Human Cysteine Cathepsins Degrade Immunoglobulin G In Vitro in a Predictable Manner

 , , , ,
, , , ,
Abstract
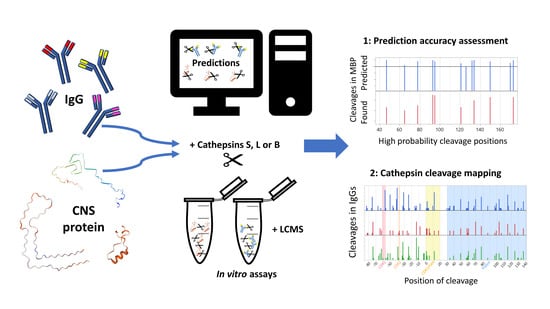
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Prediction Platform Validation: In Silico Evaluations
2.2. Prediction Platform Validation: In Vitro Findings Compared to In Silico Predictions on CNS Proteins
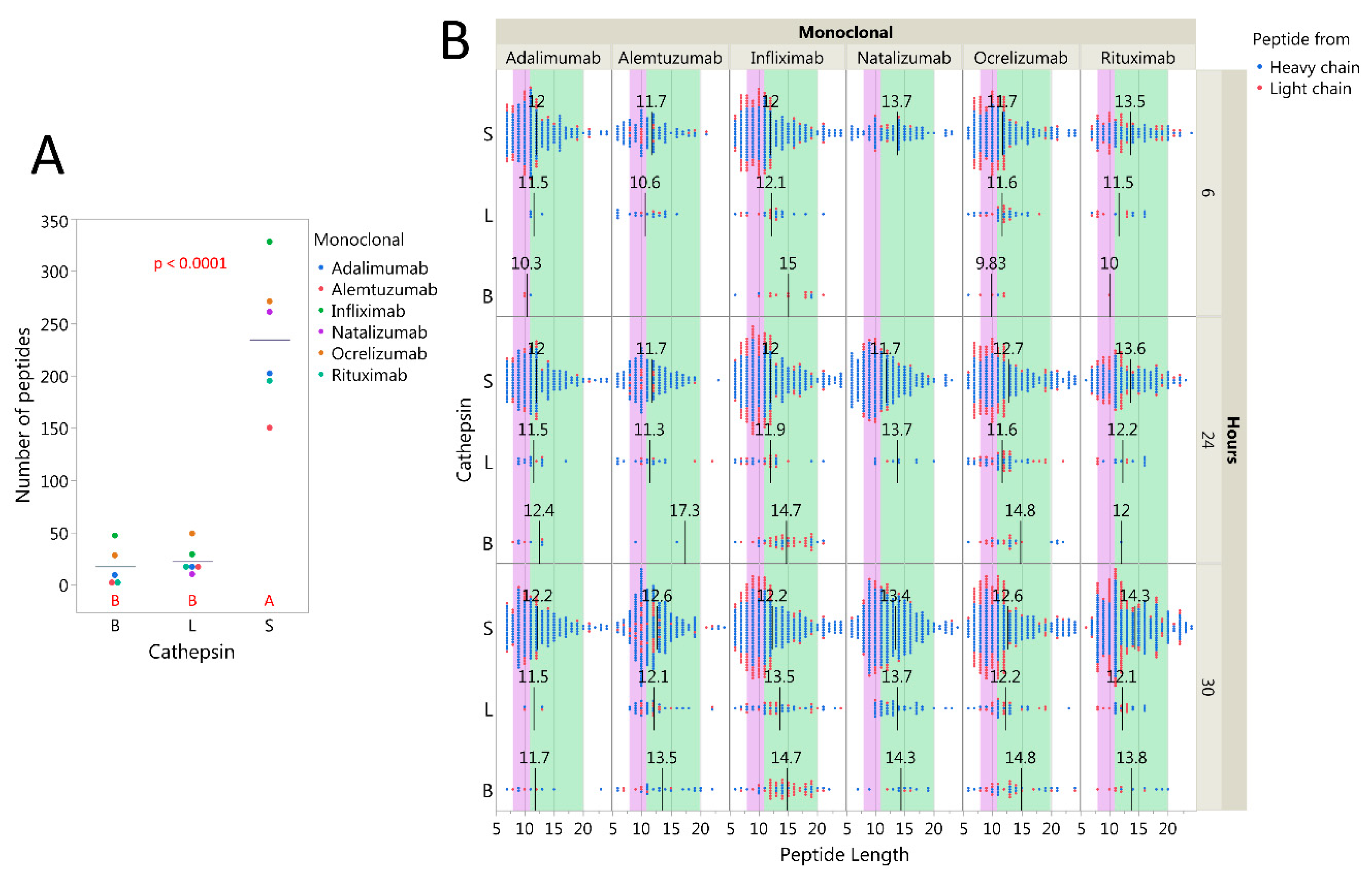
2.3. Cysteine Cathepsins Degrade Immunoglobulins In Vitro
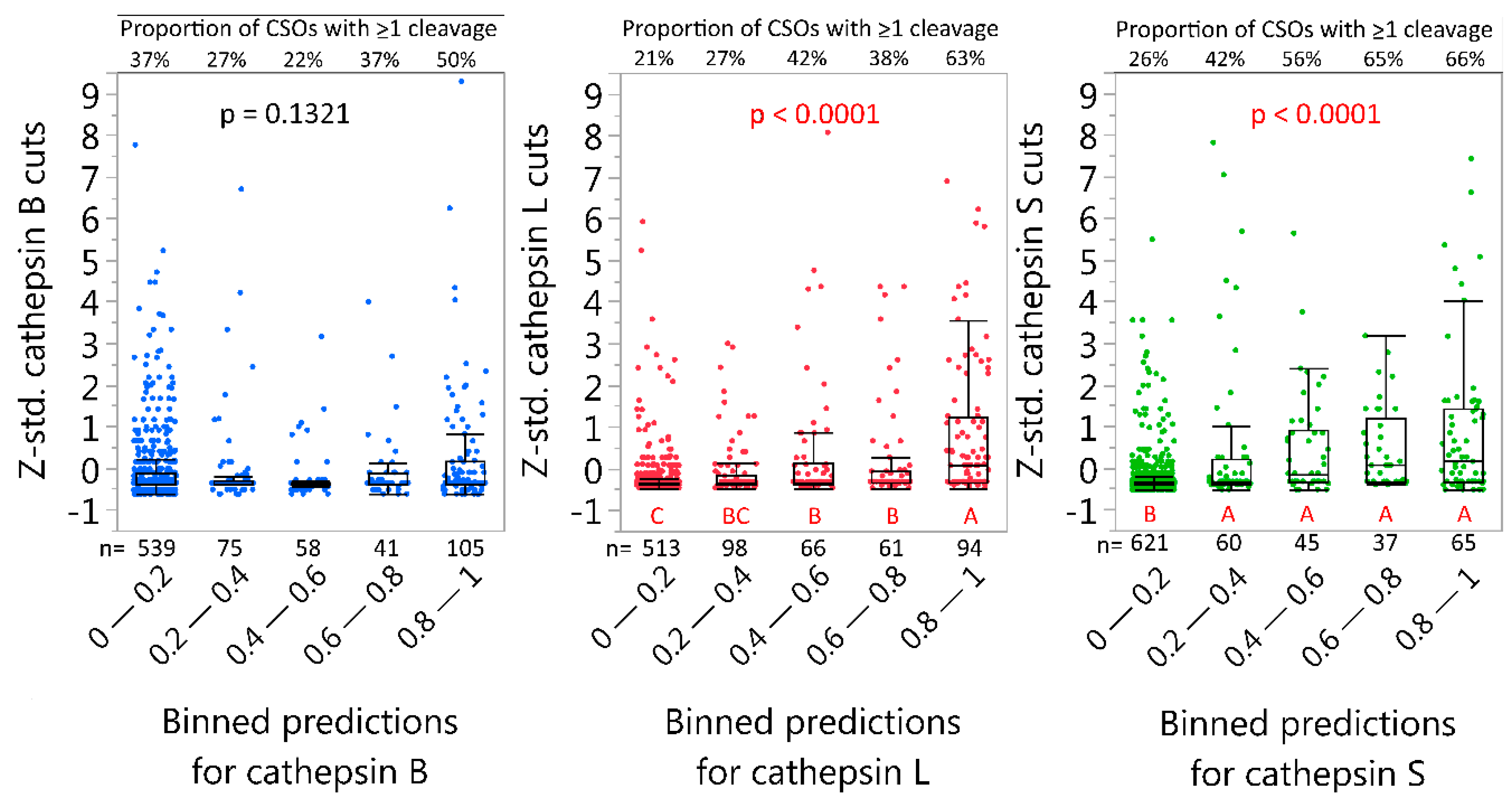
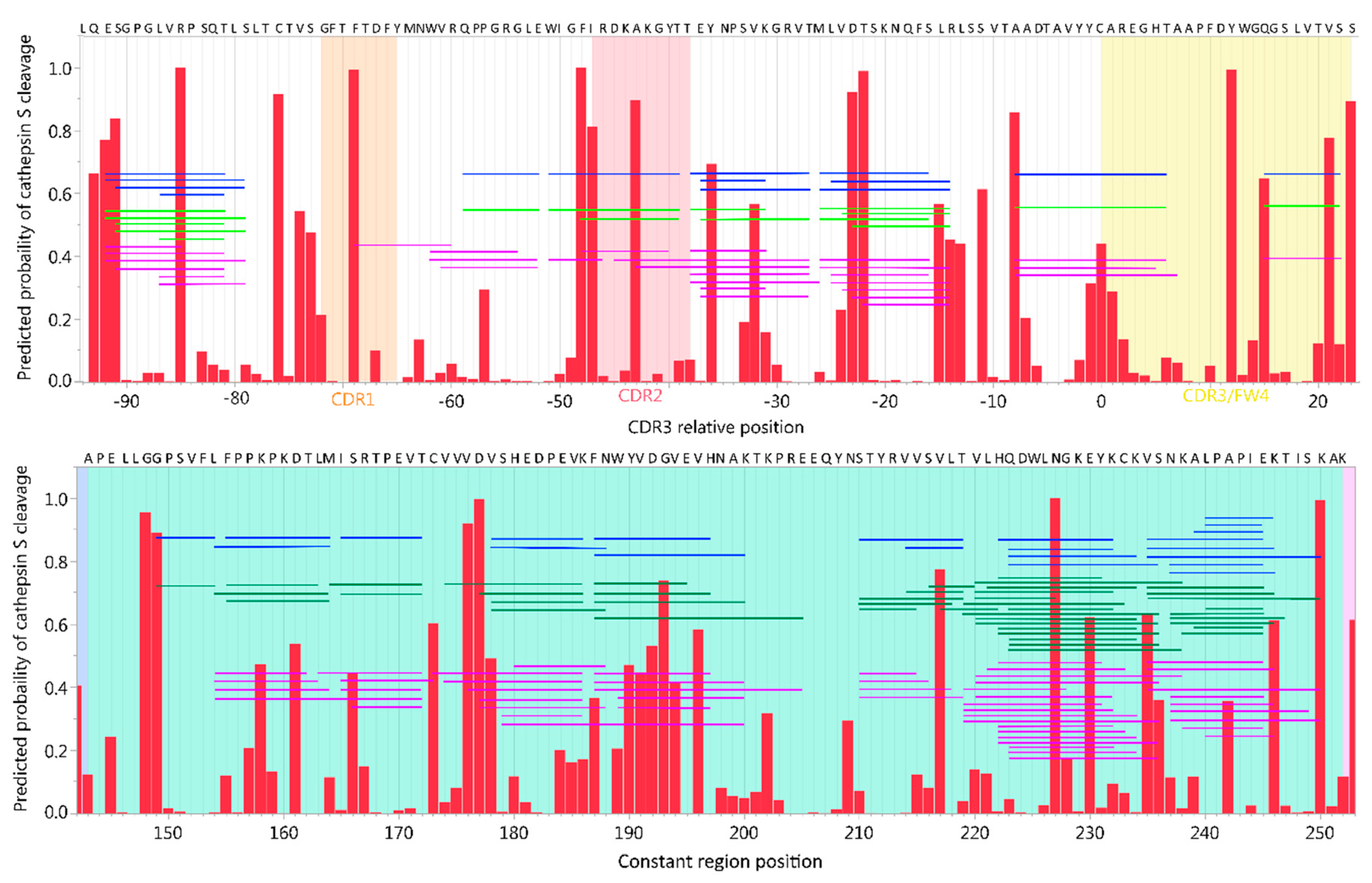
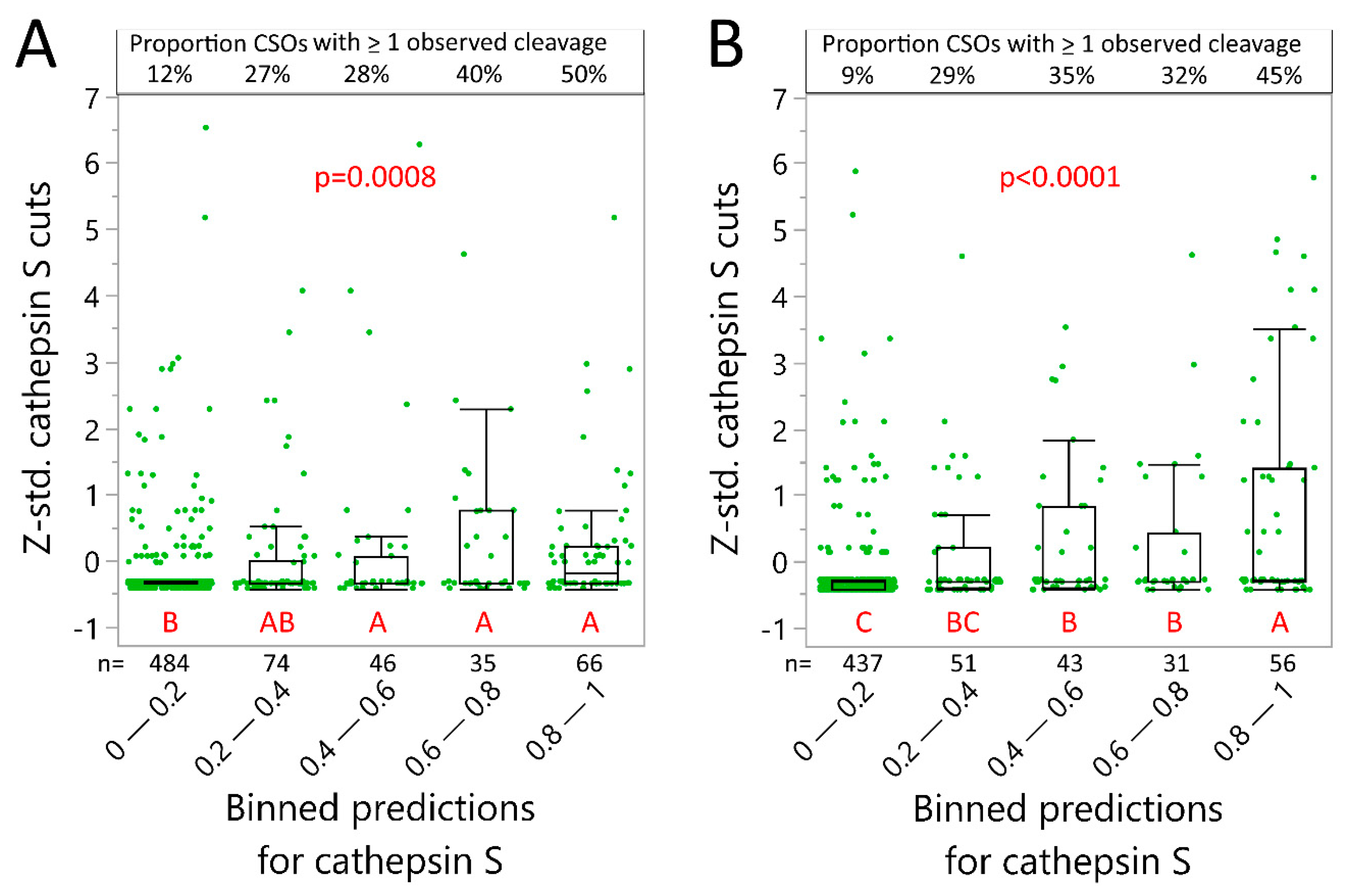
2.4. Neural Net Prediction Accuracy for Immunoglobulin Cathepsin Cleavage
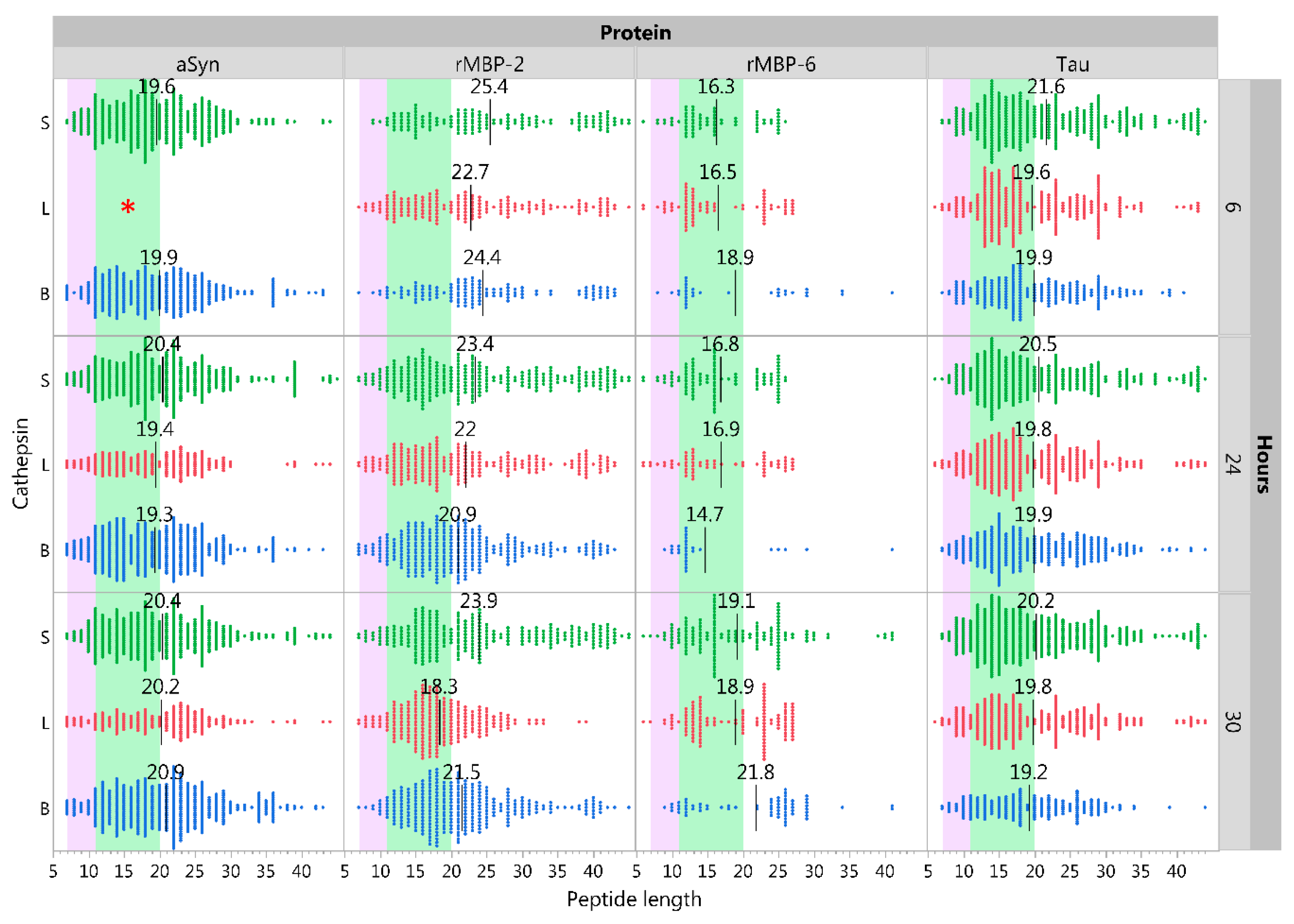
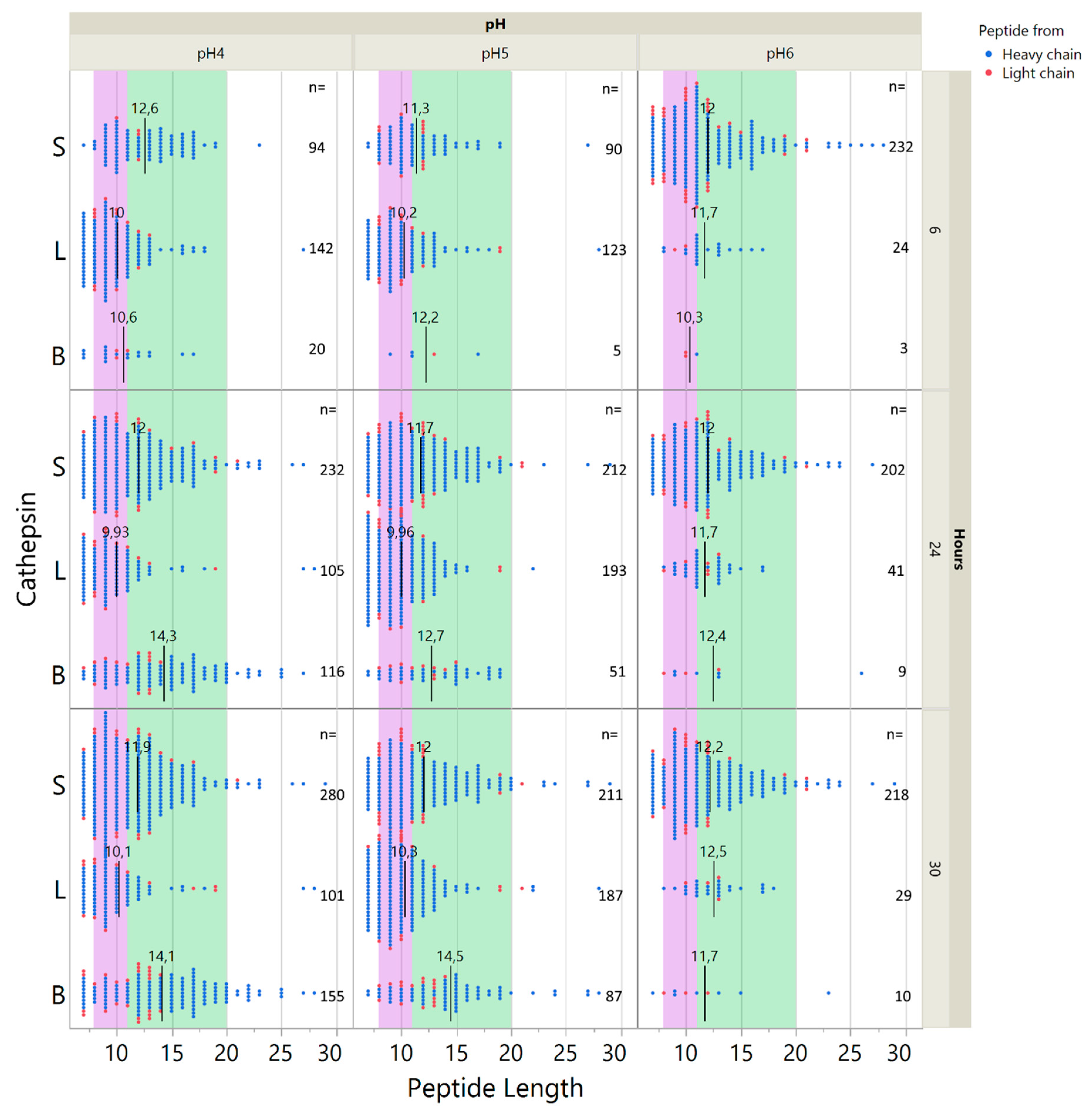
2.5. Influence of pH on Cathepsin Activity
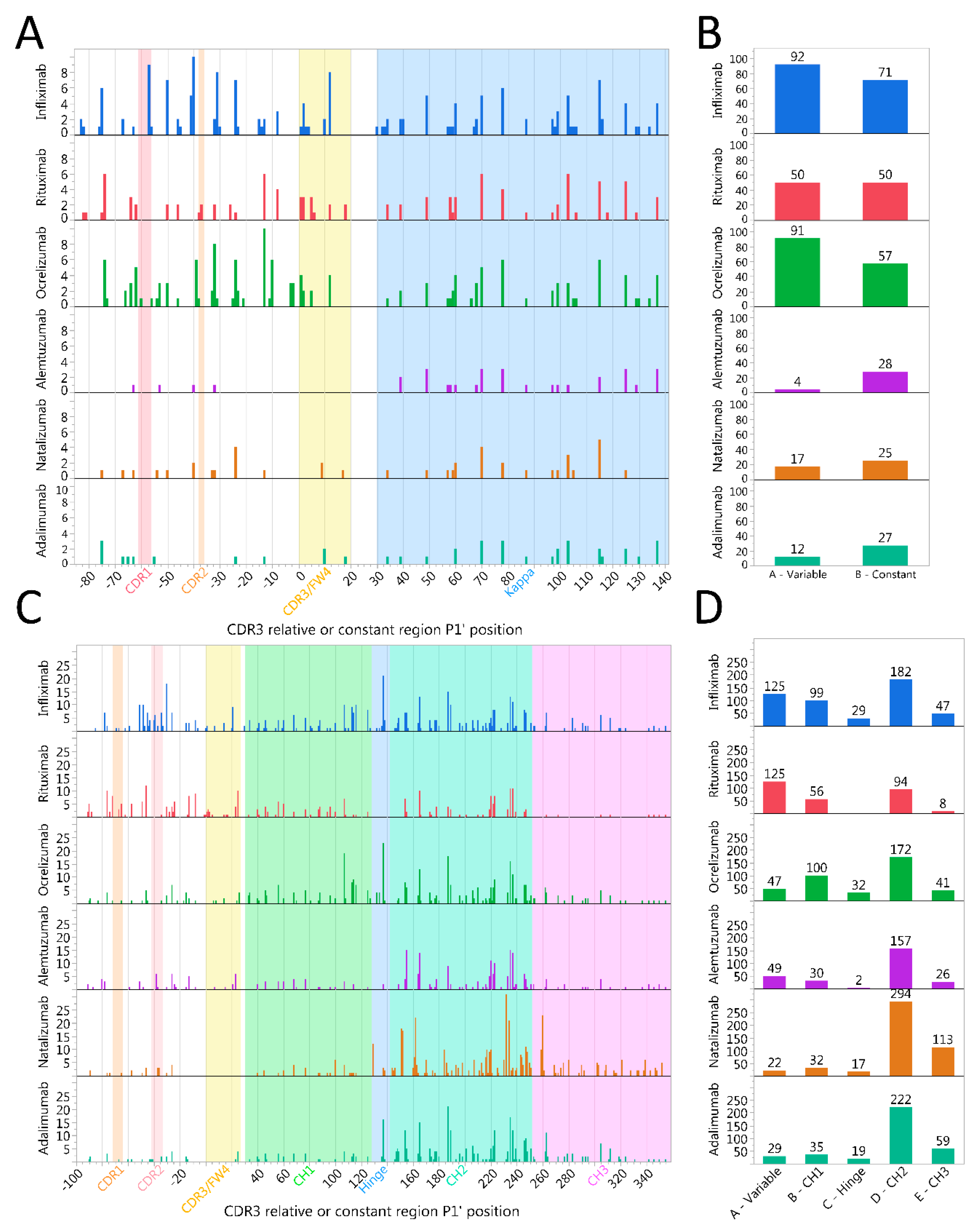
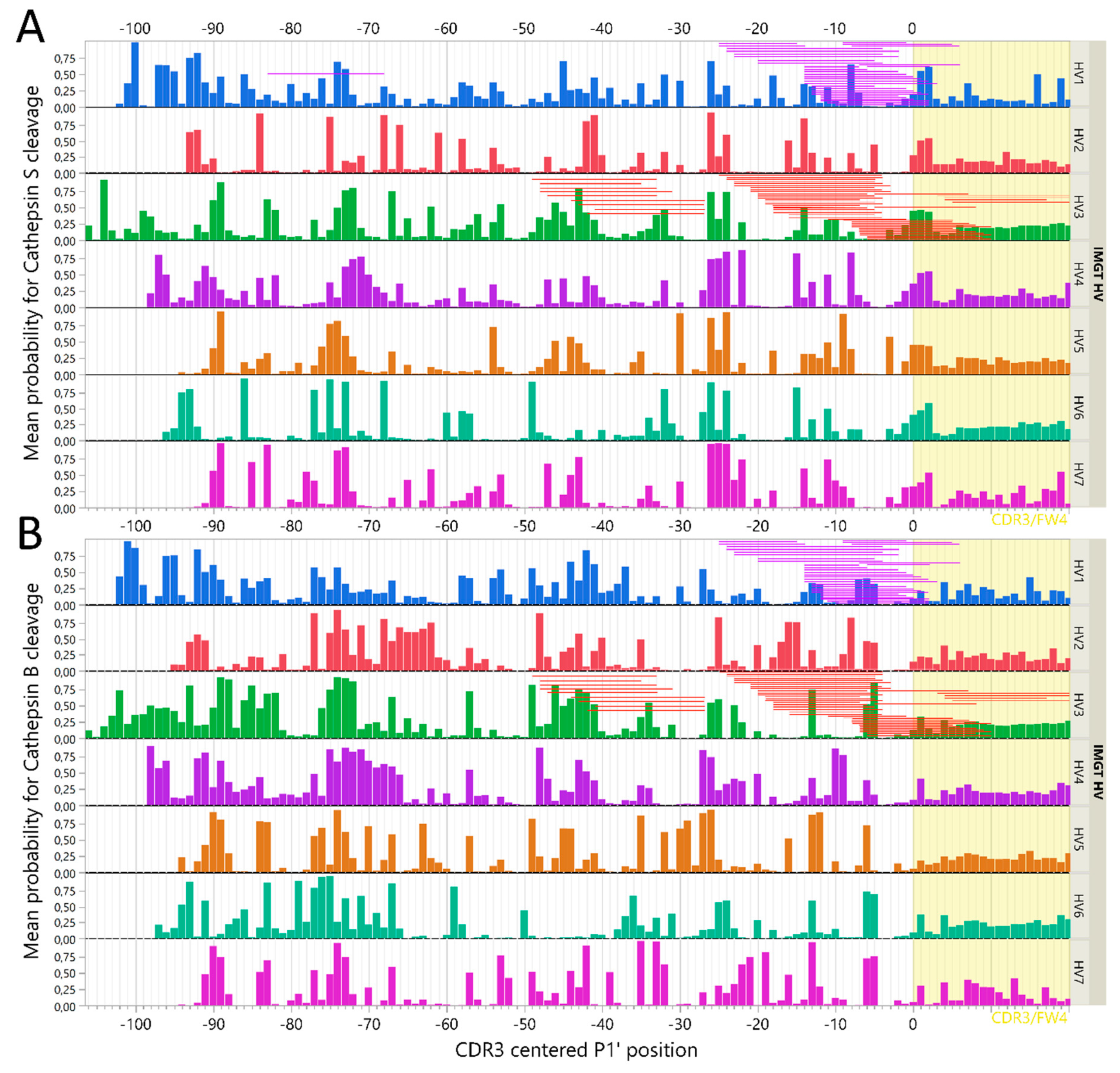
2.6. Immunoglobulin Heavy Variable Gene Family Determines Different Cleavage Patterns
3. Discussion
4. Methods
4.1. Cathepsin Cleavage Predictions
4.2. Cathepsins and Substrates of the In Vitro Cleavage Assays
4.3. Nano Liquid Chromatography Mass Spectrometry and Related Software for Data Processing
4.4. SDS-PAGE
4.5. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | antigen presenting cell |
| BCR | B cell receptor |
| CDR3 | complementarity determining region 3 |
| CNS | Central nervous system |
| CSO | cleavage site octamer |
| HLA | human leukocyte antigen |
| Ig | immunoglobulin |
| IgG | immunoglobulin G |
| IGHV | immunoglobulin heavy variable |
| mAb | monoclonal antibody |
| MHC | major histocompatibility complex |
| NN | neural network |
| nLCMS | nano liquid chromatography mass spectrometry |
| rMBP | recombinant myelin basic protein |
| SDS-PAGE | sodium dodecyl sulphate-polyacrylamide gel electrophoresis |
| SVM | support vector machine |
References
- Adler, L.N.; Jiang, W.; Bhamidipati, K.; Millican, M.; Macaubas, C.; Hung, S.C.; Mellins, E.D. The Other Function: Class II-Restricted Antigen Presentation by B Cells. Front. Immunol. 2017, 8, 319. [Google Scholar] [CrossRef] [Green Version]
- Hsing, L.C.; Rudensky, A.Y. The lysosomal cysteine proteases in MHC class II antigen presentation. Immunol. Rev. 2005, 207, 229–241. [Google Scholar] [CrossRef] [PubMed]
