Methylglyoxal and Glyoxal as Potential Peripheral Markers for MCI Diagnosis and Their Effects on the Expression of Neurotrophic, Inflammatory and Neurodegenerative Factors in Neurons and in Neuronal Derived-Extracellular Vesicles


Abstract
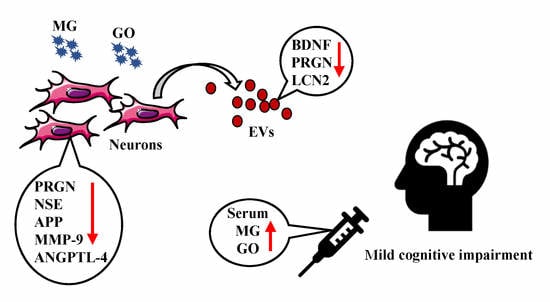
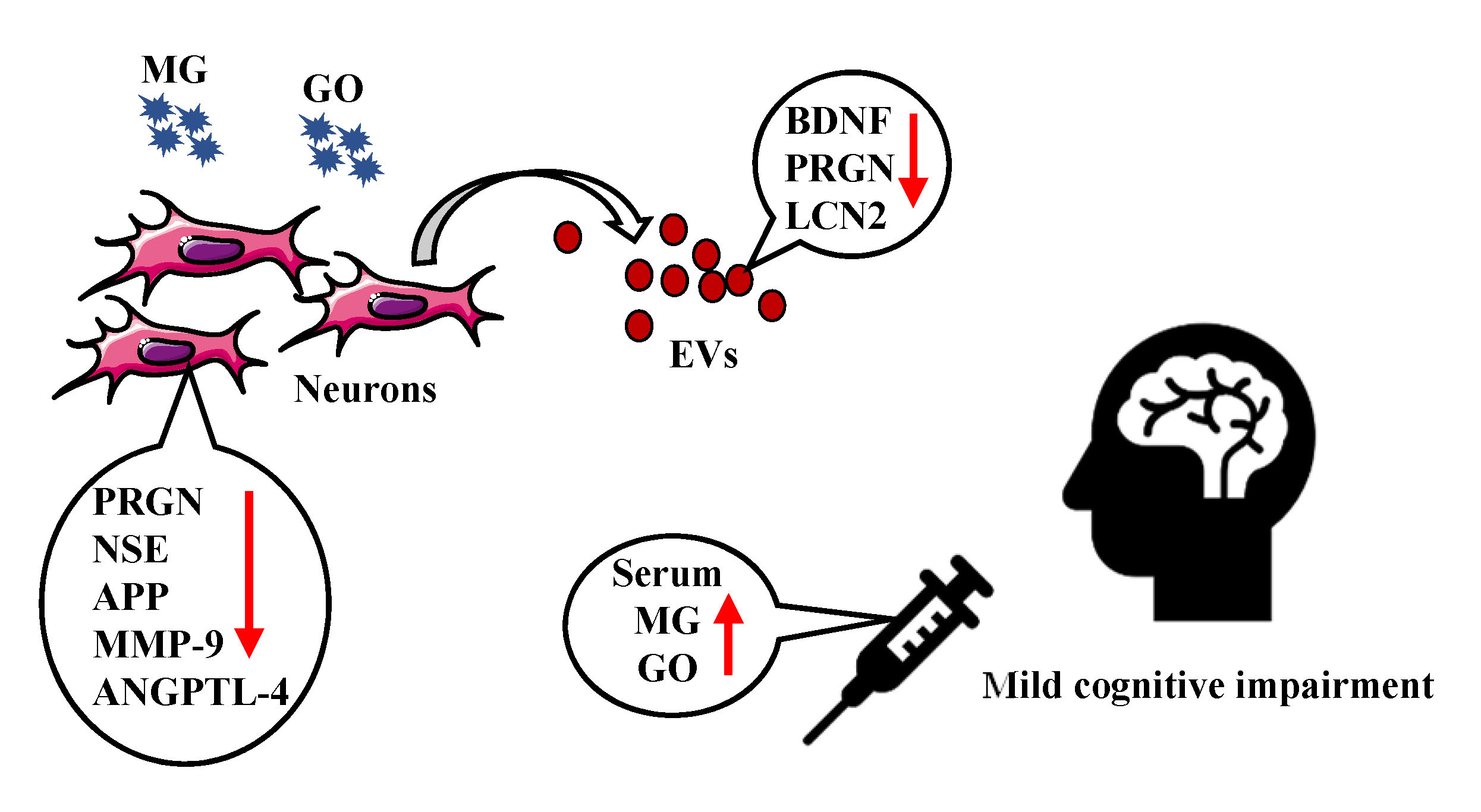
:
1. Introduction
2. Results
2.1. Main Characteristics of Study Population
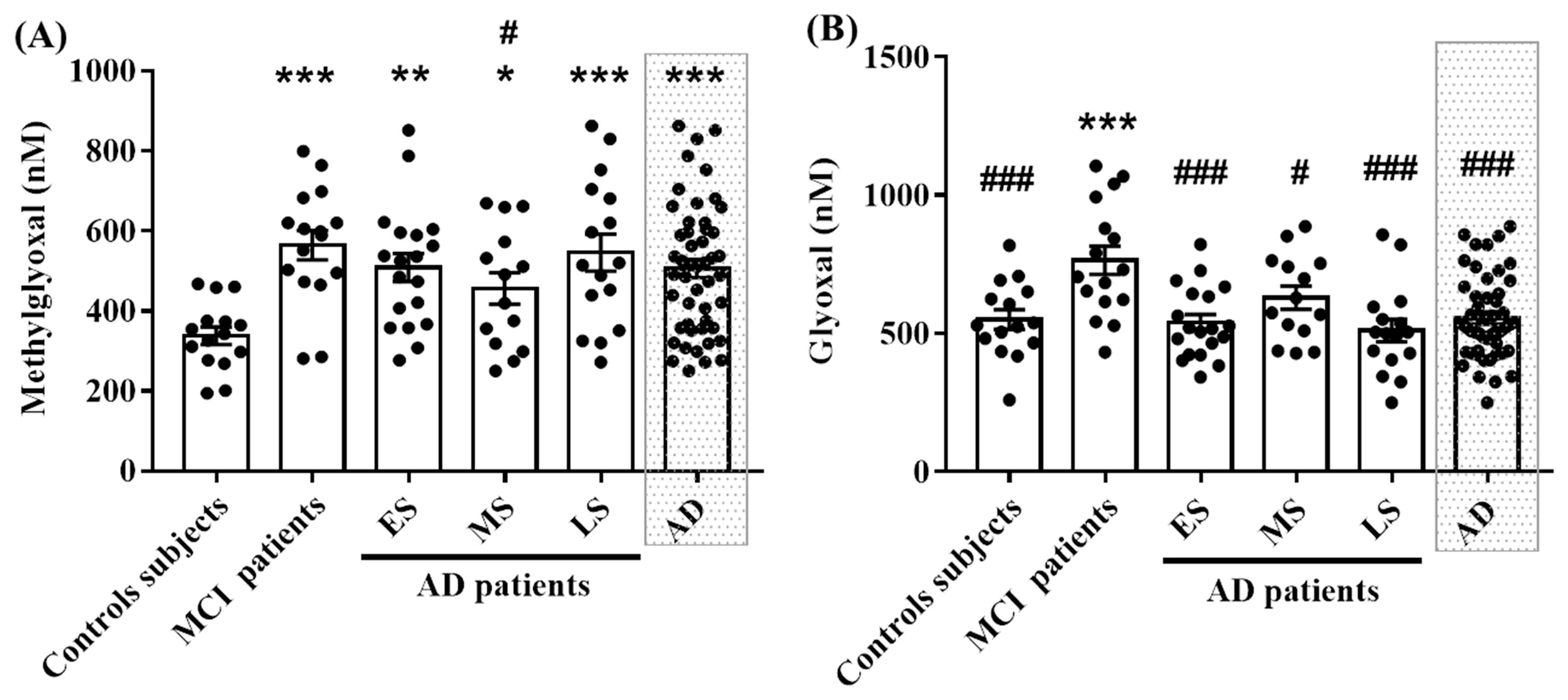
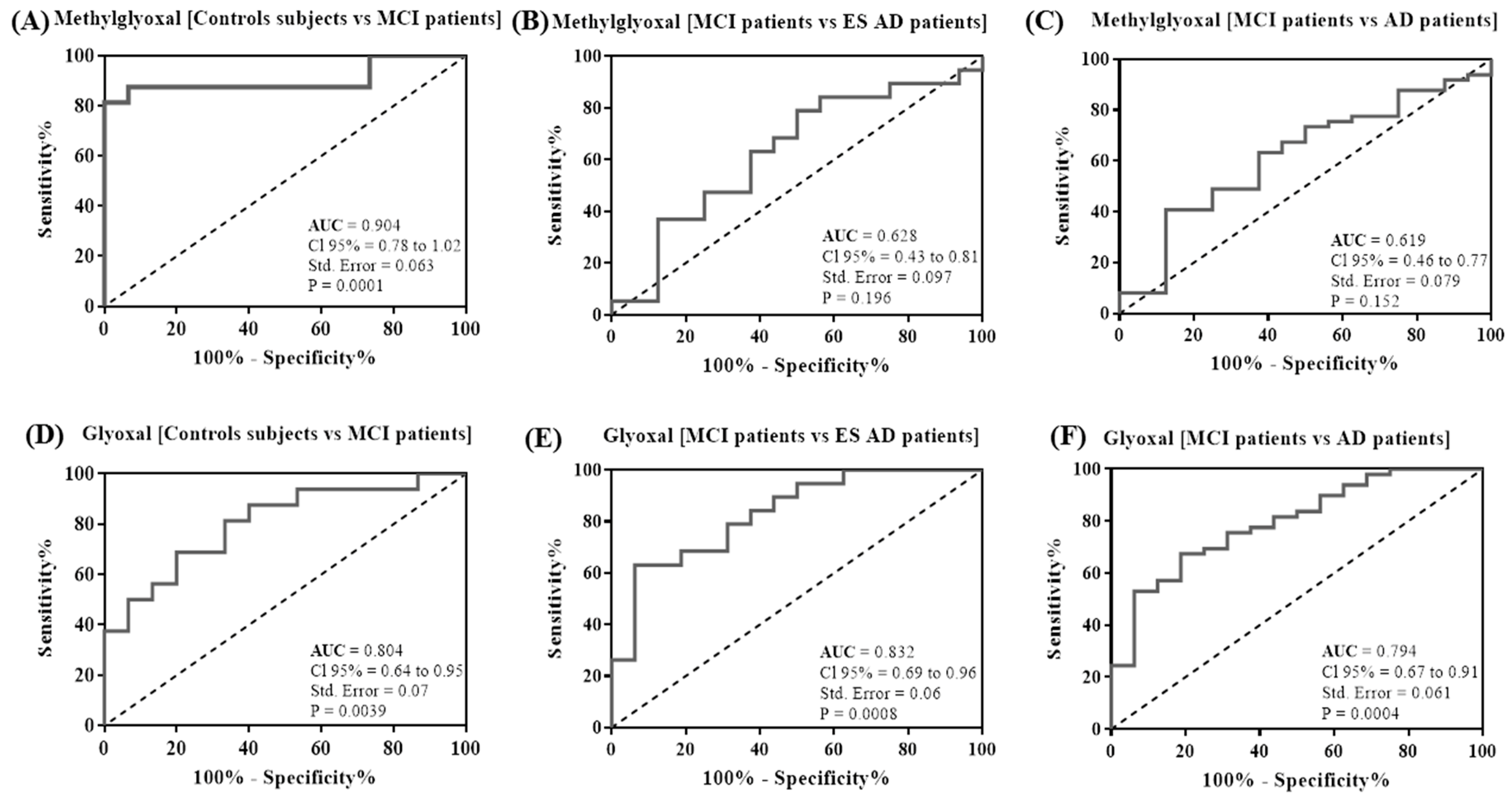
2.2. MG and GO Serum Levels in Control Subjects, in MCI and AD Patients
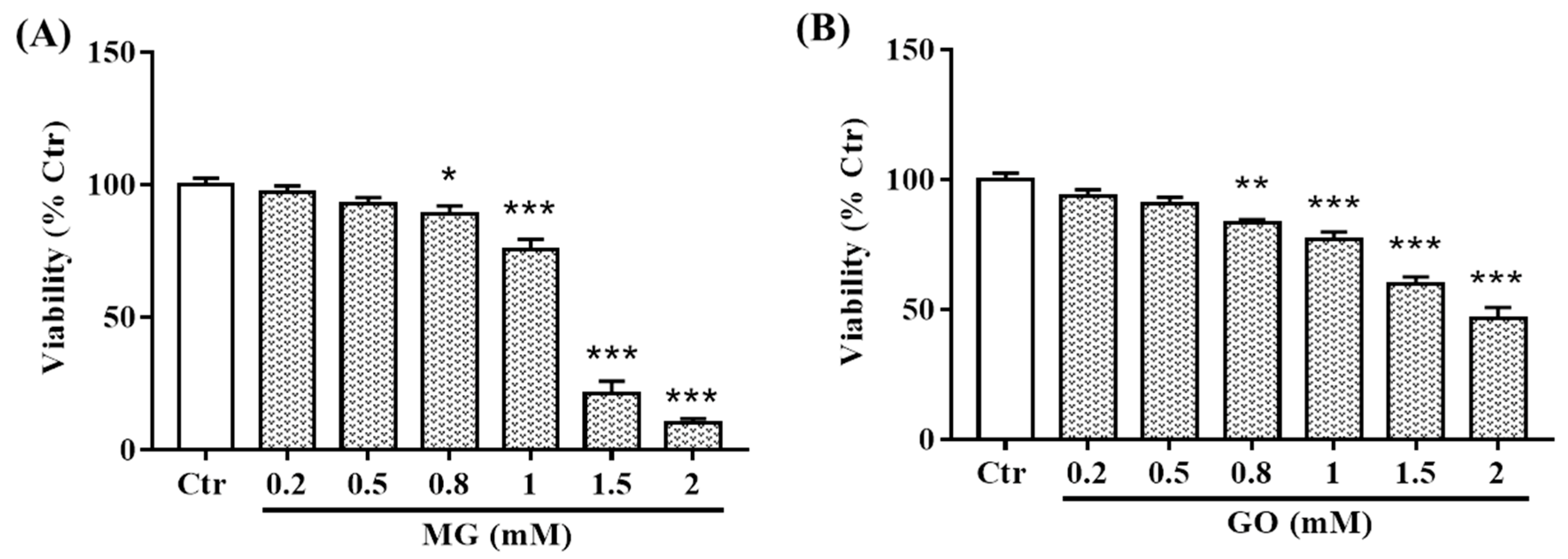
2.3. Effects of MG and GO on SK-N-SH cells Viability
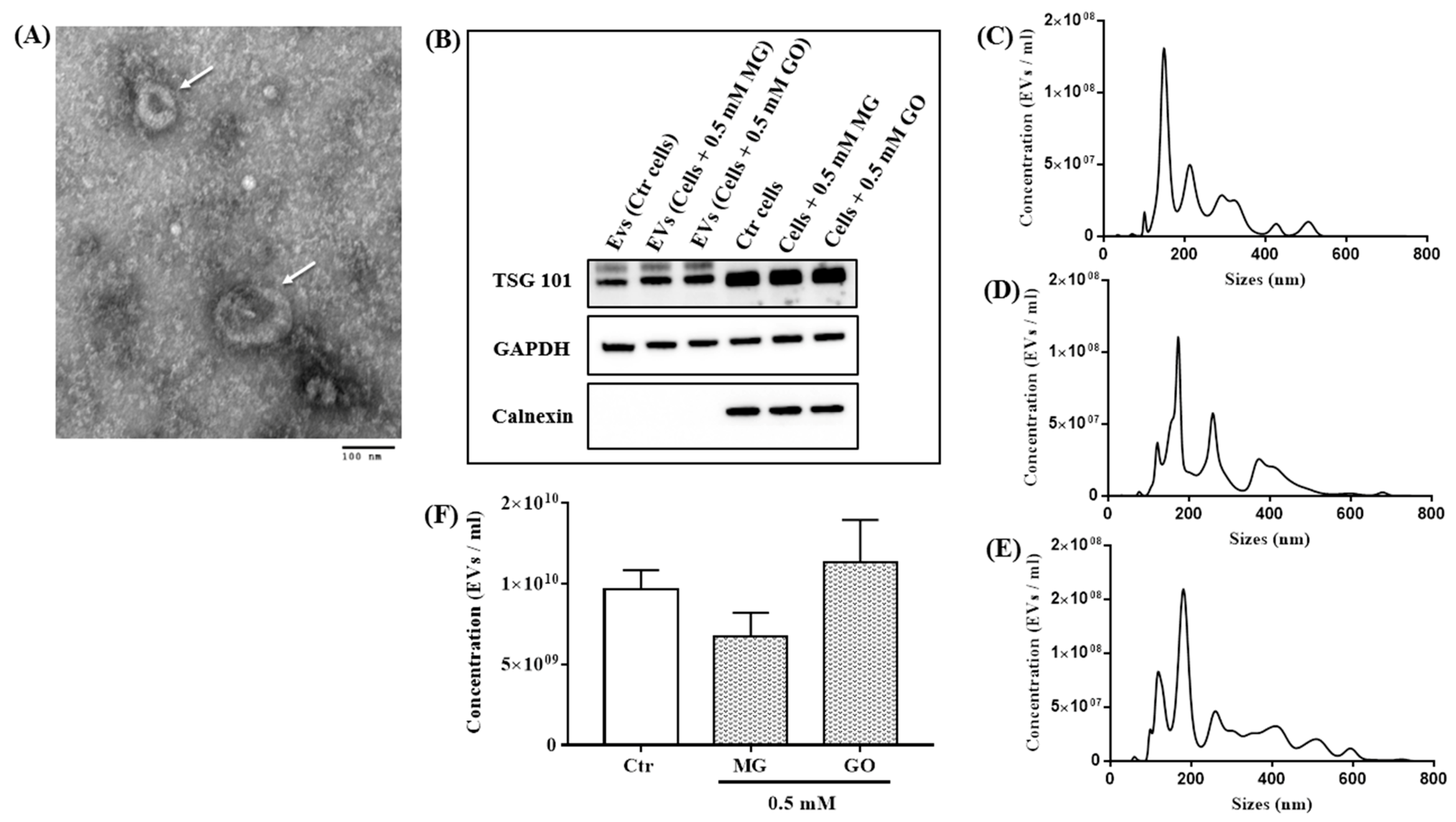
2.4. Effects of MG and GO on the Size and Density of Extracellular Vesicles Released by the SK-N-SH Neuronal Cells
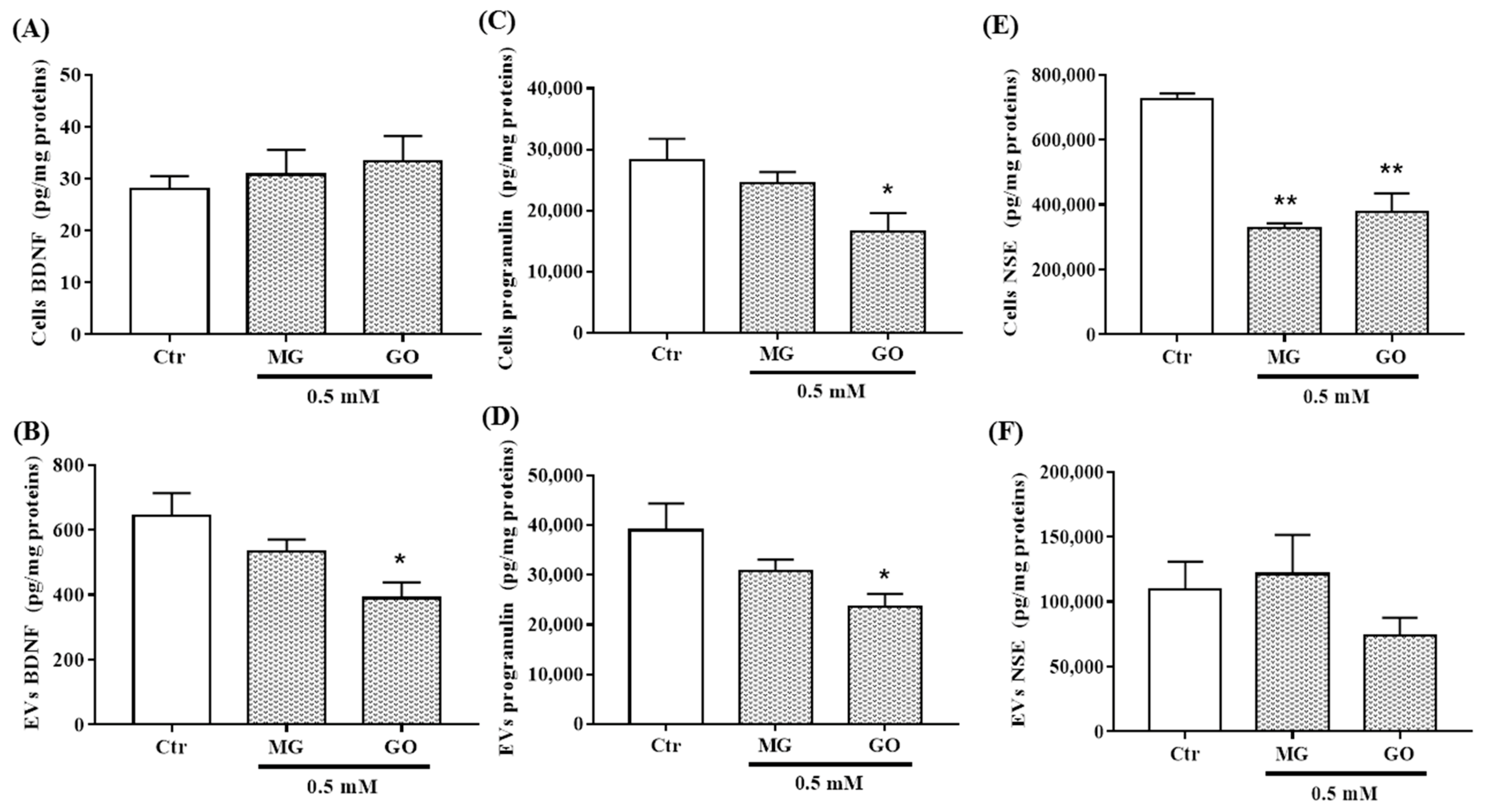
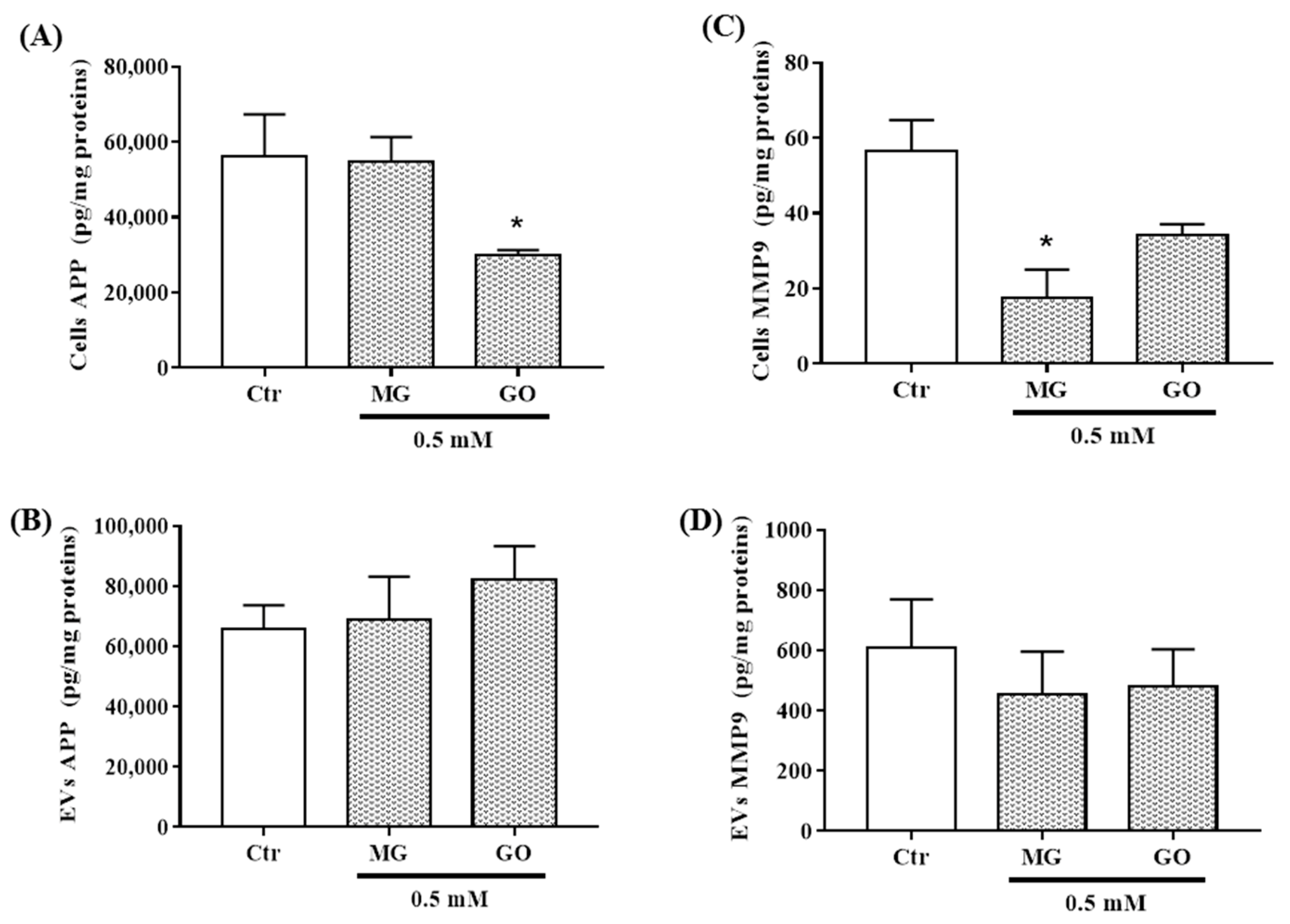
2.5. Effects of MG and GO Treatments on the Levels of BDNF, PRGN, NSE, APP and MMP-9 in Cells and in nEVs
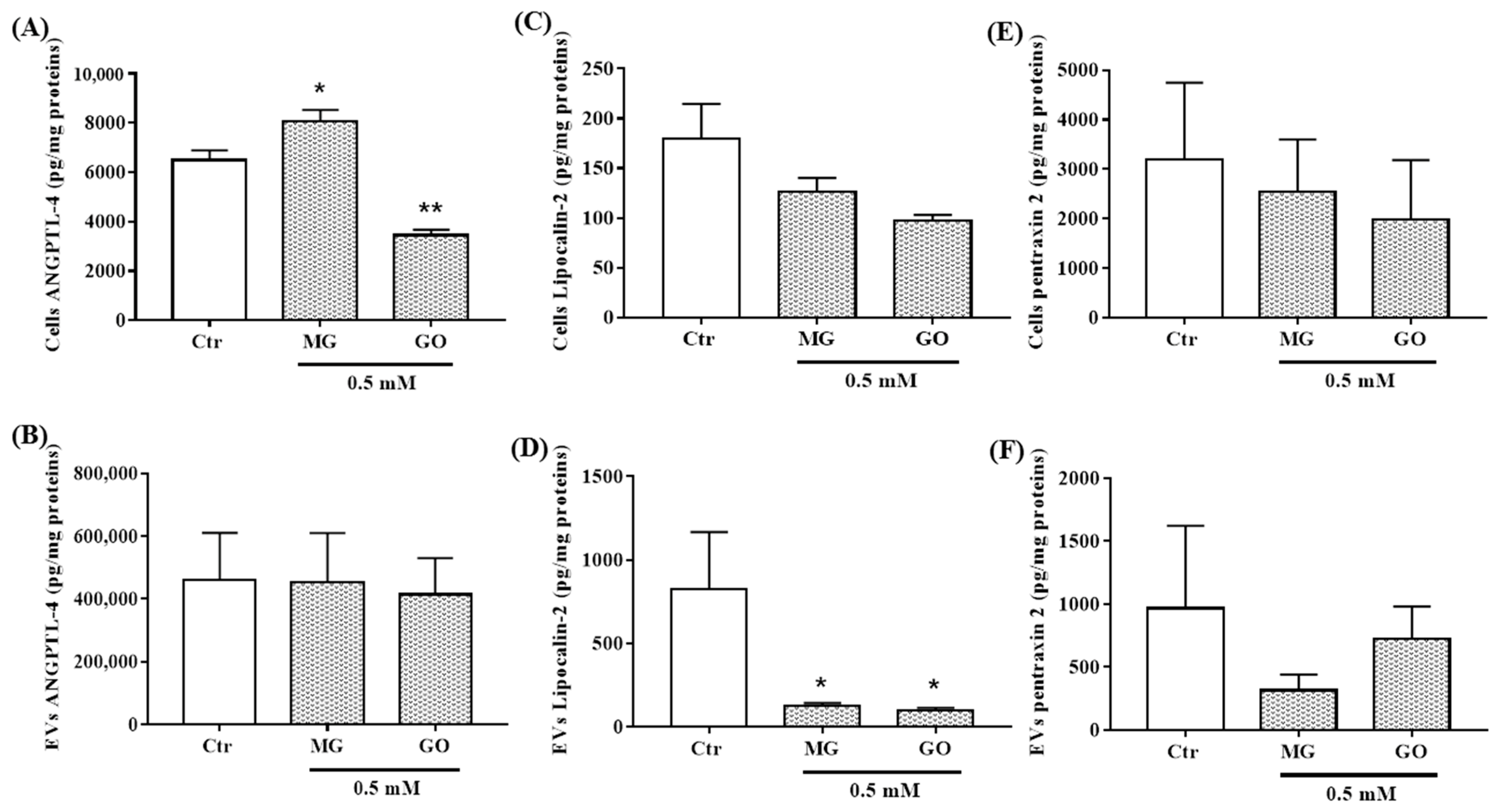
2.6. Effects of MG and GO Treatments on the Levels of ANGPTL-4, LCN2, PTX2, S100B and RAGE in Cells and nEVs
2.7. Effects of MG and GO Treatments on the Levels of Aβ1-40, Aβ1-42, pTau T181 and α-Synuclein in Cells and nEVs
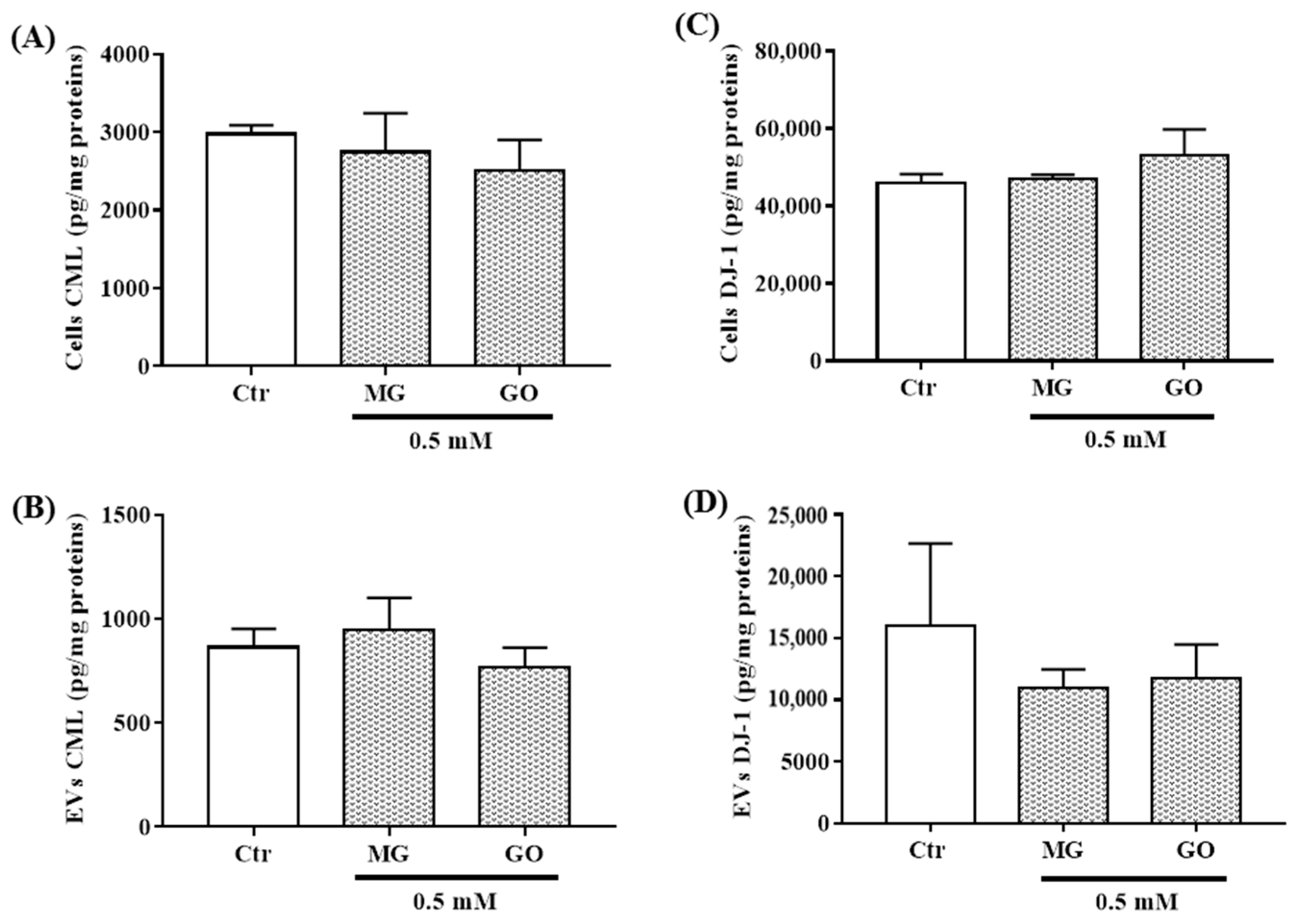
2.8. Effects of MG and GO on the Levels of the AGE Product CML and on the Deglycase Enzyme DJ-1
3. Discussion
4. Materials and Methods
4.1. Clinical Study
4.2. Serum Methylglyoxal and Glyoxal Measurement by High-Performance Liquid Chromatographic (HPLC)
4.3. Neuronal Cells Culture, Treatment and Viability Assay
4.4. Isolation of Extracellular Vesicles from Culture Media
4.5. Characterization of Extracellular Vesicles
4.5.1. Transmission Electron Microscopy
4.5.2. Nanoparticles Tracking Analysis
4.5.3. Western Blot Analysis
4.6. Determination of Cellular and nEVs Proteins
4.7. CML Determination
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | Amyloid beta peptide |
| AGEs | Advanced glycation end products |
| ALDH | Aldehyde dehydrogenase |
| ANGPTL-4 | Angiopoietin Like 4 |
| APP | Amyloid precursor protein |
| BDNF | Brain-Derived Neurotrophic Factor |
| CEL | N-(carboxyethyl)-L-lysine |
| CML | N-(1-carboxymethyl)-L-lysine |
| CSF | Cerebrospinal fluid |
| ES | Early stage of Alzheimer’s disease |
| EVs | Extracellular vesicles |
| GO | Glyoxal |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GSH | Reduced glutathione |
| GLO | Glyoxalase |
| GOLD | Glyoxal-lysine dimer |
| HPLC | High-performance liquid chromatographic |
| Icv | Intracerebroventricular |
| LCN2 | Lipocalin-2 |
| LS | Late stage of Alzheimer’s disease |
| LSD | Least significant difference |
| 2-MQ | 2-methylquinoxaline |
| 5-MQ | 5-methylquinoxaline |
| MCI | Mild cognitive impairment |
| MMP-9 | Matrix metallopeptidase 9 |
| MG | Methylglyoxal |
| MMSE | Mini-Mental State Examination |
| MoCA | Montreal Cognitive Assessment |
| MOLD | Methylglyoxal-lysine dimer |
| MS | Moderate stage of Alzheimer’s disease |
| NFTs | Neurofibrillary tangles |
| NSE | Neuron specific enolase |
| nEVs | Neuronal derived-extracellular vesicles |
| O-PD | O-Phenylenediamine |
| PRGN | Progranulin |
| PTX2 | Pentraxin 2 |
| RAGE | Receptor for advanced glycation end products |
| ROC | Receiver-operating characteristic curves |
| SSAO | Semicarbazidesensitive amine oxidase |
| TEM | Transmission electron microscopy |
| TSG101 | Tumor susceptibility gene 101 |
References
