Prognostic Factors and Biomarkers of Responses to Immune Checkpoint Inhibitors in Lung Cancer


 ,
,  ,
,
Abstract
:1. Introduction
2. The Clinical Implications of ICIs in Non-Small Cell Lung Cancer (NSCLC)
3. ICIs and Special Populations: Oncogene-Addicted Patients
4. Current Biomarkers in Immune Checkpoints Inhibitors
4.1. PD-L1 Expression and TMB
4.2. Neoantigens
4.3. STK11 Mutations
5. TME-Associated Biomarkers
5.1. TILs
5.2. IDO1
5.3. The Impact of the Microbiome on Responses to ICIs
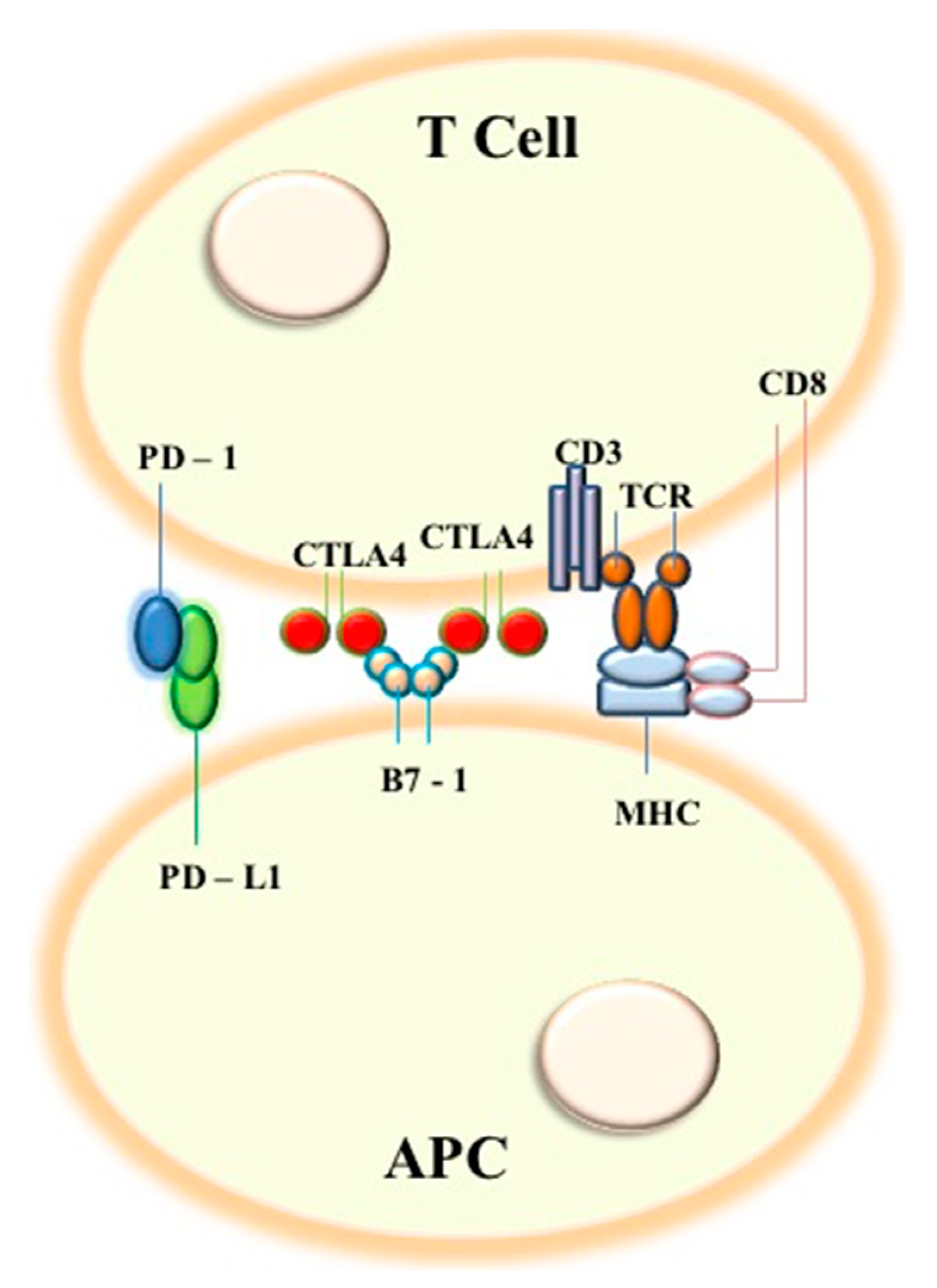
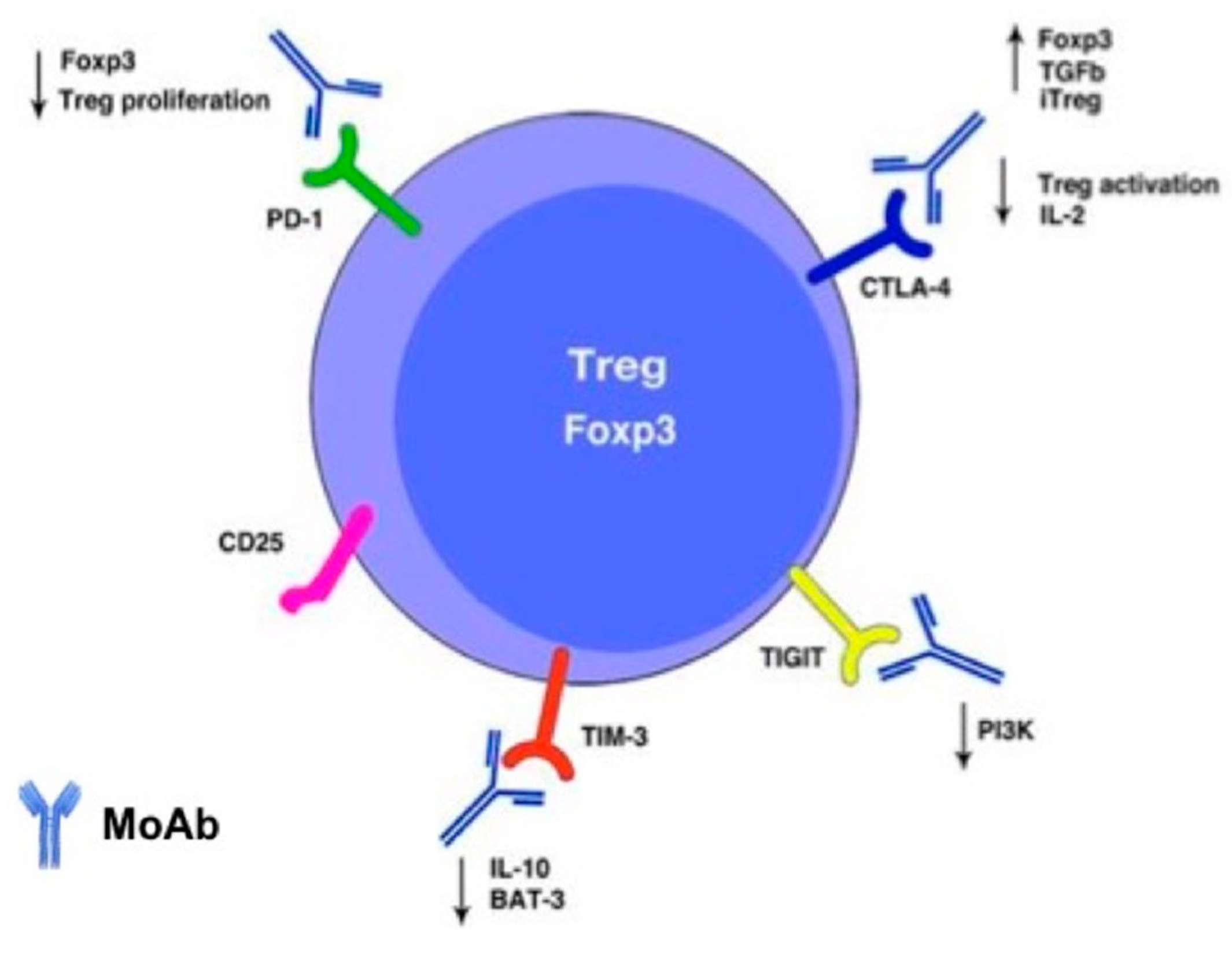
6. Expanding the Landscape: Tumor Microenvironment and T-reg Modulation in NSCLC Cancerogenesis
7. Discussion
Author Contributions
Conflicts of Interest
References
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Borst, J.; Ahrends, T.; Babala, N.; Melief, C.J.M.; Kastenmuller, W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Malapelle, U.; Rocco, D.; Perrotta, F.; Mazzarella, G. Targeting immune checkpoints in non small cell lung cancer. Curr. Opin. Pharmacol. 2018, 40, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, F.; Rocco, D.; Vitiello, F.; De Palma, R.; Guerra, G.; De Luca, A.; Navani, N.; Bianco, A. Immune Checkpoint Blockade for Advanced NSCLC: A New Landscape for Elderly Patients. Int. J. Mol. Sci. 2019, 20, 2258. [Google Scholar] [CrossRef] [PubMed]
