Cytosolic Phospholipase A2 Alpha Regulates TLR Signaling and Migration in Metastatic 4T1 Cells

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Metastatic 4T1 Cells Express Higher Levels of cPLA2α than Non-Metastatic 67NR Cells
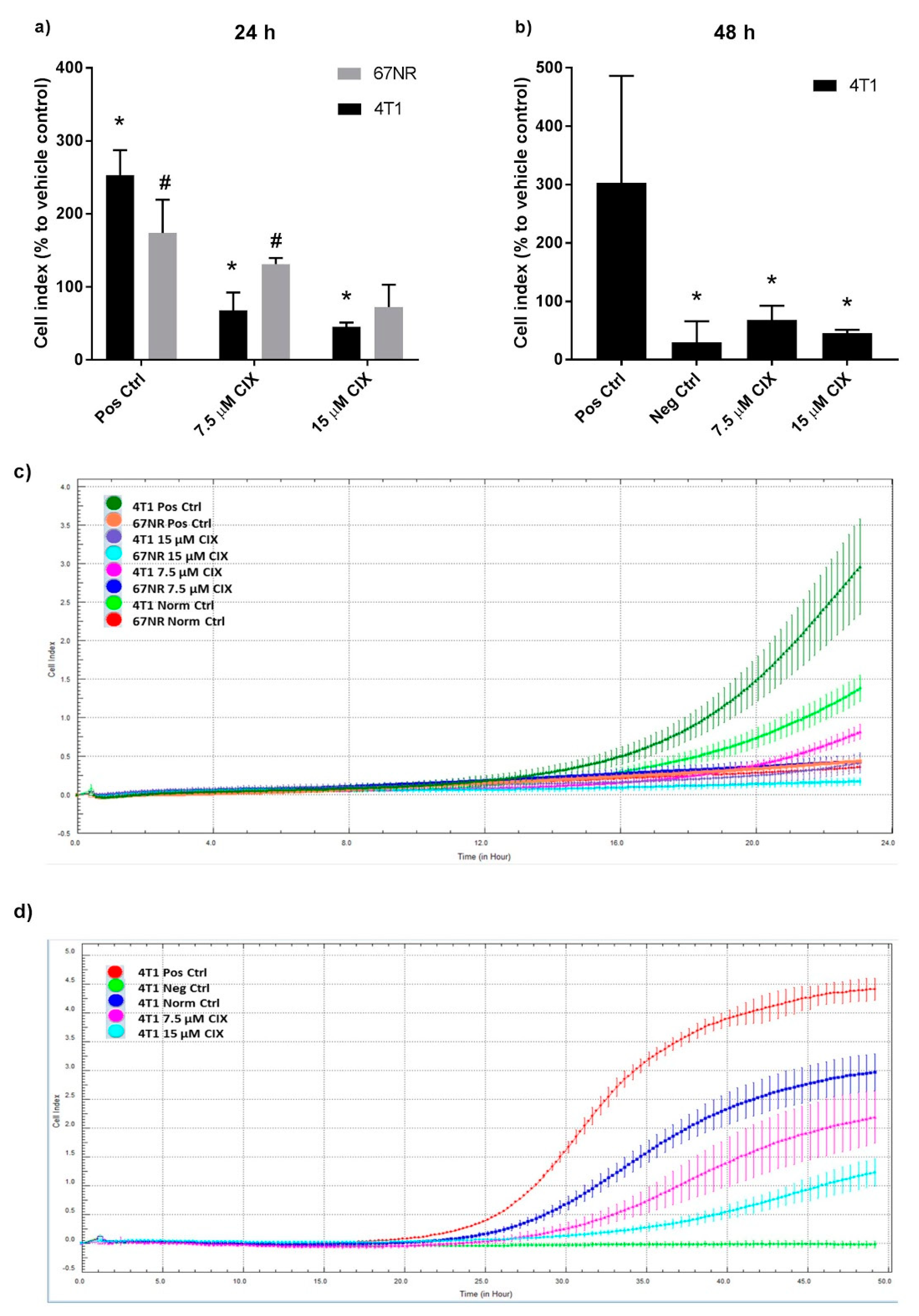
2.2. Non-Metastatic 67NR and Metastatic 4T1 Cells Show Different Sensitivity to CIX Treatment
2.3. Inhibition of cPLA2α Reduces PGE2 Production in Metastatic 4T1 Cells
2.4. cPLA2α Inhibition Impedes Migration in Metastatic 4T1 Cells
2.5. Transcriptomal Effects of cPLA2α Inhibition Include Toll-like Receptors and Type I Interferon Pathways
3. Materials and Methods
3.1. Cell Culture
3.2. XTT Assay
3.3. Proliferation Assay
3.4. Protein Extraction
3.5. Western Blotting
3.6. Enzyme Immunoassay
3.7. Migration Assay
3.8. RNA Sequencing
3.9. Gene Expression Analysis
3.10. RT-qPCR
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid |
| CIX | cPLA2α inhibitor X |
| COX2 | Cyclooxygenase-2 |
| DMEM | Dulbecco’s modified eagle medium |
| DMSO | Dimethyl sulfoxide |
| EMT | Epithelial-to-mesenchymal transition |
| FBS | Fetal bovine serum |
| GO | Gene ontology |
| IFN | Interferon |
| NF-κB | Nuclear factor kappa B |
| PGE2 | Prostaglandin E2 |
| TLR | Toll-like receptor |
Appendix A
Experimental Details for Western Blotting
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term ID | Genes |
|---|---|
| GO:0032648 | PTPRS;DDX3X;DDX58;TIRAP;IFIH1;RNF135;MAVS;IRF3;POLR3B;POLR3C;POLR3D;FLOT1;TLR9;POLR3F;IRF7;REL;TLR4;TLR3 |
| GO:0032481 | CRCP;DDX3X;DDX41;PQBP1;IFIH1;RNF135;IRAK1;FLOT1;POLR2E;STAT6;POLR2H;XRCC5;DDX58;MAVS;IRF3;POLR3B;POLR3C;POLR3D;POLR3E;TLR9;POLR3F;IRF7;TLR4;TLR3;MYD88 |
| GO:0032728 | DDX3X;DDX58;IFIH1;RNF135;MAVS;IRF3;POLR3B;POLR3C;POLR3D;FLOT1;TLR9;POLR3F;IRF7;TLR4;TLR3 |
| GO:0032479 | NLRX1;CRCP;UBA7;TRAF3IP1;GPATCH3;DDX41;PQBP1;IFIH1;RNF135;IRAK1;POLR2E;TRIM25;STAT6;POLR2H;RPS27A;TRIM21;XRCC5;DDX58;TREX1;ISG15;TIRAP;MAVS;IRF3;POLR3B;POLR3C;POLR3D;POLR3E;POLR3F;IRF7;MYD88;TRIM56 |
| GO:0000398 | DBR1;TXNL4B;DDX46;HNRNPU;WDR83;CWC25;YBX1;DDX41;PQBP1;CCAR1;CLP1;RSRC1;MAGOH;DHX16;SNRNP35;SRRM2;RBM17;PRMT7;BUD31;METTL3;PRPF40B;PLRG1;CDC40;WDR77;SRRM1;SFPQ;HNRNPUL1;FRG1;GEMIN5;NAA38;SNRPA1;HNRNPH2;PABPC1;SRSF6;SNRPB;RBM22;SRSF9;ZCCHC8;DDX1;PRCC;SRRT;DDX23;SRSF1;CSTF2T;RNPC3;HTATSF1;PRPF8;RBM15B;PUF60;PCBP1;TRA2A;ZMAT5;POLR2E;POLR2G;POLR2H;HNRNPA1;DCPS;SRSF10;HNRNPA3;SF3A1;CPSF7;PRPF38A;CDC5L;LSM2;HNRNPM;LSM6;RBM41 |
| GO:0006397 | DBR1;DDX46;HNRNPU;WDR83;CWC25;YBX1;DDX41;PQBP1;CCAR1;CLP1;RSRC1;MAGOH;DHX16;SNRNP35;CMTR2;SRRM2;RBM17;BUD31;METTL3;THOC1;PRPF40B;PLRG1;CDC40;SRRM1;SFPQ;PRPF18;HNRNPUL1;FRG1;GEMIN5;NAA38;SNRPA1;HNRNPH2;PABPC1;SRSF6;SNRPB;RBM22;SRSF9;ZCCHC8;PRCC;SRRT;DDX23;SRSF1;CSTF2T;RNPC3;HTATSF1;PRPF8;RBM15B;PUF60;PCBP1;TRA2A;ZMAT5;POLR2E;POLR2G;POLR2H;HNRNPA1;SRSF10;HNRNPA3;SF3A1;CPSF7;CHTOP;PRPF38A;CPSF6;CDC5L;GTF2H1;LSM2;HNRNPM;CDK7;LSM6;RBM41;AGGF1;MNAT1 |