- Hachinski, V. Dementia: Paradigm shifting into high gear. Alzheimers Dement. 2019, 15, 985–994. [Google Scholar] [CrossRef] [PubMed]
- World Alzheimer Report 2018 The state of the art of dementia research: New frontiers. Available online: www.alz.co.uk/research/WorldAlzheimerReport2018.pdf (accessed on 30 September 2018).
- Benussi, L.; Binetti, G.; Ghidoni, R. Loss of Neuroprotective Factors in Neurodegenerative Dementias: The End or the Starting Point? Front. Neurosci. 2017, 11, 672. [Google Scholar] [CrossRef] [PubMed]
- Diniz, B.S.; Teixeira, A.L. Brain-derived neurotrophic factor and Alzheimer’s disease: Physiopathology and beyond. Neuromolecular Med. 2011, 13, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.M.; Mergl, R.; Stach, B.; Jahn, I.; Gertz, H.J.; Schonknecht, P. Elevated levels of cerebrospinal fluid neuron-specific enolase (NSE) in Alzheimer’s disease. Neurosci. Lett. 2014, 570, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N. Y.) 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Med. Clin. North. Am. 2019, 103, 263–293. [Google Scholar] [CrossRef]
- Bruandet, A.; Richard, F.; Bombois, S.; Maurage, C.A.; Deramecourt, V.; Lebert, F.; Amouyel, P.; Pasquier, F. Alzheimer disease with cerebrovascular disease and vascular dementia: Clinical features and course compared with Alzheimer disease. J. Neurol Neurosurg Psychiatry 2009, 80, 133–139. [Google Scholar] [CrossRef]
- Sanford, A.M. Mild Cognitive Impairment. Clin. Geriatr. Med. 2017, 33, 325–337. [Google Scholar] [CrossRef]
- Unverzagt, F.W.; Gao, S.; Baiyewu, O.; Ogunniyi, A.O.; Gureje, O.; Perkins, A.; Emsley, C.L.; Dickens, J.; Evans, R.; Musick, B.; et al. Prevalence of cognitive impairment: Data from the Indianapolis Study of Health and Aging. Neurology 2001, 57, 1655–1662. [Google Scholar] [CrossRef]
- Manly, J.J.; Tang, M.X.; Schupf, N.; Stern, Y.; Vonsattel, J.P.; Mayeux, R. Frequency and course of mild cognitive impairment in a multiethnic community. Ann. Neurol. 2008, 63, 494–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguli, M.; Chang, C.C.; Snitz, B.E.; Saxton, J.A.; Vanderbilt, J.; Lee, C.W. Prevalence of mild cognitive impairment by multiple classifications: The Monongahela-Youghiogheny Healthy Aging Team (MYHAT) project. Am. J. Geriatr Psychiatry 2010, 18, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Bachurin, S.O.; Gavrilova, S.I.; Samsonova, A.; Barreto, G.E.; Aliev, G. Mild cognitive impairment due to Alzheimer disease: Contemporary approaches to diagnostics and pharmacological intervention. Pharm. Res. 2018, 129, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Lopez, O.L. Mild cognitive impairment. Contin. (Minneap. Minn.) 2013, 19, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Behrman, S.; Valkanova, V.; Allan, C.L. Diagnosing and managing mild cognitive impairment. Practitioner 2017, 261, 17–20. [Google Scholar] [PubMed]
- Maillard, L.C. Action des acides amines sur les sucres: Formation des melanoidines par voie methodique. C. R. Hebd. Seances Acad. Sci. 1912, 154, 66–68. [Google Scholar]
- Luth, H.J.; Ogunlade, V.; Kuhla, B.; Kientsch-Engel, R.; Stahl, P.; Webster, J.; Arendt, T.; Munch, G. Age- and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer’s disease brains. Cereb. Cortex 2005, 15, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Girones, X.; Guimera, A.; Cruz-Sanchez, C.Z.; Ortega, A.; Sasaki, N.; Makita, Z.; Lafuente, J.V.; Kalaria, R.; Cruz-Sanchez, F.F. N epsilon-carboxymethyllysine in brain aging, diabetes mellitus, and Alzheimer’s disease. Free Radic. Biol. Med. 2004, 36, 1241–1247. [Google Scholar] [CrossRef]