- van Kasteren, S.I.; Overkleeft, H.S. Endo-lysosomal proteases in antigen presentation. Curr. Opin. Chem. Biol. 2014, 23, 8–15. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Zoghi, S.; Liebig, T.M.; Wennhold, K.; Chemitz, J.; Draube, A.; Kochanek, M.; Blaschke, F.; Pallasch, C.; Holtick, U.; et al. Inhibition of protein geranylgeranylation specifically interferes with CD40-dependent B cell activation, resulting in a reduced capacity to induce T cell immunity. J. Immunol. 2014, 193, 5294–5305. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L. The Charcot Lecture | beating MS: A story of B cells, with twists and turns. Mult. Scler. 2015, 21, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Mathias, A.; Perriard, G.; Canales, M.; Soneson, C.; Delorenzi, M.; Schluep, M.; Du Pasquier, R.A. Increased ex vivo antigen presentation profile of B cells in multiple sclerosis. Mult. Scler. J. 2016, 23, 802–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoydahl, L.S.; Richter, L.; Frick, R.; Snir, O.; Gunnarsen, K.S.; Landsverk, O.J.B.; Iversen, R.; Jeliazkov, J.R.; Gray, J.J.; Bergseng, E.; et al. Plasma Cells Are the Most Abundant Gluten Peptide MHC-expressing Cells in Inflamed Intestinal Tissues From Patients With Celiac Disease. Gastroenterology 2019, 156, 1428–1439.e1410. [Google Scholar] [CrossRef] [PubMed]
- Honey, K.; Rudensky, A.Y. Lysosomal cysteine proteases regulate antigen presentation. Nat. Rev. Immunol. 2003, 3, 472–482. [Google Scholar] [CrossRef]
- Avalos, A.; Ploegh, H. Early BCR Events and Antigen Capture, Processing, and Loading on MHC Class II on B Cells. Front. Immunol. 2014, 5, 777–780. [Google Scholar] [CrossRef]
- Weiss, S.; Bogen, B. B-lymphoma cells process and present their endogenous immunoglobulin to major histocompatibility complex-restricted T cells. Proc. Natl. Acad. Sci. USA 1989, 86, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 2016, 32, 22–37. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Wootla, B.; Denic, A.; Rodriguez, M. Polyclonal and Monoclonal Antibodies in Clinic. In Human Monoclonal Antibodies: Methods and Protocols; Steinitz, M., Ed.; Humana Press: Totowa, NJ, USA, 2014; pp. 79–110. [Google Scholar]