| GO:0000377 | DBR1;DDX46;HNRNPU;WDR83;CWC25;YBX1;DDX41;PQBP1;CCAR1;CLP1;RSRC1;MAGOH;DHX16;SNRNP35;SRRM2;RBM17;BUD31;METTL3;PRPF40B;PLRG1;CDC40;SRRM1;SFPQ;HNRNPUL1;FRG1;GEMIN5;NAA38;SNRPA1;HNRNPH2;PABPC1;SRSF6;SNRPB;RBM22;SRSF9;ZCCHC8;PRCC;SRRT;DDX23;SRSF1;CSTF2T;RNPC3;HTATSF1;PRPF8;RBM15B;PUF60;PCBP1;TRA2A;ZMAT5;POLR2E;POLR2G;POLR2H;HNRNPA1;SRSF10;HNRNPA3;SF3A1;CPSF7;PRPF38A;CDC5L;LSM2;HNRNPM;LSM6;RBM41 |
| GO:0000086 | DYNC1I2;CDKN1A;DCTN2;DCTN1;SSNA1;CEP164;FOXM1;CCNB2;PPME1;CNTRL;RPS27A;LCMT1;CEP290;CEP76;PLK3;BORA;ODF2;CEP152;MBD1;PLK1;FBXL15;HAUS5;MASTL;DYNLL1;CDC25C;CDC25B;DNM2;HAUS1;CCNA2;CDK7;CENPJ;CDK2;MAPRE1;MNAT1;SFI1;TAF2;SDCCAG8 |
| GO:0044839 | DYNC1I2;CDKN1A;DCTN2;DCTN1;SSNA1;CEP164;FOXM1;CCNB2;PPME1;CNTRL;RPS27A;LCMT1;CEP290;CEP76;PLK3;BORA;ODF2;CEP152;MBD1;PLK1;FBXL15;HAUS5;MASTL;DYNLL1;CDC25C;CDC25B;DNM2;HAUS1;CCNA2;CDK7;CENPJ;CDK2;MAPRE1;MNAT1;SFI1;TAF2;SDCCAG8 |
| GO:0060337 | IFNAR2;IFITM3;SP100;RSAD2;STAT2;MX2;ADAR;ISG15;TYK2;IFI35;SAMHD1;IFIT1;IFIT2;ISG20;BST2;IRF3;IRAK1;IFI27;IRF7;GBP2;XAF1;MYD88;IRF9 |
| GO:0071357 | IFNAR2;IFITM3;SP100;RSAD2;STAT2;MX2;ADAR;ISG15;TYK2;IFI35;SAMHD1;IFIT1;IFIT2;ISG20;BST2;IRF3;IRAK1;IFI27;IRF7;GBP2;XAF1;MYD88;IRF9 |
| GO:0000070 | SEH1L;PHF23;VPS4A;PINX1;CDCA8;SMC4;NSL1;NCAPH;SMC2;CHMP1B;RRS1;DLGAP5;DIS3L2;NAA50;SPAG5;BOD1;PLK1;TUBG2;KNSTRN;PSRC1;KIF18B;KIFC1;CHMP2A;KIF2C;NCAPD2;CHMP7;CHMP5 |
| GO:0018205 | HDAC4;TOP2B;POM121;NUP205;PCNA;SEH1L;CDCA8;LOXL4;PLOD2;PLOD1;AAAS;AURKB;IFIH1;SENP6;MTA1;SUMO1;SENP8;NFATC2IP;NUP43;NDC1;PIAS3;ESCO1;SIRT4;MITF;RANGAP1;TOPORS;SIRT2;INCENP;CTH;NDUFAB1;NUP35;BIRC5;VIPAS39 |
| GO:0051310 | SEH1L;BOD1;VPS4A;PINX1;CDCA8;GEM;CENPF;PSRC1;KIFC1;CHMP1B;RRS1;CHMP2A;KIF2C;FAM83D;CHMP7;CENPQ;CHMP5 |
| GO:0010506 | GSK3A;CISD2;ZDHHC8;CAPNS1;MLST8;WDR6;PIP4K2C;DNM1L;ATP6V1E1;TRIM21;TRIM65;SREBF1;RUBCN;EXOC8;CSNK2A2;SIRT2;MID2;RRAGA;KAT2A;CPTP;ATP6V1B2;ULK3;ULK2;SMCR8;ATP6V1A;GBA;HTRA2;VPS26A;RASIP1;MAPK8;DHRSX;LARS;ATG101;ATP6V1H;SH3BP4;BMF;SLC38A9;ATP6V0E2;ATP6V1D;SNX5;ATP6V1C1;PLK2;TPCN1;CDK5;BCL2;LAMTOR2;LAMTOR4;LAMTOR3;CDK5R1;LAMTOR5 |
| GO:0044772 | CDKN1A;CEP164;FOXM1;PPME1;MYC;CNTRL;CEP290;BORA;HAUS5;MASTL;DYNLL1;CDC25C;CDC25B;DNM2;HAUS1;CCNA2;CCNE2;MAPRE1;SFI1;CHMP7;CACUL1;ANAPC1;SDCCAG8;DYNC1I2;DCTN2;DCTN1;SSNA1;PRIM1;CUL2;ORC5;CCNB2;RPS27A;LCMT1;CEP76;CDT1;PLK3;ODF2;UBE2C;CEP152;PLK2;MBD1;PLK1;FBXL15;CDK7;POLA2;CDK6;CENPJ;POLE3;CDK2;TACC3;CHMP2A;MNAT1;TAF2 |
| GO:0002181 | RPL4;RPL32;RPLP1;RPL22;RPL7;RPS26;RPL18A;RPS29;RPL27A;MRTO4;RPS3;RPL36;RPLP2;RPL24;RPL22L1;RPL15;RPL29;EIF4B;RPL19 |