- Horie, K.; Miyata, T.; Yasuda, T.; Takeda, A.; Yasuda, Y.; Maeda, K.; Sobue, G.; Kurokawa, K. Immunohistochemical localization of advanced glycation end products, pentosidine, and carboxymethyllysine in lipofuscin pigments of Alzheimer’s disease and aged neurons. Biochem. Biophys. Res. Commun. 1997, 236, 327–332. [Google Scholar] [CrossRef]
- Castellani, R.J.; Harris, P.L.; Sayre, L.M.; Fujii, J.; Taniguchi, N.; Vitek, M.P.; Founds, H.; Atwood, C.S.; Perry, G.; Smith, M.A. Active glycation in neurofibrillary pathology of Alzheimer disease: N(epsilon)-(carboxymethyl) lysine and hexitol-lysine. Free Radic. Biol. Med. 2001, 31, 175–180. [Google Scholar] [CrossRef]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47, 3–27. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Pharmacology of methylglyoxal: Formation, modification of proteins and nucleic acids, and enzymatic detoxification--a role in pathogenesis and antiproliferative chemotherapy. Gen. Pharm. 1996, 27, 565–573. [Google Scholar] [CrossRef]
- Li, X.H.; Xie, J.Z.; Jiang, X.; Lv, B.L.; Cheng, X.S.; Du, L.L.; Zhang, J.Y.; Wang, J.Z.; Zhou, X.W. Methylglyoxal induces tau hyperphosphorylation via promoting AGEs formation. Neuromolecular Med. 2012, 14, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Tajes, M.; Eraso-Pichot, A.; Rubio-Moscardo, F.; Guivernau, B.; Bosch-Morato, M.; Valls-Comamala, V.; Munoz, F.J. Methylglyoxal reduces mitochondrial potential and activates Bax and caspase-3 in neurons: Implications for Alzheimer’s disease. Neurosci. Lett. 2014, 580, 78–82. [Google Scholar] [CrossRef] [PubMed]
- de Arriba, S.G.; Krugel, U.; Regenthal, R.; Vissiennon, Z.; Verdaguer, E.; Lewerenz, A.; Garcia-Jorda, E.; Pallas, M.; Camins, A.; Munch, G.; et al. Carbonyl stress and NMDA receptor activation contribute to methylglyoxal neurotoxicity. Free Radic. Biol. Med. 2006, 40, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Huang, X.B.; Li, Z.X.; Yin, L.L.; Chen, W.Q.; Li, L. Tenuigenin protects cultured hippocampal neurons against methylglyoxal-induced neurotoxicity. Eur. J. Pharm. 2010, 645, 1–8. [Google Scholar] [CrossRef]
- Qi, L.; Chen, Z.; Wang, Y.; Liu, X.; Liu, X.; Ke, L.; Zheng, Z.; Lin, X.; Zhou, Y.; Wu, L.; et al. Subcutaneous liraglutide ameliorates methylglyoxal-induced Alzheimer-like tau pathology and cognitive impairment by modulating tau hyperphosphorylation and glycogen synthase kinase-3β. Am. J. Transl. Res. 2017, 9, 247–260. [Google Scholar]
- Huang, X.; Wang, F.; Chen, W.; Chen, Y.; Wang, N.; von Maltzan, K. Possible link between the cognitive dysfunction associated with diabetes mellitus and the neurotoxicity of methylglyoxal. Brain Res. 2012, 1469, 82–91. [Google Scholar] [CrossRef]
- Hansen, F.; Pandolfo, P.; Galland, F.; Torres, F.V.; Dutra, M.F.; Batassini, C.; Guerra, M.C.; Leite, M.C.; Gonçalves, C.-A. Methylglyoxal can mediate behavioral and neurochemical alterations in rat brain. Physiol. Behav. 2016, 164, 93–101. [Google Scholar] [CrossRef]
- Haddad, M.; Perrotte, M.; Landri, S.; Lepage, A.; Fulop, T.; Ramassamy, C. Circulating and Extracellular Vesicles Levels of N-(1-Carboxymethyl)-L-Lysine (CML) Differentiate Early to Moderate Alzheimer’s Disease. J. Alzheimers Dis. 2019, 69, 751–762. [Google Scholar] [CrossRef]