- Fiorelli, A.; Perrotta, F.; Mollica, M.; Santini, M.; Vitiello, F.; Gilli, M.; Calabrese, C.; Bianco, A. Endoscopic central airway recanalization to enable first line pembrolizumab treatment in a PD-L1 strongly positive non-small cell lung cancer: A case report. J. Cardiothorac. Surg. 2019, 14, 50. [Google Scholar] [CrossRef] [PubMed]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef]
- Teng, M.W.L.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef] [Green Version]
- Djenidi, F.; Adam, J.; Goubar, A.; Durgeau, A.; Meurice, G.; de Montpreville, V.; Validire, P.; Besse, B.; Mami-Chouaib, F. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J. Immunol. 2015, 194, 3475–3486. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.; Johnson, D.; Temin, S.; Baker, S., Jr.; Brahmer, J.; Ellis, P.M.; Giaccone, G.; Hesketh, P.J.; Jaiyesimi, I.; Leighl, N.B.; et al. Systemic Therapy for Stage IV Non—Small-Cell Lung Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J. Clin. Oncol. 2017, 35, 3484–3515. [Google Scholar] [CrossRef] [PubMed]
- BMS Press Release. Bristol-Myers Squibb’s Opdivo (nivolumab) Receives Expanded FDA Approval in Previously-Treated Metastatic Non-Small Cell Lung Cancer (NSCLC), Offering Improved Survival to More Patients. Available online: https://news.bms.com/press-release/bristol-myers-squibbs-opdivo-nivolumab-receives-expanded-fda-approval-previously-treat (accessed on 9 October 2015).
- BMS Press Release. FDA Approves Opdivo (nivolumab) for the Treatment of Patients with Previously Treated Metastatic Squamous Non-Small Cell Lung Cancer. Available online: https://news.bms.com/press-release/fda-approves-opdivo-nivolumab-treatment-patients-previously-treated-metastatic-squamous (accessed on 4 March 2015).