References
- Ervik, M.; Lam, F.; Ferlay, J.; Mery, L.; Soerjomataram, I.; Bray, F. Cancer Today. Available online: http://gco.iarc.fr/today/fact-sheets-cancers?cancer=15&type=0&sex=2 (accessed on 31 July 2017).
- Colzani, E.; Liljegren, A.; Johansson, A.L.V.; Adolfsson, J.; Hellborg, H.; Hall, P.F.L.; Czene, K. Prognosis of patients with breast cancer: causes of death and effects of time since diagnosis, age, and tumor characteristics. J. Clin. Oncol. 2011, 29, 4014–4021. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Do, H.T.T.; Her, J.; Kim, Y.; Seo, D.; Rhee, I. Inflammasome as a promising therapeutic target for cancer. Life Sci. 2019, 231, 116593. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, B.P. Inflammation: A driving force speeds cancer metastasis. Cell Cycle 2009, 8, 3267–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 2009, 50, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, R.M.; Sharp, J.D. Structure, function and regulation of Ca2+-sensitive cytosolic phospholipase A2 (cPLA2). FEBS Lett. 1997, 410, 49–53. [Google Scholar] [CrossRef]
- Anthonsen, M.W.; Andersen, S.; Solhaug, A.; Johansen, B. Atypical lambda/iota PKC conveys 5-lipoxygenase/leukotriene B4-mediated cross-talk between phospholipase A2s regulating NF-kappa B activation in response to tumor necrosis factor-alpha and interleukin-1beta. J. Biol. Chem. 2001, 276, 35344–35351. [Google Scholar] [CrossRef] [PubMed]
- Laye, J.P.; Gill, J.H. Phospholipase A2 expression in tumours: a target for therapeutic intervention? Drug Discov. Today 2003, 8, 710–716. [Google Scholar] [CrossRef]
- Niknami, M.; Patel, M.; Witting, P.K.; Dong, Q. Molecules in focus: Cytosolic phospholipase A2-α. Int. J. Biochem. Cell Biol. 2009, 41, 994–997. [Google Scholar] [CrossRef]
- Linkous, A.G.; Yazlovitskaya, E.M.; Hallahan, D.E. Cytosolic phospholipase A2 and lysophospholipids in tumor angiogenesis. J. Natl. Cancer Inst. 2010, 102, 1398–1412. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Rosenberg, D.W. Roles of cPLA2alpha and arachidonic acid in cancer. Biochim. Biophys. Acta 2006, 1761, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Park, J.B.; Lee, C.S.; Jang, J.-H.; Ghim, J.; Kim, Y.-J.; You, S.; Hwang, D.; Suh, P.-G.; Ryu, S.H. Phospholipase signalling networks in cancer. Nat. Rev. Cancer 2012, 12, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.C.; Kim, K.J.; Ju, H.W.; Back, H.K.; Kim, H.K.; Im, S.Y.; Lee, H.K. Cytoplasmic phospholipase A2 metabolites play a critical role in pulmonary tumor metastasis in mice. Anticancer Res. 2010, 30, 3421–3427. [Google Scholar] [PubMed]