- Iraci, N.; Leonardi, T.; Gessler, F.; Vega, B.; Pluchino, S. Focus on Extracellular Vesicles: Physiological Role and Signalling Properties of Extracellular Membrane Vesicles. Int. J. Mol. Sci. 2016, 17, 171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Yang, P. A novel cell-cell communication mechanism in the nervous system: Exosomes. J. Neurosci. Res. 2018, 96, 45–52. [Google Scholar] [CrossRef]
- Yuyama, K.; Igarashi, Y. Physiological and pathological roles of exosomes in the nervous system. Biomol. Concepts 2016, 7, 53–68. [Google Scholar] [CrossRef]
- Janas, A.M.; Sapon, K.; Janas, T.; Stowell, M.H.; Janas, T. Exosomes and other extracellular vesicles in neural cells and neurodegenerative diseases. Biochim. Biophys. Acta 2016, 1858, 1139–1151. [Google Scholar] [CrossRef]
- Coleman, B.M.; Hill, A.F. Extracellular vesicles--Their role in the packaging and spread of misfolded proteins associated with neurodegenerative diseases. Semin. Cell Dev. Biol. 2015, 40, 89–96. [Google Scholar] [CrossRef]
- Quek, C.; Hill, A.F. The role of extracellular vesicles in neurodegenerative diseases. Biochem. Biophys. Res. Commun. 2017, 483, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Karlawish, J.; Jack, C.R., Jr.; Rocca, W.A.; Snyder, H.M.; Carrillo, M.C. Alzheimer’s disease: The next frontier-Special Report 2017. Alzheimers Dement. 2017, 13, 374–380. [Google Scholar] [CrossRef]
- Munch, G.; Thome, J.; Foley, P.; Schinzel, R.; Riederer, P. Advanced glycation endproducts in ageing and Alzheimer’s disease. Brain Res. 1997, 23, 134–143. [Google Scholar] [CrossRef]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Munch, G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.Y.; Ko, H.A.; Chu, K.H.; Shieh, T.M.; Chi, T.C.; Chen, H.I.; Chang, W.C.; Chang, S.S. The Possible Mechanism of Advanced Glycation End Products (AGEs) for Alzheimer’s Disease. PLoS ONE 2015, 10, e0143345. [Google Scholar] [CrossRef]
- Vicente Miranda, H.; El-Agnaf, O.M.A.; Outeiro, T.F. Glycation in Parkinson’s disease and Alzheimer’s disease. Mov. Disord. 2016, 31, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Zhang, W.; Jiao, B.; Pan, C.-Z.; Liu, X.; Shen, L. The role of exosomes in the pathogenesis of Alzheimer’ disease. Transl. Neurodegener 2017, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Rudnicka-Nawrot, M.; Richey, P.L.; Praprotnik, D.; Mulvihill, P.; Miller, C.A.; Sayre, L.M.; Perry, G. Carbonyl-Related Posttranslational Modification of Neurofilament Protein in the Neurofibrillary Pathology of Alzheimer’s Disease. J. Neurochem. 1995, 64, 2660–2666. [Google Scholar] [CrossRef] [PubMed]
- Kuhla, B.; Haase, C.; Flach, K.; Lüth, H.-J.; Arendt, T.; Münch, G. Effect of Pseudophosphorylation and Cross-linking by Lipid Peroxidation and Advanced Glycation End Product Precursors on Tau Aggregation and Filament Formation. J. Biol. Chem. 2007, 282, 6984–6991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Ahmed, U.; Thornalley, P.J.; Hager, K.; Fleischer, G.; Münch, G. Protein glycation, oxidation and nitration adduct residues and free adducts of cerebrospinal fluid in Alzheimer’s disease and link to cognitive impairment. J. Neurochem. 2005, 92, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Beeri, M.S.; Moshier, E.; Schmeidler, J.; Godbold, J.; Uribarri, J.; Reddy, S.; Sano, M.; Grossman, H.T.; Cai, W.; Vlassara, H.; et al. Serum concentration of an inflammatory glycotoxin, methylglyoxal, is associated with increased cognitive decline in elderly individuals. Mech. Ageing Dev. 2011, 132, 583–587. [Google Scholar] [CrossRef]