- Grilo, A.L.; Mantalaris, A. The Increasingly Human and Profitable Monoclonal Antibody Market. Trends Biotechnol. 2019, 37, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, H. Human monoclonal antibodies: The residual challenge of antibody immunogenicity. Methods Mol. Biol. 2014, 1060, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, R.; Lefranc, M.-P. Human immunoglobulin allotypes: Possible implications for immunogenicity. mAbs 2009, 1, 332–338. [Google Scholar] [CrossRef] [PubMed]
- De Groot, A.S.; Scott, D.W. Immunogenicity of protein therapeutics. Trends Immunol. 2007, 28, 482–490. [Google Scholar] [CrossRef]
- Andersen, T.K.; Huszthy, P.C.; Gopalakrishnan, R.P.; Jacobsen, J.T.; Fauskanger, M.; Tveita, A.A.; Grødeland, G.; Bogen, B. Enhanced germinal center reaction by targeting vaccine antigen to major histocompatibility complex class II molecules. npj Vaccines 2019, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, J.; Haabeth, O.-A.W.; Tveita, A.A.; Schjetne, K.W.; Munthe, L.A.; Bogen, B. Naive Idiotope-Specific B and T Cells Collaborate Efficiently in the Absence of Dendritic Cells. J. Immunol. 2014, 192, 4174–4183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Groot, A.S.; Martin, W. Reducing risk, improving outcomes: Bioengineering less immunogenic protein therapeutics. Clin. Immunol. 2009, 131, 189–201. [Google Scholar] [CrossRef]
- Su, Y.; Carey, G.; Marić, M.; Scott, D.W. B Cells Induce Tolerance by Presenting Endogenous Peptide-IgG on MHC Class II Molecules via an IFN-γ-Inducible Lysosomal Thiol Reductase-Dependent Pathway. J. Immunol. 2008, 181, 1153–1160. [Google Scholar] [CrossRef]
- Hastings, K.T.; Cresswell, P. Disulfide reduction in the endocytic pathway: Immunological functions of gamma-interferon-inducible lysosomal thiol reductase. Antioxid Redox Signal. 2011, 15, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Santoro, L.; Reboul, A.; Kerblat, I.; Drouet, C.; Colomb, M.G. Monoclonal IgG as antigens: Reduction is an early intracellular event of their processing by antigen-presenting cells. Int. Immunol. 1996, 8, 211–219. [Google Scholar] [CrossRef]
- Fehr, K.; LoSpalluto, J.; Ziff, M. Degradation of immunoglobulin G by lysosomal acid proteases. J. Immunol. 1970, 105, 973–983. [Google Scholar]
