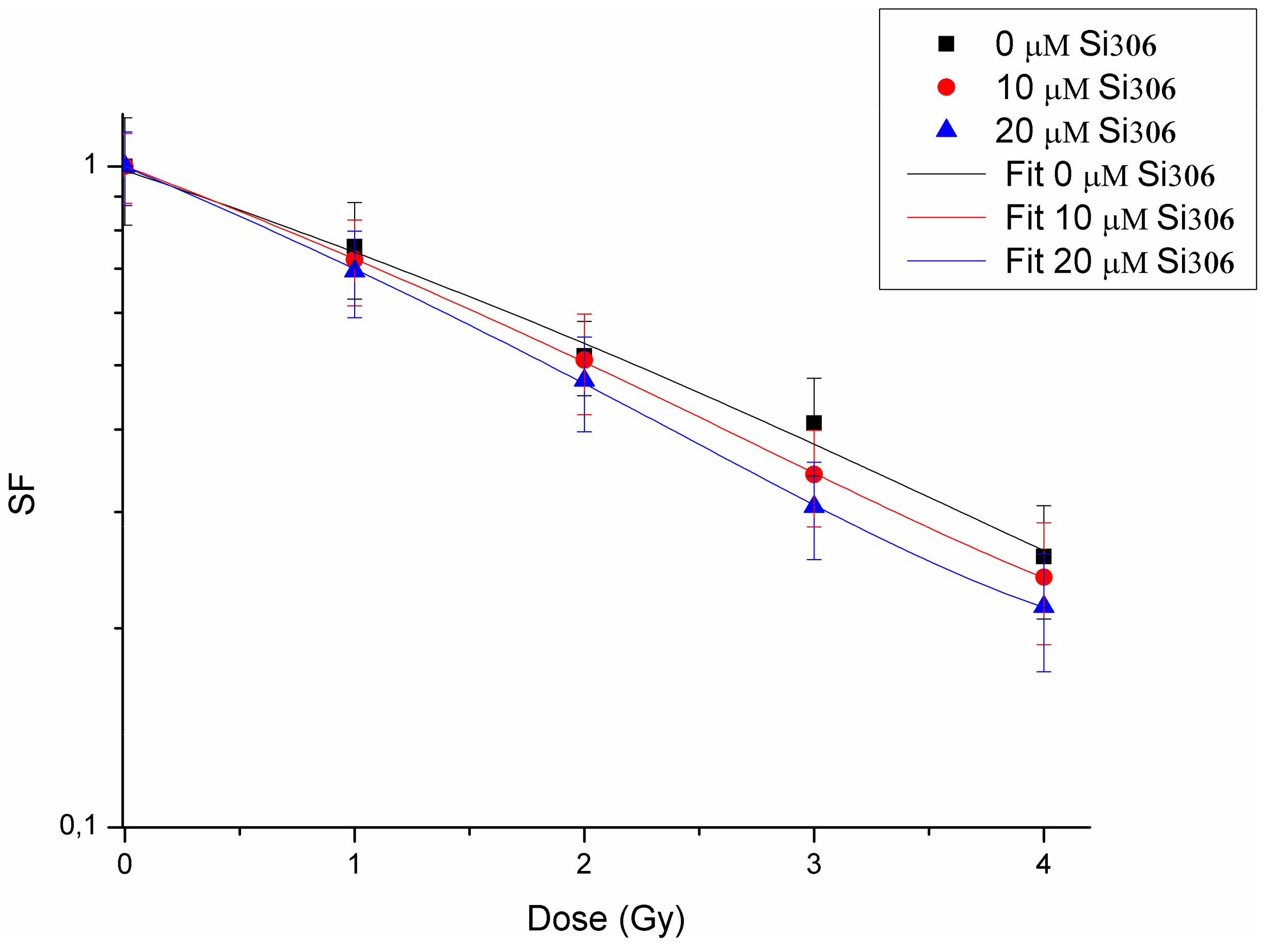
The first purpose of this study was to evaluate the radiosensitizing effects mediated by combination of the new compound, the Si306 targeting SRC proteins, with PT on the U87 human glioblastoma cell line. The IC50 evaluation showed that this cell line is sensitive to treatment with the Si306 compound. Based on the IC50 values, we tested the radiosensitizing effect of Si306, used at concentrations of 10 and 20 μM, in combination with proton irradiation at increasing doses of 1,2, 3, 4, 10, and 21 Gy, in order to generate dose/response curves for the dose configurations tested.