- Srikanth, V.; Westcott, B.; Forbes, J.; Phan, T.G.; Beare, R.; Venn, A.; Pearson, S.; Greenaway, T.; Parameswaran, V.; Munch, G. Methylglyoxal, cognitive function and cerebral atrophy in older people. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Obata, T. Diabetes and semicarbazide-sensitive amine oxidase (SSAO) activity: A review. Life Sci. 2006, 79, 417–422. [Google Scholar] [CrossRef]
- del Mar Hernandez, M.; Esteban, M.; Szabo, P.; Boada, M.; Unzeta, M. Human plasma semicarbazide sensitive amine oxidase (SSAO), beta-amyloid protein and aging. Neurosci. Lett. 2005, 384, 183–187. [Google Scholar] [CrossRef]
- Unzeta, M.; Solé, M.; Boada, M.; Hernández, M. Semicarbazide-sensitive amine oxidase (SSAO) and its possible contribution to vascular damage in Alzheimer’s disease. J. Neural. Transm. 2007, 114, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Sole, M.; Esteban-Lopez, M.; Taltavull, B.; Fabregas, C.; Fado, R.; Casals, N.; Rodriguez-Alvarez, J.; Minano-Molina, A.J.; Unzeta, M. Blood-brain barrier dysfunction underlying Alzheimer’s disease is induced by an SSAO/VAP-1-dependent cerebrovascular activation with enhanced Abeta deposition. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2189–2202. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Langborg, A.; Minhas, H.S. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem. J. 1999, 344 Pt 1, 109–116. [Google Scholar] [CrossRef]
- Kalapos, M.P. Where does plasma methylglyoxal originate from? Diabetes Res. Clin. Pr. 2013, 99, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Sousa Silva, M.; Gomes, R.A.; Ferreira, A.E.; Ponces Freire, A.; Cordeiro, C. The glyoxalase pathway: The first hundred years. and beyond. Biochem. J. 2013, 453, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, P.; Martin-Aragon, S.; Benedi, J.; Susin, C.; Felici, E.; Gil, P.; Ribera, J.M.; Villar, A.M. Peripheral levels of glutathione and protein oxidation as markers in the development of Alzheimer’s disease from Mild Cognitive Impairment. Free Radic. Res. 2008, 42, 162–170. [Google Scholar] [CrossRef]
- Puertas, M.C.; Martinez-Martos, J.M.; Cobo, M.P.; Carrera, M.P.; Mayas, M.D.; Ramirez-Exposito, M.J. Plasma oxidative stress parameters in men and women with early stage Alzheimer type dementia. Exp. Gerontol. 2012, 47, 625–630. [Google Scholar] [CrossRef]
- Kuhla, B.; Boeck, K.; Luth, H.J.; Schmidt, A.; Weigle, B.; Schmitz, M.; Ogunlade, V.; Munch, G.; Arendt, T. Age-dependent changes of glyoxalase I expression in human brain. Neurobiol. Aging 2006, 27, 815–822. [Google Scholar] [CrossRef]
- Kuhla, B.; Boeck, K.; Schmidt, A.; Ogunlade, V.; Arendt, T.; Munch, G.; Luth, H.J. Age- and stage-dependent glyoxalase I expression and its activity in normal and Alzheimer’s disease brains. Neurobiol. Aging 2007, 28, 29–41. [Google Scholar] [CrossRef]
- Munch, G.; Westcott, B.; Menini, T.; Gugliucci, A. Advanced glycation endproducts and their pathogenic roles in neurological disorders. Amino Acids 2012, 42, 1221–1236. [Google Scholar] [CrossRef]