- Driessen, C.; Lennon-Dumenil, A.M.; Ploegh, H.L. Individual cathepsins degrade immune complexes internalized by antigen-presenting cells via Fcgamma receptors. Eur. J. Immunol. 2001, 31, 1592–1601. [Google Scholar] [CrossRef]
- Hoglund, R.A.; Lossius, A.; Johansen, J.N.; Homan, J.; Benth, J.S.; Robins, H.; Bogen, B.; Bremel, R.D.; Holmoy, T. In Silico Prediction Analysis of Idiotope-Driven T-B Cell Collaboration in Multiple Sclerosis. Front. Immunol. 2017, 8, 1255. [Google Scholar] [CrossRef] [PubMed]
- Holmoy, T.; Vartdal, F.; Hestvik, A.L.; Munthe, L.; Bogen, B. The idiotype connection: Linking infection and multiple sclerosis. Trends Immunol. 2010, 31, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Orozco, C.; Boyer, J.; Leglise, M.; Goodale, J.; Batalov, S.; Hodge, C.L.; Haase, J.; Janes, J.; Huss, J.W.; et al. BioGPS: An extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009, 10, R130. [Google Scholar] [CrossRef] [PubMed]
- Rock, R.B.; Hu, S.; Deshpande, A.; Munir, S.; May, B.J.; Baker, C.A.; Peterson, P.K.; Kapur, V. Transcriptional response of human microglial cells to interferon-gamma. Genes Immun. 2005, 6, 712–719. [Google Scholar] [CrossRef]
- Bogen, B.; Weiss, S. Processing and presentation of idiotypes to MHC-restricted T cells. Int. Rev. Immunol. 1993, 10, 337–355. [Google Scholar] [CrossRef]
- Weiss, S.; Bogen, B. MHC class II-restricted presentation of intracellular antigen. Cell 1991, 64, 767–776. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Olsson, N.; Chen, B.; Sworder, B.; Shree, T.; Liu, C.L.; Zhang, L.; Czerwinski, D.K.; Davis, M.M.; Levy, R.; et al. B-cell lymphomas present immunoglobulin neoantigens. Blood 2019, 133, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Haabeth, O.A.W.; Chen, B.; Swaminathan, K.; Rawson, K.; Liu, C.L.; Steiner, D.; Lund, P.; et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature 2017, 543, 723–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reth, M. Antigen Receptors on B Lymphocytes. Ann. Rev. Immunol. 1992, 10, 97–121. [Google Scholar] [CrossRef] [PubMed]
- Bremel, R.D.; Homan, E.J. Recognition of higher order patterns in proteins: Immunologic kernels. PLoS ONE 2013, 8, e70115. [Google Scholar] [CrossRef] [PubMed]