- Fu, H.; He, Y.; Qi, L.; Chen, L.; Luo, Y.; Chen, L.; Li, Y.; Zhang, N.; Guo, H. cPLA2α activates PI3K/AKT and inhibits Smad2/3 during epithelial–mesenchymal transition of hepatocellular carcinoma cells. Cancer Lett. 2017, 403, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Caiazza, F.; Harvey, B.J.; Thomas, W. Cytosolic phospholipase A2 activation correlates with HER2 overexpression and mediates estrogen-dependent breast cancer cell growth. Mol. Endocrinol. 2010, 24, 953–968. [Google Scholar] [CrossRef]
- Caiazza, F.; McCarthy, N.S.; Young, L.; Hill, A.D.K.; Harvey, B.J.; Thomas, W. Cytosolic phospholipase A2-α expression in breast cancer is associated with EGFR expression and correlates with an adverse prognosis in luminal tumours. Br. J. Cancer 2011, 104, 338–344. [Google Scholar] [CrossRef]
- Grinde, M.T.; Skrbo, N.; Moestue, S.A.; Rødland, E.A.; Borgan, E.; Kristian, A.; Sitter, B.; Bathen, T.F.; Børresen-Dale, A.-L.; Mælandsmo, G.M.; et al. Interplay of choline metabolites and genes in patient-derived breast cancer xenografts. Breast Cancer Res. 2014, 16, R5. [Google Scholar] [CrossRef]
- Wen, Z.-H.; Su, Y.-C.; Lai, P.-L.; Zhang, Y.; Xu, Y.-F.; Zhao, A.; Yao, G.-Y.; Jia, C.-H.; Lin, J.; Xu, S.; et al. Critical role of arachidonic acid-activated mTOR signaling in breast carcinogenesis and angiogenesis. Oncogene 2013, 32, 160–170. [Google Scholar] [CrossRef]
- Kim, E.; Tunset, H.M.; Cebulla, J.; Vettukattil, R.; Helgesen, H.; Feuerherm, A.J.; Engebråten, O.; Mælandsmo, G.M.; Johansen, B.; Moestue, S.A. Anti-vascular effects of the cytosolic phospholipase A2 inhibitor AVX235 in a patient-derived basal-like breast cancer model. BMC Cancer 2016, 16, 191. [Google Scholar] [CrossRef]
- You, J.; Mi, D.; Zhou, X.; Qiao, L.; Zhang, H.; Zhang, X.; Ye, L. Extracellularly Regulated Kinase and Cyclooxygenase / Lipoxygenase Maintains Proliferation and Migration of Breast Cancer Cells. Cancer Oncog. 2009, 150, 1607–1617. [Google Scholar]
- Chen, L.; Fu, H.; Luo, Y.; Chen, L.; Cheng, R.; Zhang, N.; Guo, H. cPLA2α mediates TGF-β-induced epithelial–mesenchymal transition in breast cancer through PI3k/Akt signaling. Cell Death Dis. 2017, 8, e2728. [Google Scholar] [CrossRef]
- Subbaramaiah, K.; Dannenberg, A.J. Cyclooxygenase 2: A molecular target for cancer prevention and treatment. Trends Pharmacol. Sci. 2003, 24, 96–102. [Google Scholar] [CrossRef]
- Vendramini-Costa, D.B.; Carvalho, J.E. Molecular Link Mechanisms between Inflammation and Cancer. Curr. Pharm. Des. 2012, 3831–3852. [Google Scholar] [CrossRef]
- Timoshenko, A.V.; Xu, G.; Chakrabarti, S.; Lala, P.K.; Chakraborty, C. Role of prostaglandin E2 receptors in migration of murine and human breast cancer cells. Exp. Cell Res. 2003, 289, 265–274. [Google Scholar] [CrossRef]
