BET Proteins Are Required for Transcriptional Activation of the Senescent Islet Cell Secretome in Type 1 Diabetes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
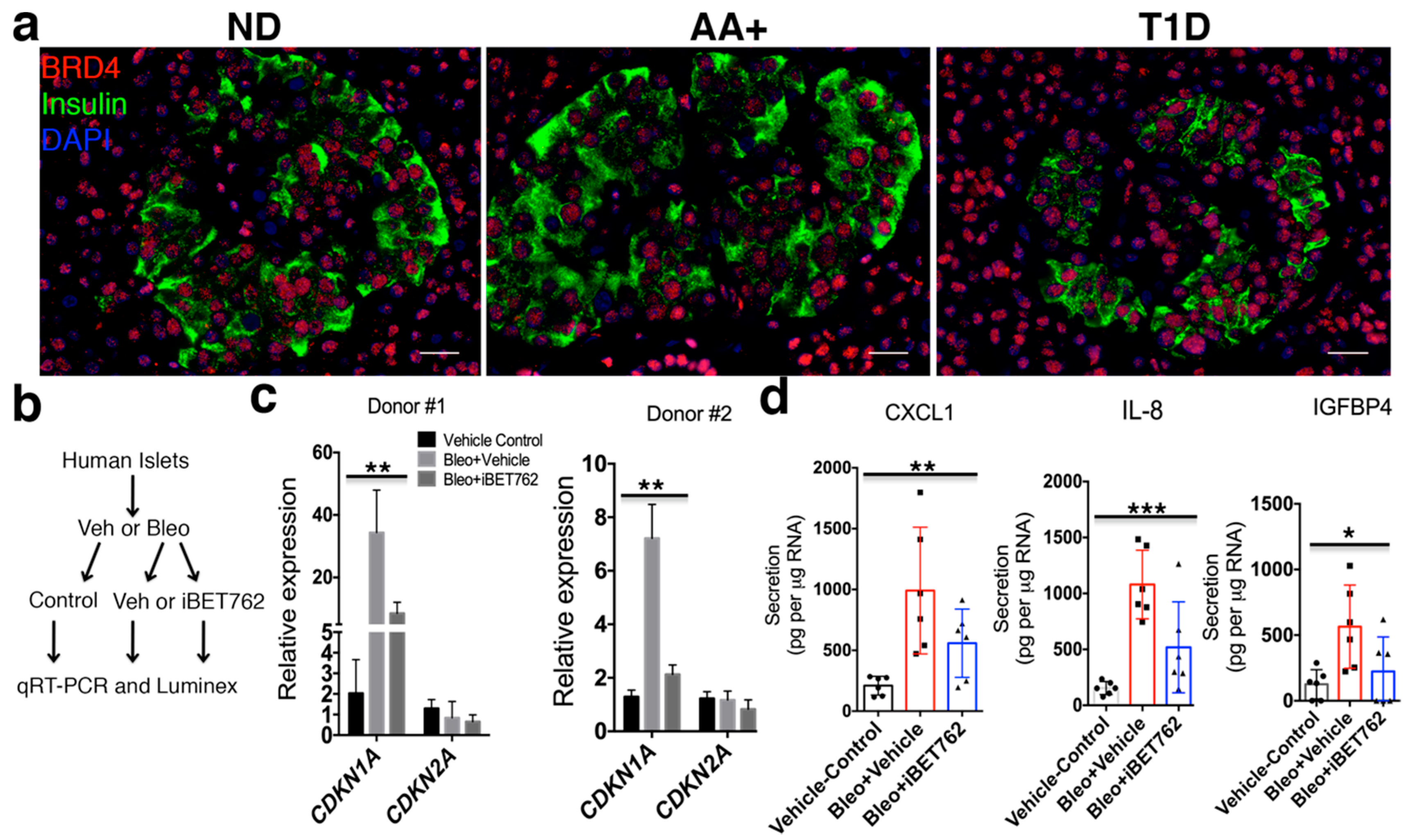
2. Results
2.1. BRD4 Is Required for SASP Gene Activation in NOD Islets
2.2. BET Inhibition Blunts SASP Secretion and Paracrine Activities
2.3. BET Proteins Are Required for SASP in Human Islets
2.4. BET Protein Inhibition Diminishes SASP In Vivo
3. Discussion
4. Materials and Methods
4.1. Animal Husbandry and Procedures
4.2. Human Pancreas Sections and Islets
4.3. Culture of Mouse Islets, Inhibitors and RNAi
4.4. Culture of Human Islets and Inhibitors
4.5. Immunohistochemistry and Quantification
4.6. THP-1 Chemotaxis Assay
4.7. Paracrine Senescence Assays
4.8. ChIP and qPCR
4.9. Quantitative Reverse-Transcription PCR
4.10. Luminex Assays
4.11. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BET | Bromodomain ExtraTerminal domain |
| NOD | Nonobese diabetic |
| qRT-PCR | Quantitative reverse transcription polymerase chain reaction |
| ChIP | Chromatin immunoprecipitation |
| DMSO | Dimethyl sulfoxide |
| DDR | DNA damage response |
| SASP | Senescence-associated secretory phenotype |
| ATM | Ataxia Telangiectasia mutated |
| NF-κB | Nuclear Factor kappa B |
| PARP1 | Poly-(ADP) ribose polymerase 1 |
| macroH2A1 | Macro Histone H2A1 |
| Bcl-2 | B cell lymphoma 2 |
| HMGB2 | High mobility group protein B2 |
| CEBPβ | CAAT enhancer binding protein beta |
| MLL1 | Mixed lineage leukemia 1 |
| Mitf | Melanocyte inducing transcription factor |
| H3K27ac | Histone H3 Lysine 27 acetylation |
| dbSUPER | Database of Superenhancers |
| BRD4 | Bromodomain-containing protein 4 |
| siRNA | Small interfering RNA |
| IL-6 | Interleukin 6 |
| Flnb | Filamin B |
| Mmp2 | Matrix metalloprotease 2 |
| Igfbp4 | Insulin-like growth factor binding protein 4 |
| Ppia | Peptidyl prolyl isomerase A |
| Cdkn1a | Cyclin dependent kinase inhibitor 1A |
| Cdkn2a | Cyclin dependent kinase inhibitor 2A |
| CD45 | Cluster of differentiation 45 |
| FITC | Fluorescein isothiocyanate |
| T1D | Type 1 Diabetes |
| Cxcl1 | CXC Chemokine ligand 1 |
| IL-8 | Interleukin 8 |
References
- Bluestone, J.A.; Herold, K.; Eisenbarth, G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010, 464, 1293–1300. [Google Scholar] [CrossRef] [Green Version]
- Christoffersson, G.; Rodriguez-Calvo, T.; von Herrath, M. Recent advances in understanding Type 1 Diabetes. F1000Research 2016, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.J.; Shah, A.; Ntranos, V.; Van Gool, F.; Atkinson, M.; Bhushan, A. Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes. Cell Metab. 2019, 29, 1045–1060. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.W.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özcan, S.; Alessio, N.; Acar, M.B.; Mert, E.; Omerli, F.; Peluso, G.; Galderisi, U. Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging (Albany. NY). 2016, 8, 1316–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef] [Green Version]
- Tasdemir, N.; Banito, A.; Roe, J.-S.; Alonso-Curbelo, D.; Camiolo, M.; Tschaharganeh, D.F.; Huang, C.-H.; Aksoy, O.; Bolden, J.E.; Chen, C.-C.; et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov. 2016, 6, 612–629. [Google Scholar] [CrossRef]