- Biniossek, M.L.; Nagler, D.K.; Becker-Pauly, C.; Schilling, O. Proteomic identification of protease cleavage sites characterizes prime and non-prime specificity of cysteine cathepsins B, L, and S. J. Proteome Res. 2011, 10, 5363–5373. [Google Scholar] [CrossRef] [PubMed]
- Mort, J.S. Chapter 406—Cathepsin B. In Handbook of Proteolytic Enzymes (Third Edition); Rawlings, N.D., Salvesen, G., Eds.; Academic Press, Elsevier: London, UK, 2013; pp. 1784–1791. [Google Scholar]
- Han, J.C.; Han, G.Y. A procedure for quantitative determination of tris(2-carboxyethyl)phosphine, an odorless reducing agent more stable and effective than dithiothreitol. Anal. Biochem. 1994, 220, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Kirschke, H. Chapter 413—Cathepsin S. In Handbook of Proteolytic Enzymes (Third Edition); Rawlings, N.D., Salvesen, G., Eds.; Academic Press, Elsevier: London, UK, 2013; pp. 1824–1830. [Google Scholar]
- Kirschke, H. Chapter 410—Cathepsin L. In Handbook of Proteolytic Enzymes (Third Edition); Rawlings, N.D., Salvesen, G., Eds.; Academic Press, Elsevier: London, UK, 2013; pp. 1808–1817. [Google Scholar]
- Yates, R.M.; Hermetter, A.; Taylor, G.A.; Russell, D.G. Macrophage Activation Downregulates the Degradative Capacity of the Phagosome. Traffic 2007, 8, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Bremel, R.D.; Homan, E.J. Frequency Patterns of T-Cell Exposed Amino Acid Motifs in Immunoglobulin Heavy Chain Peptides Presented by MHCs. Front Immunol. 2014, 5, 541. [Google Scholar] [CrossRef]
- Alamyar, E.; Duroux, P.; Lefranc, M.-P.; Giudicelli, V. IMGT® Tools for the Nucleotide Analysis of Immunoglobulin (IG) and T Cell Receptor (TR) V-(D)-J Repertoires, Polymorphisms, and IG Mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. In Immunogenetics: Methods and Applications in Clinical Practice; Christiansen, F.T., Tait, B.D., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 569–604. [Google Scholar]
- Lowry, J.R.; Klegeris, A. Emerging roles of microglial cathepsins in neurodegenerative disease. Brain Res. Bull. 2018, 139, 144–156. [Google Scholar] [CrossRef]
- Beck, H.; Schwarz, G.; Schroter, C.J.; Deeg, M.; Baier, D.; Stevanovic, S.; Weber, E.; Driessen, C.; Kalbacher, H. Cathepsin S and an asparagine-specific endoprotease dominate the proteolytic processing of human myelin basic protein in vitro. Eur. J. Immunol. 2001, 31, 3726–3736. [Google Scholar] [CrossRef]