The radiosensitizing effect was evaluated by calculating the DMF, obtained at the SF of 50%, in order to highlight the combined treatment capacity of enhancing tumor cells killing in respect of irradiation only [
39]. Our data show that pretreatment with Si306 at both concentrations leads to a synergic cytotoxic effect with PT on the U87 cell line, further suggesting this compound as a new possible candidate to treat GBM in combination with PT. Indeed, the possibility to use drug/IR combined treatments, permits to increase the tumor control probability (TCP) even for radioresistant tumors, such as GBM. In addition, we also analyzed the U87 cell radiosensitivity by applying the radiobiological LQ model calculating the α, β parameters, and
ratio, which predict the radiosensitivity of normal and tumor cells [
32]. The LQ model is considered to be the best-fitting model to describe cell survival and, therefore, is of great interest in radiation oncology to highlight the link existing between the
ratio and the following RT-induced tissue reactions [
34,
40,
41]. The
ratio obtained on U87 cell line is in line with the
ratio calculated for a population of glioma cells reported by Barazzuol et al., who used a mathematical model to extract radiobiological information from clinical GBM patients data [
42]. In addition, our results showed a higher
ratio by using combined treatments of Si306 and protons. Therefore, we speculate that the clinical effect of using combined treatments of PT/Si306 administration, with an optimized Si306 pharmacological quantity for the patients, could be translated into the possibility of modifying the PT schedule treatment. Thus, all of this gains an efficacy in TCP, by using a more tolerable fractionated PT treatment plan and a reduced total dose delivered to the tumor [
43,
44].
As a second aim of this work, we carried out a transcriptomic study in order to define gene signatures as biomarkers of treatment response. GEP by whole genome cDNA microarray was firstly performed to analyze the gene expression changes uniquely induced by proton irradiation with 2 and 10 Gy of IR doses, which represent two clinical doses of interest and also for comparison with high-dose GEP studies of our research group [
23,
24,
36,
45].
In particular, the treatment of U87 with 2 Gy revealed that a large number of genes were deregulated and involved in the regulation of specific cellular processes (
Table 6). One of the activated pathways was the Hippo signaling pathway, an emerging growth control and tumor suppressor pathway that regulates cell proliferation and stem cell functions; the hyperactivation of its downstream effectors (such as TAZ protein, up regulated in U87 2 Gy with a fold change of 1,89) contributes to the development of cancer including GBM, suggesting that pharmacological inhibition of these factors may be an effective anticancer strategy [
46,
47]. In turn, in GBM cells Yang et al. recently reported that the Hippo transducer TAZ promotes cell proliferation and tumor formation through the EGFR pathway [
48]. In addition, Hippo and Wnt signaling, up regulated in U87 2 Gy cells, reciprocally regulate each other’s activity through a variety of mechanisms that needs to be better clarified in GBM cells [
49]. As known, Wnt/β-catenin signaling plays important roles in maintaining the stemness of cancer stem cells in various cancer types and in promoting cellular invasiveness. Multimodality in vivo and in vitro studies revealed a key role of Wnt activation in GBM radiation resistance. In turn, literature data report a pivotal role of the Wnt/β-catenin signaling pathway in IR-induced invasion of U87 GBM cells, indicating that β-catenin is a potential therapeutic target for overcoming evasive radioresistance [
50,
51].