- Kokotou, M.G.; Limnios, D.; Nikolaou, A.; Psarra, A.; Kokotos, G. Inhibitors of phospholipase A2 and their therapeutic potential: an update on patents (2012-2016). Expert Opin. Ther. Pat. 2017, 27, 217–225. [Google Scholar] [CrossRef]
- Anthonsen, M.W.; Solhaug, A.; Johansen, B. Functional coupling between secretory and cytosolic phospholipase A2 modulates tumor necrosis factor-alpha- and interleukin-1beta-induced NF-kappa B activation. J. Biol. Chem. 2001, 276, 30527–30536. [Google Scholar] [CrossRef]
- Omland, S.H.; Habicht, A.; Damsbo, P.; Wilms, J.; Johansen, B.; Gniadecki, R. A randomized, double-blind, placebo-controlled, dose-escalation first-in-man study (phase 0) to assess the safety and efficacy of topical cytosolic phospholipase A2 inhibitor, AVX001, in patients with mild to moderate plaque psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1161–1167. [Google Scholar] [CrossRef]
- Sommerfelt, R.M.; Feuerherm, A.J.; Skuland, T.; Johansen, B. Cytosolic phospholipase A2 modulates TLR2 signaling in synoviocytes. PLoS ONE 2015, 10, e0119088. [Google Scholar] [CrossRef]
- Feuerherm, A.J.; Dennis, E.A.; Johansen, B. Cytosolic group IVA phospholipase A2 inhibitors, AVX001 and AVX002, ameliorate collagen-induced arthritis. Arthritis Res. Ther. 2019, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Huwiler, A.; Feuerherm, A.J.; Sakem, B.; Pastukhov, O.; Filipenko, I.; Nguyen, T.; Johansen, B. The ω3-polyunsaturated fatty acid derivatives AVX001 and AVX002 directly inhibit cytosolic phospholipase A(2) and suppress PGE(2) formation in mesangial cells. Br. J. Pharmacol. 2012, 167, 1691–1701. [Google Scholar] [CrossRef]
- Aslakson, C.J.; Miller, F.R. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992, 52. [Google Scholar]
- Kramer, R.M.; Roberts, E.F.; Manetta, J.V.; Hyslop, P.A.; Jakubowski, J.A. Thrombin-Induced Phosphorylation and Activation of Ca2+- Sensitive Cytosolic Phospholipase-A(2) in Human Platelets. J. Biol. Chem. 1993, 268, 26796–26804. [Google Scholar]
- Simões, R.V.; Serganova, I.S.; Kruchevsky, N.; Leftin, A.; Shestov, A.A.; Thaler, H.T.; Sukenick, G.; Locasale, J.W.; Blasberg, R.G.; Koutcher, J.A.; et al. Metabolic plasticity of metastatic breast cancer cells: adaptation to changes in the microenvironment. Neoplasia 2015, 17, 671–684. [Google Scholar] [CrossRef]
- Li, Z.; Qu, M.; Sun, Y.; Wan, H.; Chai, F.; Liu, L.; Zhang, P. Blockage of cytosolic phospholipase A2 alpha sensitizes aggressive breast cancer to doxorubicin through suppressing ERK and mTOR kinases. Biochem. Biophys. Res. Commun. 2018, 496, 153–158. [Google Scholar] [CrossRef]