- Bielekova, B.; Sung, M.-H.; Kadom, N.; Simon, R.; McFarland, H.; Martin, R. Expansion and Functional Relevance of High-Avidity Myelin-Specific CD4+ T Cells in Multiple Sclerosis. J. Immunol. 2004, 172, 3893. [Google Scholar] [CrossRef]
- Tiller, K.E.; Tessier, P.M. Advances in Antibody Design. Annu. Rev. Biomed. Eng. 2015, 17, 191–216. [Google Scholar] [CrossRef] [Green Version]
- Baker, M.P.; Jones, T.D. Identification and removal of immunogenicity in therapeutic proteins. Curr. Opin. Drug Discov. Dev. 2007, 10, 219–227. [Google Scholar]
- Jawa, V.; Cousens, L.P.; Awwad, M.; Wakshull, E.; Kropshofer, H.; De Groot, A.S. T-cell dependent immunogenicity of protein therapeutics: Preclinical assessment and mitigation. Clin. Immunol. 2013, 149, 534–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flobakk, M.; Rasmussen, I.B.; Lunde, E.; Frigstad, T.; Berntzen, G.; Michaelsen, T.E.; Bogen, B.; Sandlie, I. Processing of an Antigenic Sequence from IgG Constant Domains for Presentation by MHC Class II. J. Immunol. 2008, 181, 7062. [Google Scholar] [CrossRef] [PubMed]
- Bogen, B.; Ruffini, P. Review: To what extent are T cells tolerant to immunoglobulin variable regions? Scand J. Immunol. 2009, 70, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Munthe, L.A.; Corthay, A.; Os, A.; Zangani, M.; Bogen, B. Systemic autoimmune disease caused by autoreactive B cells that receive chronic help from Ig V region-specific T cells. J. Immunol. 2005, 175, 2391–2400. [Google Scholar] [CrossRef] [PubMed]
- Munthe, L.A.; Os, A.; Zangani, M.; Bogen, B. MHC-restricted Ig V region-driven T-B lymphocyte collaboration: B cell receptor ligation facilitates switch to IgG production. J. Immunol. 2004, 172, 7476–7484. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Hartman, I.Z.; Poore, B.; Boronina, T.; Cole, R.N.; Song, N.; Ciudad, M.T.; Caspi, R.R.; Jaraquemada, D.; Sadegh-Nasseri, S. Divergent paths for the selection of immunodominant epitopes from distinct antigenic sources. Nat. Commun. 2014, 5, 5369. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- Collado, J.A.; Alvarez, I.; Ciudad, M.T.; Espinosa, G.; Canals, F.; Pujol-Borrell, R.; Carrascal, M.; Abian, J.; Jaraquemada, D. Composition of the HLA-DR-associated human thymus peptidome. Eur. J. Immunol. 2013, 43, 2273–2282. [Google Scholar] [CrossRef]