- Capell, B.C.; Drake, A.M.; Zhu, J.; Shah, P.P.; Dou, Z.; Dorsey, J.; Simola, D.F.; Donahue, G.; Sammons, M.; Rai, T.S.; et al. MLL1 is essential for the senescence- associated secretory phenotype. Genes Dev. 2016, 30, 321–336. [Google Scholar] [CrossRef]
- Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Tanizawa, H.; Fatkhutdinov, N.; Bitler, B.G.; Le, L.; Alicea, G.; Yang, T.L.; Johnson, F.B.; et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J. Cell Biol. 2016, 215, 325–334. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol. Cell 2015, 59, 719–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohanna, M.; Giuliano, S.; Bonet, C.; Imbert, V.; Hofman, V.; Zangari, J.; Bille, K.; Robert, C.; Bressac-de Paillerets, B.; Hofman, P.; et al. Senescent cells develop a PARP-1 and nuclear factor- k B-associated secretome (PNAS). Genes Dev. 2011, 25, 1245–1261. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.A.; Wong, F.S.; Wen, L. The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes. J. Autoimmun. 2016, 66, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Zhang, X. DbSUPER: A database of Super-enhancers in mouse and human genome. Nucleic Acids Res. 2016, 44, D164–D171. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Fu, W.; Farache, J.; Clardy, S.M.; Hattori, K.; Mander, P.; Lee, K.; Rioja, I.; Weissleder, R.; Prinjha, R.K.; Benoist, C.; et al. Epigenetic modulation of type-1 diabetes via a dual effect on pancreatic macrophages and Beta cells. Elife 2014, 3, e04631. [Google Scholar] [CrossRef]
- Ferri, E.; Petosa, C.; McKenna, C.E. Bromodomains: Structure, function and pharmacology of inhibition. Biochem. Pharmacol. 2016, 106, 1–18. [Google Scholar] [CrossRef]
- Malaquin, N.; Martinez, A.; Rodier, F. Keeping the senescence secretome under control: Molecular reins on the senescence-associated secretory phenotype. Exp. Gerontol. 2016, 82, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Thompson, M. Organ donor specimens: What can they tell us about type 1 diabetes? Pediatr. Diabetes 2015, 16, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, S.; Tschen, S.; Zeng, C.; Guo, T.; Hebrok, M.; Matveyenko, A.; Bhushan, A. DNA methylation directs functional maturation of pancreatic β cells. J. Clin. Investig. 2015, 125, 2851–2860. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thompson, P.J.; Shah, A.; Apostolopolou, H.; Bhushan, A. BET Proteins Are Required for Transcriptional Activation of the Senescent Islet Cell Secretome in Type 1 Diabetes. Int. J. Mol. Sci. 2019, 20, 4776. https://doi.org/10.3390/ijms20194776
Thompson PJ, Shah A, Apostolopolou H, Bhushan A. BET Proteins Are Required for Transcriptional Activation of the Senescent Islet Cell Secretome in Type 1 Diabetes. International Journal of Molecular Sciences. 2019; 20(19):4776. https://doi.org/10.3390/ijms20194776
Chicago/Turabian StyleThompson, Peter J., Ajit Shah, Hara Apostolopolou, and Anil Bhushan. 2019. "BET Proteins Are Required for Transcriptional Activation of the Senescent Islet Cell Secretome in Type 1 Diabetes" International Journal of Molecular Sciences 20, no. 19: 4776. https://doi.org/10.3390/ijms20194776
APA StyleThompson, P. J., Shah, A., Apostolopolou, H., & Bhushan, A. (2019). BET Proteins Are Required for Transcriptional Activation of the Senescent Islet Cell Secretome in Type 1 Diabetes. International Journal of Molecular Sciences, 20(19), 4776. https://doi.org/10.3390/ijms20194776