- Di Loreto, S.; Caracciolo, V.; Colafarina, S.; Sebastiani, P.; Gasbarri, A.; Amicarelli, F. Methylglyoxal induces oxidative stress-dependent cell injury and up-regulation of interleukin-1beta and nerve growth factor in cultured hippocampal neuronal cells. Brain Res. 2004, 1006, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Di Loreto, S.; Zimmitti, V.; Sebastiani, P.; Cervelli, C.; Falone, S.; Amicarelli, F. Methylglyoxal causes strong weakening of detoxifying capacity and apoptotic cell death in rat hippocampal neurons. Int. J. Biochem. Cell Biol. 2008, 40, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Maley, J.; Yu, P.H. Potential inplications of endogenous aldehydes in beta-amyloid misfolding, oligomerization and fibrillogenesis. J. Neurochem. 2006, 99, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Emendato, A.; Milordini, G.; Zacco, E.; Sicorello, A.; Dal Piaz, F.; Guerrini, R.; Thorogate, R.; Picone, D.; Pastore, A. Glycation affects fibril formation of Abeta peptides. J. Biol. Chem. 2018, 293, 13100–13111. [Google Scholar] [CrossRef]
- Angeloni, C.; Zambonin, L.; Hrelia, S. Role of methylglyoxal in Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 238485. [Google Scholar] [CrossRef] [PubMed]
- Acheson, A.; Conover, J.C.; Fandl, J.P.; DeChiara, T.M.; Russell, M.; Thadani, A.; Squinto, S.P.; Yancopoulos, G.D.; Lindsay, R.M. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature 1995, 374, 450–453. [Google Scholar] [CrossRef]
- Lu, B.; Nagappan, G.; Guan, X.; Nathan, P.J.; Wren, P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 401–416. [Google Scholar] [CrossRef]
- Fragkouli, A.; Tsilibary, E.C.; Tzinia, A.K. Neuroprotective role of MMP-9 overexpression in the brain of Alzheimer’s 5xFAD mice. Neurobiol. Dis. 2014, 70, 179–189. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Haque, A.; Polcyn, R.; Matzelle, D.; Banik, N.L. New Insights into the Role of Neuron-Specific Enolase in Neuro-Inflammation, Neurodegeneration, and Neuroprotection. Brain Sci. 2018, 8, 33. [Google Scholar] [CrossRef]
- Zheng, H.; Koo, E.H. The amyloid precursor protein: Beyond amyloid. Mol. Neurodegener 2006, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Kaminari, A.; Tsilibary, E.C.; Tzinia, A. A New Perspective in Utilizing MMP-9 as a Therapeutic Target for Alzheimer’s Disease and Type 2 Diabetes Mellitus. J. Alzheimer’s Dis. 2018, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mendsaikhan, A.; Tooyama, I.; Walker, D.G. Microglial Progranulin: Involvement in Alzheimer’s Disease and Neurodegenerative Diseases. Cells 2019, 8, 230. [Google Scholar] [CrossRef] [PubMed]
- Vienberg, S.G.; Kleinridders, A.; Suzuki, R.; Kahn, C.R. Differential effects of angiopoietin-like 4 in brain and muscle on regulation of lipoprotein lipase activity. Mol. Metab. 2014, 4, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Kim, O.Y. Perspectives in Lipocalin-2: Emerging Biomarker for Medical Diagnosis and Prognosis for Alzheimer’s Disease. Clin. Nutr. Res. 2018, 7, 1–10. [Google Scholar] [CrossRef]
- Ishii, T.; Warabi, E.; Mann, G.E. Circadian control of BDNF-mediated Nrf2 activation in astrocytes protects dopaminergic neurons from ferroptosis. Free Radic. Biol. Med. 2019, 133, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, W.H.; Lee, M.S.; Mori, K.; Suk, K. Regulation by lipocalin-2 of neuronal cell death, migration, and morphology. J. Neurosci. Res. 2012, 90, 540–550. [Google Scholar] [CrossRef]
- Lee, S.; Kim, J.H.; Kim, J.H.; Seo, J.W.; Han, H.S.; Lee, W.H.; Mori, K.; Nakao, K.; Barasch, J.; Suk, K. Lipocalin-2 Is a chemokine inducer in the central nervous system: Role of chemokine ligand 10 (CXCL10) in lipocalin-2-induced cell migration. J. Biol. Chem. 2011, 286, 43855–43870. [Google Scholar] [CrossRef]