In U87 2 Gy the involvement of cAMP signaling pathway was also observed. Existing evidence suggests that intracellular cAMP level and signaling may affect the survival of cancer cells, including resistant cancer cells to standard chemotherapeutic drugs. Suppression of the cAMP pathway is a common feature across different cancers including GBM. [
52,
53]. In addition, IR is known to be able to activate the transcription of genes, through the presence of cAMP responsive elements (CREs) in their promoters, in order to guide cell response and survival after radiation exposure [
54].
Moreover, the activation of antigen processing and presentation pathway after proton exposure with dose of 2 Gy in GBM cells is sustained by an up regulation of genes belonging to the human leukocyte antigen (HLA) class family (probably activated by β-catenin), factors involved in antigen presentation. As reported by Ghosh et al., HLA genes increasing level, often caused by a hypoxic tumor microenvironment, is associated with evasion of immune responses in cancer cells [
55]. Finally, an overall activation of several cell adhesion molecules was highlighted in U87 2 Gy cells, involved in the activation of inflammation process and in the regulation of cancer invasiveness.
On the other hand, U87 cells exposed to 10 Gy of proton irradiation activate specific cell pathways, including the phosphatidylinositol-3-kinase (PI3K)-protein kinase B (Akt) signaling pathway (
Table 6). As known, the PI3K/AKT pathway is commonly activated in cancer initiation and progression, including GBM, as it regulates different processes, such as proliferation, apoptosis, and migration [
56], therefore representing a key target for cancer therapeutics. Moreover, the activation of TP53 pathway was observed in U87 10 Gy and driven by TP53 gene that was significantly altered by 1.77-fold. As described, TP53, exerts a crucial role following IR-induced DNA damage because it is able to cause cell cycle arrest, DNA repair, and apoptosis processes. Moreover, the influence of TP53 status on DNA damage repair after cell irradiation has been studied in several malignancies and also reported by our group in breast cancer cells after a high dose of electron irradiation [
45,
57]. Finally, in U87 10 Gy, an activation of proteoglycan signaling was observed. Proteoglycans are known to have many roles in tumor progression and are the main extracellular matrix (ECM) components of normal brain tissue, playing an important role in brain development; an overproduction of different molecules of this family were found in GBM cells [
58,
59].
Interestingly, in U87 10 Gy Hippo and cAMP signaling pathways were activated, as above described in U87 2 Gy configuration, underling once again the important role of these processes in GBM cells after proton exposure.
In a second step, we evaluated the GEPs induced by Si306 molecule in U87 cells irradiated with 2 and 10 Gy of proton doses and we selected a large number of deregulated genes, grouped according to their involvement in specific biological pathways (
Table 7). In particular, in U87 Si306 + 2 Gy combined treatments a deregulated expression level of genes controlling phagosome was observed.
In GBM an intensive autophagic activity regulated by several signaling pathways was described [
60]. As recently reported by Yasui et al., an altered autophagic flux was described in GBM cell lines exposed to 10 Gy of γ-rays. Our data also confirms this trend after proton exposure. These altered fluxes represent a useful biomarker of metabolic stress induced by IR and provide a metabolic context for radiation sensitization [
61]. Here the Si306 radiosensitization effect seems to act by stressing this molecular mechanism. In addition, in U87 Si306 + 2 Gy configuration the involvement of antigen processing and presentation and cell adhesion molecules pathways were observed, similarly to that shown in U87 cells proton treated with only 2 Gy. Therefore, the Si306 treatment seems to cause an overall down regulation of HLA molecules (up regulated in U87 2 Gy), suggesting the activation of immune surveillance escaping mechanism induced by Si306 [
55,
62].