- MSD Press Release. FDA Approves Merck’s KEYTRUDA® (pembrolizumab) in Metastatic NSCLC for First-Line Treatment of Patients Whose Tumors Have High PD-L1 Expression (Tumor Proportion Score [TPS] of 50 Percent or More) with No EGFR or ALK Genomic T. Available online: https://www.mrknewsroom.com/news-release/prescription-medicine-news/fda-approves-mercks-keytruda-pembrolizumab-metastatic-nsclc (accessed on 18 June 2019).
- AscoPost. FDA Approves Pembrolizumab as First-Line Treatment for PD-L1–Positive Non–Small Cell Lung Cancer. Available online: https://www.ascopost.com/issues/november-10-2016/fda-approves-pembrolizumab-as-first-line-treatment-for-pd-l1-positive-non-small-cell-lung-cancer/ (accessed on 10 November 2016).
- Roche Media Release. FDA Approves Roche’s Cancer Immunotherapy TECENTRIQ (atezolizumab) for People with a Specific Type of Metastatic Lung Cancer. Available online: https://www.roche.com/media/releases/med-cor-2016-10-19.htm (accessed on 19 October 2016).
- FDA Press Release. FDA Grants Regular Approval for Pembrolizumab in Combination with Chemotherapy for First-Line Treatment of Metastatic Non-squamous NSCLC. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm617471.htm (accessed on 20 August 2018).
- FDA Press Release. FDA Approves Pembrolizumab in Combination with Chemotherapy for First-Line Treatment of Metastatic Squamous NSCLC. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm624659.htm (accessed on 14 December 2018).