- Sorde, L.; Spindeldreher, S.; Palmer, E.; Karle, A. Tregitopes and impaired antigen presentation: Drivers of the immunomodulatory effects of IVIg? Immun. Inflamm. Dis. 2017, 5, 400–415. [Google Scholar] [CrossRef]
- Heyder, T.; Kohler, M.; Tarasova, N.K.; Haag, S.; Rutishauser, D.; Rivera, N.V.; Sandin, C.; Mia, S.; Malmström, V.; Wheelock, Å.M.; et al. Approach for Identifying Human Leukocyte Antigen (HLA)-DR Bound Peptides from Scarce Clinical Samples. Mol. Cell Proteom. 2016, 15, 3017–3029. [Google Scholar] [CrossRef] [Green Version]
- Seward, R.J.; Drouin, E.E.; Steere, A.C.; Costello, C.E. Peptides presented by HLA-DR molecules in synovia of patients with rheumatoid arthritis or antibiotic-refractory Lyme arthritis. Mol. Cell Proteom. 2011, 10, M110.002477–M002110.002477. [Google Scholar] [CrossRef]
- Hamze, M.; Meunier, S.; Karle, A.; Gdoura, A.; Goudet, A.; Szely, N.; Pallardy, M.; Carbonnel, F.; Spindeldreher, S.; Mariette, X.; et al. Characterization of CD4 T Cell Epitopes of Infliximab and Rituximab Identified from Healthy Donors. Front. Immunol. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dall, E.; Brandstetter, H. Structure and function of legumain in health and disease. Biochimie 2016, 122, 126–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoury, B.; Hewitt, E.W.; Morrice, N.; Dando, P.M.; Barrett, A.J.; Watts, C. An asparaginyl endopeptidase processes a microbial antigen for class II MHC presentation. Nature 1998, 396, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.B.; Luo, C.; O’Connell, D.J.; Lefkovith, A.; Brown, E.M.; Yassour, M.; Varma, M.; Abelin, J.G.; Conway, K.L.; Jasso, G.J.; et al. Antigen discovery and specification of immunodominance hierarchies for MHCII-restricted epitopes. Nat. Med. 2018, 24, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, D503–D509. [Google Scholar] [CrossRef]