The latest two pathways deregulated in U87 Si306 + 2 Gy were linked to inflammation and calcium signaling. As known, the inflammation process is often activated in cell exposed to radiation, affecting cell fate by the activation of key transcription factors (TFs), such as NF-KB and STATs (i.e., STAT1 and STAT3) [
63]. Interestingly, the combined Si306 + 2 Gy treatment induced a down regulation of STAT1 and STAT3 proteins. Thus, we speculate that this inhibition could promote radiation sensitivity decreasing angiogenesis and cell survival as hypothesized in other malignancies by several authors [
64,
65]. Indeed, a number of studies confirm that selective inhibitors of these proinflammatory pathways driven by STAT TFs, could be combined to conventional radiation or chemotherapy to increase their effectiveness [
66,
67].
On the other hand, the combined treatment with Si306 and 2 Gy PT seem to affect survival/death balance by modulating the intracellular calcium levels, a mechanism known to be involved in regulating IR-induced cell cycle arrest, apoptosis, and chromatin structure modifications [
45,
68,
69].
Some of these pathways were also deregulated in U87 cells exposed to Si306 + 10 Gy, such as: Proteoglycans in cancer, leukocyte transendothelial migration, phagosome, cell adhesion molecules, and autoimmune disease. Three of the five pathways (proteoglycans in cancer, phagosome, and cell adhesion molecules), were also deregulated in the other configurations analyzed, suggesting once again their important role in U87 cells in response to radiation and/or to Si306 molecule. The other two selected pathways in U87 