- Law, I.K.M.; Xu, A.; Lam, K.S.L.; Berger, T.; Mak, T.W.; Vanhoutte, P.M.; Liu, J.T.C.; Sweeney, G.; Zhou, M.; Yang, B.; et al. Lipocalin-2 deficiency attenuates insulin resistance associated with aging and obesity. Diabetes 2010, 59, 872–882. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Pinto, V.; Da Mesquita, S.; Novais, A.; Sousa, J.C.; Correia-Neves, M.; Sousa, N.; Palha, J.A.; Marques, F. Lipocalin-2 is involved in emotional behaviors and cognitive function. Front. Cell Neurosci. 2013, 7, 122. [Google Scholar] [CrossRef]
- Fleitas, C.; Pinol-Ripoll, G.; Marfull, P.; Rocandio, D.; Ferrer, I.; Rampon, C.; Egea, J.; Espinet, C. proBDNF is modified by advanced glycation end products in Alzheimer’s disease and causes neuronal apoptosis by inducing p75 neurotrophin receptor processing. Mol. Brain 2018, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Mihoub, M.; Abdallah, J.; Gontero, B.; Dairou, J.; Richarme, G. The DJ-1 superfamily member Hsp31 repairs proteins from glycation by methylglyoxal and glyoxal. Biochem. Biophys. Res. Commun. 2015, 463, 1305–1310. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Rao, S.P.; Kalivendi, S.V. The deglycase activity of DJ-1 mitigates alpha-synuclein glycation and aggregation in dopaminergic cells: Role of oxidative stress mediated downregulation of DJ-1 in Parkinson’s disease. Free Radic. Biol. Med. 2019, 135, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Kalani, A.; Tyagi, A.; Tyagi, N. Exosomes: Mediators of Neurodegeneration, Neuroprotection and Therapeutics. Mol. Neurobiol. 2014, 49, 590–600. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D.; Schwartz, J.B. Declining levels of functionally specialized synaptic proteins in plasma neuronal exosomes with progression of Alzheimer’s disease. FASEB J. 2018, 32, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.H.; Chen, Z.T.; Zhou, R.L.; Zhang, X.; Ye, Q.Y.; Wang, Y.Z. Increased DJ-1 and alpha-Synuclein in Plasma Neural-Derived Exosomes as Potential Markers for Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 438. [Google Scholar] [CrossRef]
- Baba, S.P.; Barski, O.A.; Ahmed, Y.; O’Toole, T.E.; Conklin, D.J.; Bhatnagar, A.; Srivastava, S. Reductive metabolism of AGE precursors: A metabolic route for preventing AGE accumulation in cardiovascular tissue. Diabetes 2009, 58, 2486–2497. [Google Scholar] [CrossRef]
- Nomi, Y.; Aizawa, H.; Kurata, T.; Shindo, K.; Nguyen, C.V. Glutathione reacts with glyoxal at the N-terminal. Biosci. Biotechnol. Biochem. 2009, 73, 2408–2411. [Google Scholar] [CrossRef]
- Yang, K.; Qiang, D.; Delaney, S.; Mehta, R.; Bruce, W.R.; O’Brien, P.J. Differences in glyoxal and methylglyoxal metabolism determine cellular susceptibility to protein carbonylation and cytotoxicity. Chem. Biol. Interact. 2011, 191, 322–329. [Google Scholar] [CrossRef]
- da Silva, G. Hydroxyl radical regeneration in the photochemical oxidation of glyoxal: Kinetics and mechanism of the HC(O)CO + O2 reaction. Phys. Chem. Chem. Phys. 2010, 12, 6698–6705. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Ldaboy, J.R.; Ruiz-Santoyo, M.E.; Vivier-Bunge, A. Mechanism and kinetics of the reaction of OH radicals with glyoxal and methylglyoxal: A quantum chemistry + CVT/SCT approach. Chemphyschem 2004, 5, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C.; Smith, G.E.; Waring, S.C.; Ivnik, R.J.; Tangalos, E.G.; Kokmen, E. Mild cognitive impairment: Clinical characterization and outcome. Arch. Neurol. 