- Balsinde, J.; Winstead, M.V.; Dennis, E.A. Phospholipase A 2 regulation of arachidonic acid mobilization. FEBS Lett. 2002, 531, 2–6. [Google Scholar] [CrossRef]
- Eckhardt, B.L.; Parker, B.S.; van Laar, R.K.; Restall, C.M.; Natoli, A.L.; Tavaria, M.D.; Stanley, K.L.; Sloan, E.K.; Moseley, J.M.; Anderson, R.L. Genomic Analysis of a Spontaneous Model of Breast Cancer Metastasis to Bone Reveals a Role for the Extracellular Matrix. Mol. Cancer Res. 2005, 3, 1–13. [Google Scholar]
- Varone, A.; Mariggiò, S.; Patheja, M.; Maione, V.; Varriale, A.; Vessichelli, M.; Spano, D.; Formiggini, F.; Lo Monte, M.; Brancati, N.; et al. A signalling cascade involving receptor-activated phospholipase A2, glycerophosphoinositol 4-phosphate, Shp1 and Src in the activation of cell motility. Cell Commun. Signal. 2019, 17, 20. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-S.; Sengupta, S.; Berk, M.; Kwak, Y.-G.; Escobar, P.F.; Benlinson, J.; Mok, S.C.; Xu, Y. Hypoxia Enhances Lysophosphatidic Acid Responsiveness in Ovarian Cancer Cells and Lysophosphatidic Acid Induces Ovarian Tumor Metastasis In vivo. Cancer Res. 2006, 66, 7983–7990. [Google Scholar] [CrossRef]
- Lou, Y.; Preobrazhenska, O.; Auf Dem Keller, U.; Sutcliffe, M.; Barclay, L.; McDonald, P.C.; Roskelley, C.; Overall, C.M.; Dedhar, S. Epithelial-Mesenchymal Transition (EMT) is not sufficient for spontaneous murine breast cancer metastasis. Dev. Dyn. 2008, 237, 2755–2768. [Google Scholar] [CrossRef]
- Villegas-Comonfort, S.; Castillo-Sanchez, R.; Serna-Marquez, N.; Cortes-Reynosa, P.; Salazar, E.P. Arachidonic acid promotes migration and invasion through a PI3K/Akt-dependent pathway in MDA-MB-231 breast cancer cells. Prostaglandins Leukot. Essent. Fat. Acids 2014, 90, 169–177. [Google Scholar] [CrossRef]
- Lee, M.S.; Kim, Y.-J. Signaling Pathways Downstream of Pattern-Recognition Receptors and Their Cross Talk. Annu. Rev. Biochem. 2007, 76, 447–480. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, N. Toll-like receptor mediated regulation of breast cancer: a case of mixed blessings. Front. Immunol. 2014, 5, 224. [Google Scholar] [PubMed]
- Li, J.; Yin, J.; Shen, W.; Gao, R.; Liu, Y.; Chen, Y.; Li, X.; Liu, C.; Xiang, R.; Luo, N. TLR4 Promotes Breast Cancer Metastasis via Akt/GSK3β/β-Catenin Pathway upon LPS Stimulation. Anat. Rec. 2017, 300, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Zandi, Z.; Kashani, B.; Poursani, E.M.; Bashash, D.; Kabuli, M.; Momeny, M.; Mousavi-pak, S.H.; Sheikhsaran, F.; Alimoghaddam, K.; Mousavi, S.A.; et al. TLR4 blockade using TAK-242 suppresses ovarian and breast cancer cells invasion through the inhibition of extracellular matrix degradation and epithelial-mesenchymal transition. Eur. J. Pharmacol. 