- Bremel, R.D.; Homan, E.J. An integrated approach to epitope analysis II: A system for proteomic-scale prediction of immunological characteristics. Immunome Res. 2010, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Bremel, R.D.; Homan, E.J. An integrated approach to epitope analysis I: Dimensional reduction, visualization and prediction of MHC binding using amino acid principal components and regression approaches. Immunome Res. 2010, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367. [Google Scholar] [CrossRef]
- Consortium, T.U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2018, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.W.C.F.A., (Mountain View, CA, 94043, US), Chan, Andrew C. (1201 Cloud Avenue, Menlo Park, CA, 94025, US), Crowley, Craig W. (151 Durazno Way, Portola Valley, CA, 94028, US), Lowman, Henry B. (400 San Juan Avenue, P.O. Box 2556 El Granada, CA, 94018, US), Nakamura, Gerald R. (1529 Portola Drive, San Francisco, CA, 94127, US), Presta, Leonard G. (1900 Gough Street, #206 San Francisco, CA, 94109, US) IMMUNOGLOBULIN VARIANTS AND USES THEREOF. 2004. Available online: https://worldwide.espacenet.com/publicationDetails/originalDocument?CC=WO&NR=2004056312A2&KC=A2&FT=D&ND=&date=20040708&DB=&locale=# (accessed on 15 November 2018).
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Høglund, R.A.; Torsetnes, S.B.; Lossius, A.; Bogen, B.; Homan, E.J.; Bremel, R.; Holmøy, T. Human Cysteine Cathepsins Degrade Immunoglobulin G In Vitro in a Predictable Manner. Int. J. Mol. Sci. 2019, 20, 4843. https://doi.org/10.3390/ijms20194843
Høglund RA, Torsetnes SB, Lossius A, Bogen B, Homan EJ, Bremel R, Holmøy T. Human Cysteine Cathepsins Degrade Immunoglobulin G In Vitro in a Predictable Manner. International Journal of Molecular Sciences. 2019; 20(19):4843. https://doi.org/10.3390/ijms20194843
Chicago/Turabian StyleHøglund, Rune Alexander, Silje Bøen Torsetnes, Andreas Lossius, Bjarne Bogen, E. Jane Homan, Robert Bremel, and Trygve Holmøy. 2019. "Human Cysteine Cathepsins Degrade Immunoglobulin G In Vitro in a Predictable Manner" International Journal of Molecular Sciences 20, no. 19: 4843. https://doi.org/10.3390/ijms20194843
APA StyleHøglund, R. A., Torsetnes, S. B., Lossius, A., Bogen, B., Homan, E. J., Bremel, R., & Holmøy, T. (2019). Human Cysteine Cathepsins Degrade Immunoglobulin G In Vitro in a Predictable Manner. International Journal of Molecular Sciences, 20(19), 4843. https://doi.org/10.3390/ijms20194843