1999, 56, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G.; Ferguson, F.G.; Wikby, A. The SENIEUR protocol after 16 years. Mech. Ageing Dev. 2001, 122, 132–134. [Google Scholar] [CrossRef]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Nasreddine, Z.S.; Phillips, N.A.; Bedirian, V.; Charbonneau, S.; Whitehead, V.; Collin, I.; Cummings, J.L.; Chertkow, H. The Montreal Cognitive Assessment, MoCA: A brief screening tool for mild cognitive impairment. J. Am. Geriatr. Soc. 2005, 53, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Dhar, A.; Desai, K.; Liu, J.; Wu, L. Methylglyoxal, protein binding and biological samples: Are we getting the true measure? J. Chromatogr. B 2009, 877, 1093–1100. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Controls Subjects (n = 15) | MCI Patients (n = 16) | AD Patients | ||
|---|---|---|---|---|---|
| ES (AD) (n = 19) | MS (AD) (n = 14) | LS (AD) (n = 16) | |||
| Sex (M/F) | 3/12 | 4/12 | 3/16 | 2/12 | 5/11 |
| Age (years) | 69.4 ± 1.17 | 73.63 ± 1.24 * | 79.68 ± 1.2 *** | 79.73 ± 1.3 *** | 80.13 ± 1.6 *** |
| MMSE scores (/30) | 29.67 ± 0.15 | 27.19 ± 0.47 * | 23.79 ± 0.8 *** | 21.73 ± 1.16 *** | ND |
| MoCA scores (/30) | 27.53 ± 0.51 | 22.94 ± 0.79 ** | 17.06 ± 0.9 *** | 12.55 ± 1.6 *** | ND |
| Glucose (mmol/l) | 4.64 ± 0.15 | 4.95 ± 0.17 | 4.85 ± 0.13 | 5.3 ± 0.28 * | 4.45 ± 0.13 |
| Groups | Cutt off (nM) | Sensitivity (%) | Specificity (%) |
|---|---|---|---|
| Methylglyoxal [Controls subjects vs MCI patients] | >463.2 | 87.5 | 93.33 |
| Methylglyoxal [MCI patients vs ES AD patients] | <545.1 | 63.16 | 62.5 |
| Methylglyoxal [MCI patients vs AD patients] | <545.1 | 63.27 | 62.5 |
| Glyoxal [Controls subjects vs MCI patients] | >652.2 | 68.75 | 80 |
| Glyoxal [MCI patients vs ES AD patients] | <588.6 | 68.42 | 81.25 |
| Glyoxal [MCI patients vs AD patients] | <605 | 67.35 | 81.25 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haddad, M.; Perrotte, M.; Khedher, M.R.B.; Demongin, C.; Lepage, A.; Fülöp, T.; Ramassamy, C. Methylglyoxal and Glyoxal as Potential Peripheral Markers for MCI Diagnosis and Their Effects on the Expression of Neurotrophic, Inflammatory and Neurodegenerative Factors in Neurons and in Neuronal Derived-Extracellular Vesicles. Int. J. Mol. Sci. 2019, 20, 4906. https://doi.org/10.3390/ijms20194906
Haddad M, Perrotte M, Khedher MRB, Demongin C, Lepage A, Fülöp T, Ramassamy C. Methylglyoxal and Glyoxal as Potential Peripheral Markers for MCI Diagnosis and Their Effects on the Expression of Neurotrophic, Inflammatory and Neurodegenerative Factors in Neurons and in Neuronal Derived-Extracellular Vesicles. International Journal of Molecular Sciences. 2019; 20(19):4906. https://doi.org/10.3390/ijms20194906
Chicago/Turabian StyleHaddad, Mohamed, Morgane Perrotte, Mohamed Raâfet Ben Khedher, Clément Demongin, Aurélie Lepage, Tamás Fülöp, and Charles Ramassamy. 2019. "Methylglyoxal and Glyoxal as Potential Peripheral Markers for MCI Diagnosis and Their Effects on the Expression of Neurotrophic, Inflammatory and Neurodegenerative Factors in Neurons and in Neuronal Derived-Extracellular Vesicles" International Journal of Molecular Sciences 20, no. 19: 4906. https://doi.org/10.3390/ijms20194906
APA StyleHaddad, M., Perrotte, M., Khedher, M. R. B., Demongin, C., Lepage, A., Fülöp, T., & Ramassamy, C. (2019). Methylglyoxal and Glyoxal as Potential Peripheral Markers for MCI Diagnosis and Their Effects on the Expression of Neurotrophic, Inflammatory and Neurodegenerative Factors in Neurons and in Neuronal Derived-Extracellular Vesicles. International Journal of Molecular Sciences, 20(19), 4906. https://doi.org/10.3390/ijms20194906