Si306 + 10 Gy (i.e., leukocyte transendothelial migration and autoimmune disease), highlight the involvement of a complex immunological response induced by IR, as known from the literature, and by the Si306 compound addition, as observed in this study.
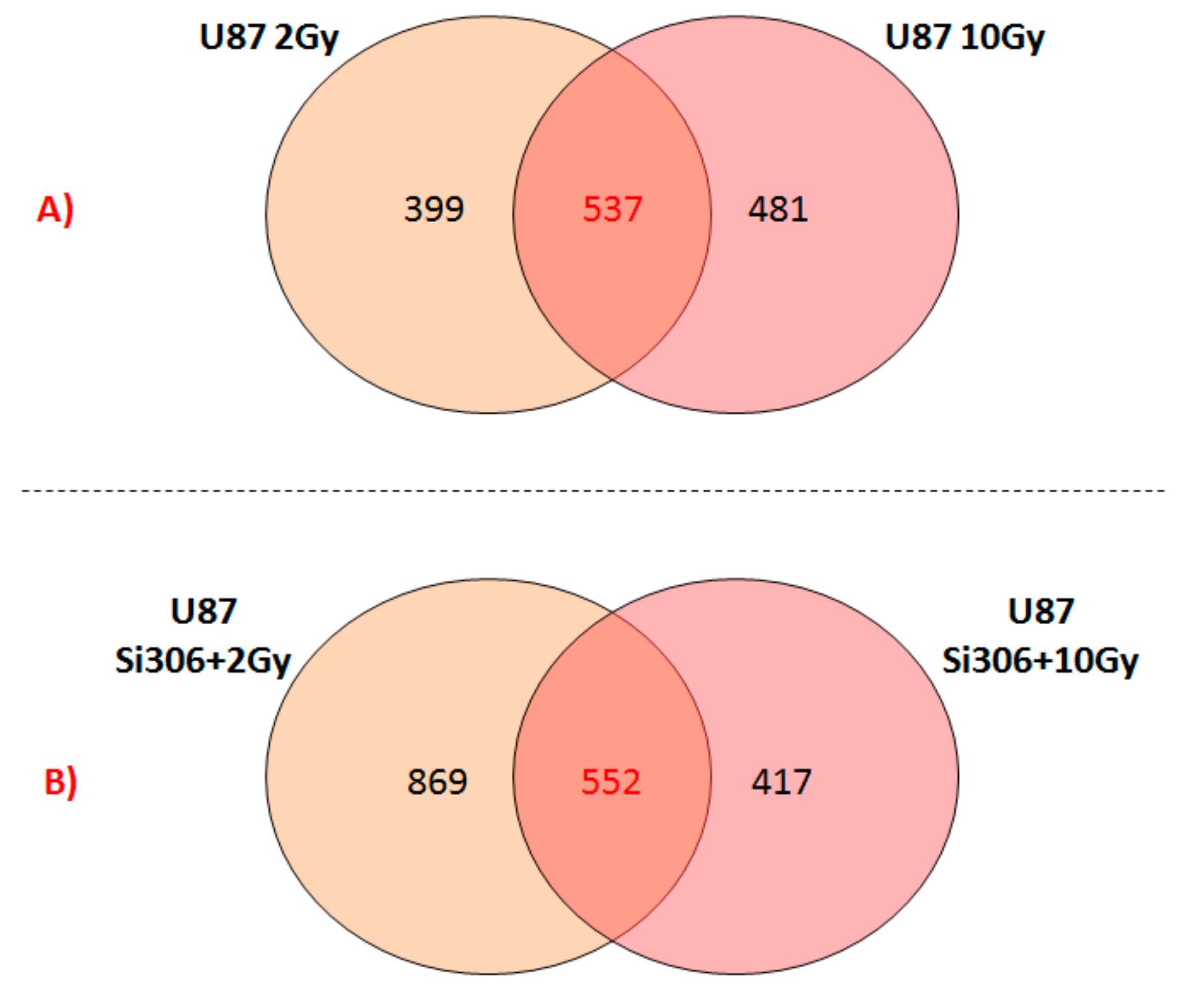
Finally, we reported the number of overlapping deregulated genes between the two configurations of the combined treatments, such as U87 Si306 + 2 Gy and U87 Si306 + 10 Gy (
Figure 2B). The top-five statistically relevant pathways selected and displayed in
Table 7, were previously described.
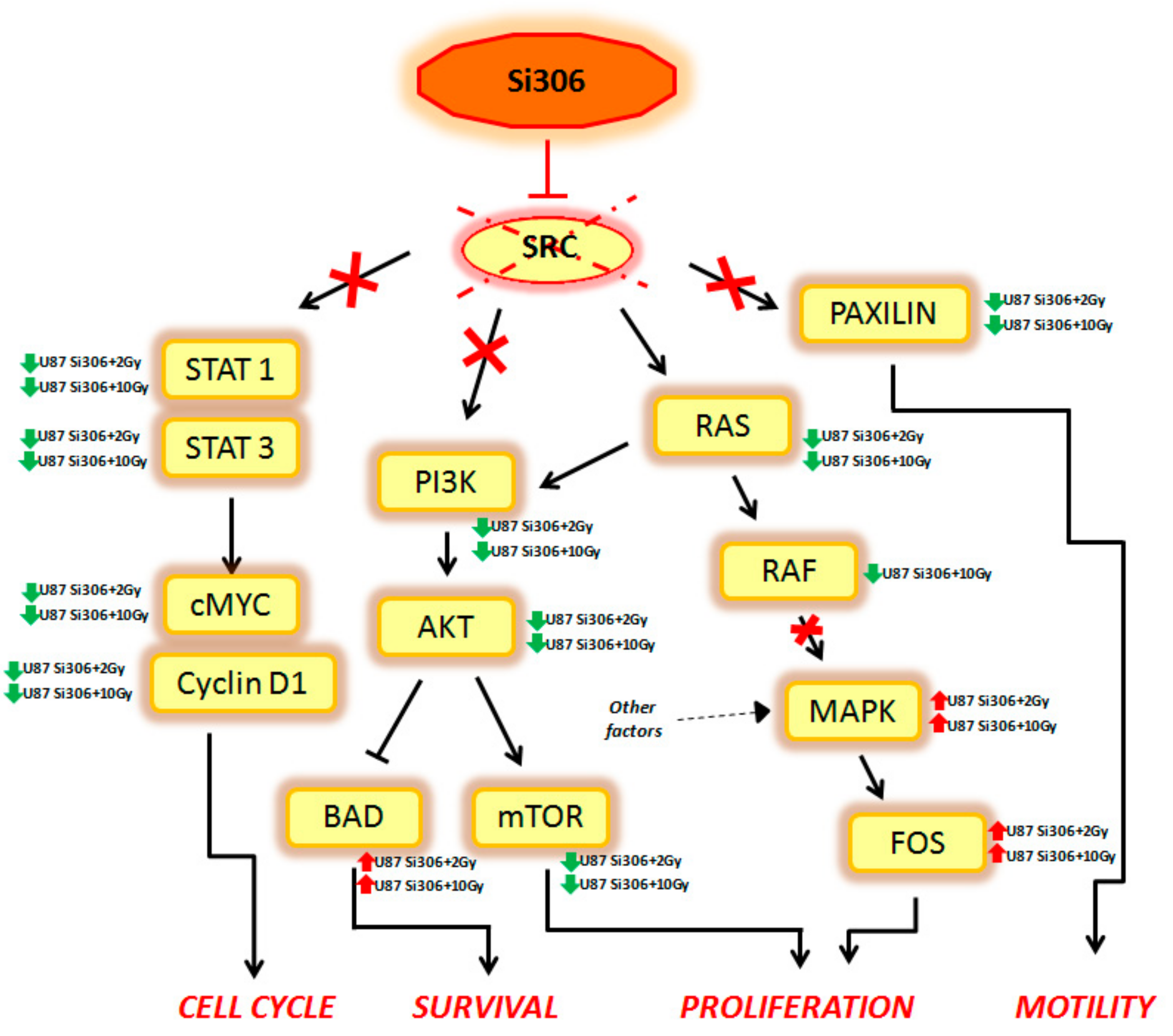
Summarizing, our GEP results show that combined treatments on U87 cells can activate multiple signal transduction pathways described, to our knowledge, for the first time, to be new targets of Si306. Finally, considering that the main target of Si306 is the tyrosine kinase SRC, we analyzed the known cellular target downstream to this transducer, in order to better clarify its role as molecular radiosensitizing. Thus, we observed that the combined treatment Si306 + protons (with 2 and 10 Gy) in U87 cells, is able to inhibit several signal transduction pathways, normally regulated by SRC as shown in
Figure 3.
In particular, the STAT1, STAT3, c-MYC, and Cyclin D1 genes, which are able to control the cell cycle, were downregulated in our analysis. Cell survival was negatively regulated by the downstream PI3K, AKT, and mTOR downregulation and by the BAD upregulation. In addition, Si306 is able to cause a partial inhibition of cell proliferation, downregulating RAS and RAF gene expression. However, the MAPK