- FDA Press Release. FDA Approves Atezolizumab with Chemotherapy and Bevacizumab for First-Line Treatment of Metastatic Non-Squamous NSCLC. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm627874.htm (accessed on 20 Sepember 2002).
- FDA Press Release. FDA Approves Durvalumab after Chemoradiation for Unresectable Stage III NSCLC. Available online: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm597248.htm (accessed on 20 February 2018).
- Guo, L.; Zhang, H.; Chen, B. Nivolumab as Programmed Death-1 (PD-1) Inhibitor for Targeted Immunotherapy in Tumor. J. Cancer 2017, 8, 410–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, J.; Jimeno, A. Pembrolizumab: PD-1 inhibition as a therapeutic strategy in cancer. Drugs Today 2015, 51, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Lee, J.Y.; Lim, H.; Lee, S.H.; Moon, Y.J.; Pyo, H.J.; Ryu, S.E.; Shin, W.; Heo, Y.S. Molecular mechanism of PD-1/PD-L1 blockade via anti-PD-L1 antibodies atezolizumab and durvalumab. Sci. Rep. 2017, 7, 5532. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Campbell, S.F. Atezolizumab plus platinum-based regimen and bevacizumab: Is it time to consider immunotherapy in a concurrent approach for lung cancer? Transl. Cancer Res. 2019, 8, S103–S105. [Google Scholar] [CrossRef]
- Gandhi, L.; Garassino, M.C. Pembrolizumab plus Chemotherapy in Lung Cancer. N. Engl. J. Med. 2018, 379, e18. [Google Scholar]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gumus, M.; Mazieres, J.; Hermes, B.; Cay Senler, F.; Csoszi, T.; Fulop, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Tsakonas, G.; Ekman, S. Oncogene-addicted non-small cell lung cancer and immunotherapy. J. Thorac. Dis. 2018, 10, S1547–S1555. [Google Scholar] [CrossRef]
- Miura, Y.; Sunaga, N. Role of Immunotherapy for Oncogene-Driven Non-Small Cell Lung Cancer. Cancers 2018, 10, 245. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.; Zhu, X.; Sun, Y.; Zhang, Y. The efficacy of anti-PD-1/PD-L1 therapy and its comparison with EGFR-TKIs for advanced non-small-cell lung cancer. Oncotarget 2017, 8, 57826–57835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.Y.; Zhang, J.T.; Liu, S.Y.; Su, J.; Zhang, C.; Xie, Z.; Zhou, Q.; Tu, H.Y.; Xu, C.R.; Yan, L.X.; et al. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD-1 blockade in non-small cell lung cancer. Oncoimmunology 2017, 6, e1356145. [Google Scholar] [CrossRef] [PubMed]
- Garassino, M.C.; Cho, B.C.; Kim, J.H.; Mazieres, J.; Vansteenkiste, J.; Lena, H.; Corral Jaime, J.; Gray, J.E.; Powderly, J.; Chouaid, C.; et al. Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (ATLANTIC): An open-label, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 521–536. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.; Chow, L.Q.; Borghaei, H.; Shen, Y.; Harbison, C.; Chen, A.C.; Rizvi, N.A. Safety and Response with Nivolumab (Anti-PD-1; BMS-936558, ONO-4538) Plus Erlotinib in Patients (Pts) with Epidermal Growth Factor Receptor Mutant (EGFR MT) Advanced Non-Small Cell Lung Cancer (NSCLC): Metastatic Non-Small Cell Lung Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2014, 90, S34–S35. [Google Scholar] [CrossRef]