2019, 853, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.; Fiegl, H.; Goebel, G.; Obexer, P.; Ausserlechner, M.; Doppler, W.; Hauser-Kronberger, C.; Reitsamer, R.; Egle, D.; Reimer, D.; et al. Toll-like receptor 9 expression in breast and ovarian cancer is associated with poorly differentiated tumors. Cancer Sci. 2010, 101, 1059–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Zhang, H.; Fu, Y.; Zhu, Y.; Kong, L.; Chen, L.; Zhao, F.; Yu, L.; Chen, X. TLR4/MyD88 signaling determines the metastatic potential of breast cancer cells. Mol. Med. Rep. 2018, 18, 3411–3420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Huang, J.; Gong, W.; Iribarren, P.; Dunlop, N.M.; Wang, J.M. Toll-like receptors in inflammation, infection and cancer. Int. Immunopharmacol. 2007, 7, 1271–1285. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Jiang, Q.; Zhang, Y.; Liang, H.; Ren, H.; Zhang, D. Toll-like receptors and breast cancer. Integr. Cancer Sci. Ther. 2016, 3, 432–436. [Google Scholar] [CrossRef] [Green Version]
- Snell, L.M.; McGaha, T.L.; Brooks, D.G. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017, 38, 542–557. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Ueno, N.T.; Finetti, P.; Vermeulen, P.; Lucci, A.; Robertson, F.M.; Marsan, M.; Iwamoto, T.; Krishnamurthy, S.; Masuda, H.; et al. Gene expression profiles of inflammatory breast cancer: correlation with response to neoadjuvant chemotherapy and metastasis-free survival. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2014, 25, 358–365. [Google Scholar] [CrossRef]
- Buess, M.; Nuyten, D.S.A.; Hastie, T.; Nielsen, T.; Pesich, R.; Brown, P.O. Characterization of heterotypic interaction effects in vitro to deconvolute global gene expression profiles in cancer. Genome Biol. 2007, 8, R191. [Google Scholar] [CrossRef] [PubMed]
- Ogony, J.; Choi, H.J.; Lui, A.; Cristofanilli, M.; Lewis-Wambi, J. Interferon-induced transmembrane protein 1 (IFITM1) overexpression enhances the aggressive phenotype of SUM149 inflammatory breast cancer cells in a signal transducer and activator of transcription 2 (STAT2)-dependent manner. Breast Cancer Res. 2016, 18, 25. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.; Zhang, Q.; Wang, F.; Zhang, D. Toll-like receptors and prostate cancer. Front. Immunol. 2014, 5, 352. [Google Scholar] [CrossRef] [PubMed]
- Scudiero, D.A.; Shoemaker, R.H.; Paull, K.D.; Monks, A.; Tierney, S.; Nofziger, T.H.; Currens, M.J.; Seniff, D.; Boyd, M.R. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988, 48, 4827–4833. [Google Scholar] [PubMed]
- GraphPad Software GraphPad for Windows 2018. Available online: www.graphpad.com (accessed on 1 May 2018).