and FOS genes were not targets of Si306, so these factors (up regulated in our data), were probably activated by other cellular pathways. Finally, Si306 is also able to negatively regulate cell motility, through the downregulation of the paxilin gene.
These data confirm the SRC role as a main target of Si306 compound and highlight the transcriptional events occurring downstream of SRC inhibition by the combined treatments. The SRC blockage observed after Si306 and PT combined treatments seems to increase the single treatments effectiveness, thus promoting a radiosensitizing effect.
Today, very little data is available regarding the combination of molecularly targeted drugs and PT. Indeed, many studies debate about chemotherapeutic agents combined with high-linear energy transfer (LET) particle beams or protons for GBM treatment, overlooking the clinical perspective of target therapy [
70,
71].
The results obtained from this work have highlighted the radiosensitizing capacity of the Si306 targeted compound on U87 GBM cell line, acting in tandem with PT. Taking into account previously in vivo pharmacokinetic data, demonstrating that Si306 was able to reach the brain, overcoming the hurdle represented by the blood–brain barrier [
31], this compound can be considered a new candidate for combined treatments of GBM. In addition, our GEP results confirm the important role of SRC as the main Si306 target and highlight new genes and pathways modulated by the combined action of Si306 and PT, which can be further explored as new radiosensitizing therapeutic targets in GBM.

 ,
, 












{kind=link}
{kind=link}
{kind=link}