- Planchard, D.; Barlesi, F.; Gomez-Roca, C.; Mazieres, J.; Varga, A.; Greillier, L.; Chaput, N.; Parlavecchio, C.; Malekzadeh, K.; Ngocamus, M.; et al. Phase I, safety, tolerability and preliminary efficacy study of tremelimumab (Trem) in combination with gefitinib (Gef) in EGFR-mutant (EGFR-mut) NSCLC (GEFTREM). Ann. Oncol. 2016, 27, vi416–vi454. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Bellosillo, B.; Caux, C.; de Langen, A.; Mazieres, J.; Normanno, N.; Preusser, M.; Provencio, M.; Rojo, F.; Wolf, J.; et al. Immune checkpoint inhibitor treatment in patients with oncogene- addicted non-small cell lung cancer (NSCLC): Summary of a multidisciplinary round-table discussion. ESMO Open 2019, 4, e000498. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Aisner, D.L.; Wood, D.E.; Akerley, W.; Bauman, J.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; Dilling, T.J.; Dobelbower, M.; et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 5.2018. J. Natl. Compr. Canc. Netw. 2018, 16, 807–821. [Google Scholar] [CrossRef]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment with Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch. Pathol. Lab. Med. 2018, 142, 321–346. [Google Scholar]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; McElhinny, A.; Stanforth, D.; Ranger-Moore, J.; Jansson, M.; Kulangara, K.; Richardson, W.; Towne, P.; Hanks, D.; Vennapusa, B.; et al. PD-L1 Immunohistochemistry Assays for Lung Cancer: Results from Phase 1 of the Blueprint PD-L1 IHC Assay Comparison Project. J. Thorac. Oncol. 2017, 12, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Sul, J.; Blumenthal, G.M.; Jiang, X.; He, K.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Patients With Metastatic Non-Small Cell Lung Cancer Whose Tumors Express Programmed Death-Ligand 1. Oncologist 2016, 21, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Tsao, M.S.; Kerr, K.M.; Kockx, M.; Beasley, M.B.; Borczuk, A.C.; Botling, J.; Bubendorf, L.; Chirieac, L.; Chen, G.; Chou, T.Y.; et al. PD-L1 Immunohistochemistry Comparability Study in Real-Life Clinical Samples: Results of Blueprint Phase 2 Project. J. Thorac. Oncol. 2018, 13, 1302–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottrell, T.R.; Taube, J.M. PD-L1 and Emerging Biomarkers in Immune Checkpoint Blockade Therapy. Cancer J. 2018, 24, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- BMS Press Release. Bristol-Myers Squibb Provides Update on the Ongoing Regulatory Review of Opdivo Plus Low-Dose Yervoy in First-Line Lung Cancer Patients with Tumor Mutational Burden ≥10 mut/Mb. Available online: https://news.bms.com/press-release/corporatefinancial-news/bristol-myers-squibb-provides-update-ongoing-regulatory-review (accessed on 19 October 2018).
- Khagi, Y.; Goodman, A.M.; Daniels, G.A.; Patel, S.P.; Sacco, A.G.; Randall, J.M.; Bazhenova, L.A.; Kurzrock, R. Hypermutated Circulating Tumor DNA: Correlation with Response to Checkpoint Inhibitor-Based Immunotherapy. Clin. Cancer Res. 2017, 23, 5729–5736. [Google Scholar] [CrossRef]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef]
- Wang, Z.; Duan, J.; Cai, S.; Han, M.; Dong, H.; Zhao, J.; Zhu, B.; Wang, S.; Zhuo, M.; Sun, J.; et al. Assessment of Blood Tumor Mutational Burden as a Potential Biomarker for Immunotherapy in Patients With Non-Small Cell Lung Cancer With Use of a Next-Generation Sequencing Cancer Gene Panel. JAMA Oncol. 2019, 5, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Ghorani, E.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Lynch, M.; Peggs, K.S.; Swanton, C.; Quezada, S.A. Differential binding affinity of mutated peptides for MHC class I is a predictor of survival in advanced lung cancer and melanoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, F.; Bluthgen, M.V.; Tergemina-Clain, G.; Faivre, L.; Pignon, J.P.; Planchard, D.; Remon, J.; Soria, J.C.; Lacroix, L.; Besse, B. LKB1/STK11 mutations in non-small cell lung cancer patients: Descriptive analysis and prognostic value. Lung Cancer 2017, 112, 62–68. [Google Scholar] [CrossRef]
- Pecuchet, N.; Laurent-Puig, P.; Mansuet-Lupo, A.; Legras, A.; Alifano, M.; Pallier, K.; Didelot, A.; Gibault, L.; Danel, C.; Just, P.A.; et al. Different prognostic impact of STK11 mutations in non-squamous non-small-cell lung cancer. Oncotarget 2017, 8, 23831–23840. [Google Scholar] [CrossRef]