- MyAssays Ltd Prostaglandin E2 Monoclonal from Cayman Chemical. Available online: https://www.myassays.com/prostaglandin-e2-monoclonal.assay (accessed on 15 February 2016).
- Barbraham Bioinformatics FastQC. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 May 2018).
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patro, R.; Duggal, G.; Love, M.; Irizarry, R.; Kingsford, C. Salmon. Available online: http://salmon.readthedocs.io/en/latest/salmon.html (accessed on 1 June 2018).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.; Soneson, C.; Robinson, M.; Patro, R.; Morgan, A.P.; Thompson, R.C.; Shirley, M. Import and summarize transcript-level estimates for transcript- and gene-level analysis. Available online: http://bioconductor.org/packages/release/bioc/html/tximport.html (accessed on 3 March 2018).
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research 2016, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.; Hu, Y.; Ritchie, M.; Silver, J.; Wettenhall, J.; McCarthy, D.; Wu, D.; Shi, W.; Phipson, B.; Lun, A.; et al. Linear Models for Microarray Data. Available online: https://bioconductor.org/packages/release/bioc/html/limma.html (accessed on 10 June 2018).
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 2013, 14, 128. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, 90–97. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]




| Term ID | Description | Overlap | False Rate Discovery |
|---|---|---|---|
| GO:0032648 | regulation of interferon-beta production | 18/35 | 0.000859 |
| GO:0032481 | positive regulation of type I interferon production | 25/63 | 0.000859 |
| GO:0032728 | positive regulation of interferon-beta production | 15/27 | 0.000859 |
| GO:0032479 | regulation of type I interferon production | 31/86 | 0.000859 |
| GO:0000398 | mRNA splicing, via spliceosome | 67/262 | 0.000993 |
| GO:0006397 | mRNA processing | 71/284 | 0.000993 |
| GO:0000377 | RNA splicing, via transesterification reactions with bulged adenosine as nucleophile | 62/237 | 0.000993 |
| GO:0000086 | G2/M transition of mitotic cell cycle | 37/127 | 0.007793 |
| GO:0044839 | cell cycle G2/M phase transition | 37/128 | 0.008405 |
| GO:0060337 | type I interferon signaling pathway | 23/66 | 0.009802 |
| GO:0071357 | cellular response to type I interferon | 23/66 | 0.009802 |
| GO:0000070 | mitotic sister chromatid segregation | 27/83 | 0.009802 |
| GO:0018205 | peptidyl-lysine modification | 33/116 | 0.022896 |
| GO:0051310 | metaphase plate congression | 17/45 | 0.027307 |
| GO:0010506 | regulation of autophagy | 50/204 | 0.027307 |
| GO:0044772 | mitotic cell cycle phase transition | 53/222 | 0.03186 |
| GO:0002181 | cytoplasmic translation | 19/55 | 0.038663 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tunset, H.M.; Feuerherm, A.J.; Selvik, L.-K.M.; Johansen, B.; Moestue, S.A. Cytosolic Phospholipase A2 Alpha Regulates TLR Signaling and Migration in Metastatic 4T1 Cells. Int. J. Mol. Sci. 2019, 20, 4800. https://doi.org/10.3390/ijms20194800
Tunset HM, Feuerherm AJ, Selvik L-KM, Johansen B, Moestue SA. Cytosolic Phospholipase A2 Alpha Regulates TLR Signaling and Migration in Metastatic 4T1 Cells. International Journal of Molecular Sciences. 2019; 20(19):4800. https://doi.org/10.3390/ijms20194800
Chicago/Turabian StyleTunset, Hanna Maja, Astrid Jullumstrø Feuerherm, Linn-Karina Myrland Selvik, Berit Johansen, and Siver Andreas Moestue. 2019. "Cytosolic Phospholipase A2 Alpha Regulates TLR Signaling and Migration in Metastatic 4T1 Cells" International Journal of Molecular Sciences 20, no. 19: 4800. https://doi.org/10.3390/ijms20194800
APA StyleTunset, H. M., Feuerherm, A. J., Selvik, L. -K. M., Johansen, B., & Moestue, S. A. (2019). Cytosolic Phospholipase A2 Alpha Regulates TLR Signaling and Migration in Metastatic 4T1 Cells. International Journal of Molecular Sciences, 20(19), 4800. https://doi.org/10.3390/ijms20194800