- Skoulidis, F.; Arbour, K.C.; Hellmann, M.D.; Patil, P.D.; Marmarelis, M.E.; Awad, M.M.; Murray, J.C.; Hellyer, J.; Gainor, J.F.; Dimou, A.; et al. Association of STK11/LKB1 genomic alterations with lack of benefit from the addition of pembrolizumab to platinum doublet chemotherapy in non-squamous non-small cell lung cancer. J. Clin. Oncol. 2019, 37, 102. [Google Scholar] [CrossRef]
- Willard, M.D.; Nash Smyth, E.N.; Tiu, R.V.; Beyrer, J.; Zhu, Y.E.; Bowman, L.; Sheffield, K.M.; Han, Y.; Brastianos, P. Genomic characterization of lung tumors and metastatic (Met) sites in advanced (Adv) NSCLC. J. Clin. Oncol. 2019, 37, 2014. [Google Scholar] [CrossRef]
- Schalper, K.A.; Brown, J.; Carvajal-Hausdorf, D.; McLaughlin, J.; Velcheti, V.; Syrigos, K.N.; Herbst, R.S.; Rimm, D.L. Objective measurement and clinical significance of TILs in non-small cell lung cancer. J. Natl. Cancer Inst. 2015, 107, dju435. [Google Scholar] [CrossRef] [PubMed]
- Uryvaev, A.; Passhak, M.; Hershkovits, D.; Sabo, E.; Bar-Sela, G. The role of tumor-infiltrating lymphocytes (TILs) as a predictive biomarker of response to anti-PD1 therapy in patients with metastatic non-small cell lung cancer or metastatic melanoma. Med. Oncol. 2018, 35, 25. [Google Scholar] [CrossRef] [PubMed]
- Hornyak, L.; Dobos, N.; Koncz, G.; Karanyi, Z.; Pall, D.; Szabo, Z.; Halmos, G.; Szekvolgyi, L. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 2018, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Brochez, L.; Chevolet, I.; Kruse, V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Botticelli, A.; Cerbelli, B.; Lionetto, L.; Zizzari, I.; Salati, M.; Pisano, A.; Federica, M.; Simmaco, M.; Nuti, M.; Marchetti, P. Can IDO activity predict primary resistance to anti-PD-1 treatment in NSCLC? J. Transl. Med. 2018, 16, 219. [Google Scholar] [CrossRef] [PubMed]
- Stingele, F.; Corthesy, B.; Kusy, N.; Porcelli, S.A.; Kasper, D.L.; Tzianabos, A.O. Zwitterionic polysaccharides stimulate T cells with no preferential V beta usage and promote anergy, resulting in protection against experimental abscess formation. J. Immunol. 2004, 172, 1483–1490. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chehrazi-Raffle, A.; Placencio-Hickok, V.; Guan, M.; Hendifar, A.; Salgia, R. The gut microbiome and response to immune checkpoint inhibitors: Preclinical and clinical strategies. Clin. Transl. Med. 2019, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Banat, G.A.; Tretyn, A.; Pullamsetti, S.S.; Wilhelm, J.; Weigert, A.; Olesch, C.; Ebel, K.; Stiewe, T.; Grimminger, F.; Seeger, W.; et al. Immune and Inflammatory Cell Composition of Human Lung Cancer Stroma. PLoS ONE 2015, 10, e0139073. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef]
- Friedrich, M.; Jasinski-Bergner, S.; Lazaridou, M.-F.; Subbarayan, K.; Massa, C.; Tretbar, S.; Mueller, A.; Handke, D.; Biehl, K.; Bukur, J.; et al. Tumor-induced escape mechanisms and their association with resistance to checkpoint inhibitor therapy. Cancer Immunol. Immunother. 2019, 1–12. [Google Scholar] [CrossRef]
- Chraa, D.; Naim, A.; Olive, D.; Badou, A. T lymphocyte subsets in cancer immunity: Friends or foes. J. Leukoc. Biol. 2019, 105, 243–255. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Brunner, M.C.; Chambers, C.A.; Chan, F.K.; Hanke, J.; Winoto, A.; Allison, J.P. CTLA-4-Mediated inhibition of early events of T cell proliferation. J. Immunol. 1999, 162, 5813–5820. [Google Scholar] [PubMed]
- Zheng, S.G.; Wang, J.H.; Stohl, W.; Kim, K.S.; Gray, J.D.; Horwitz, D.A. TGF-beta requires CTLA-4 early after T cell activation to induce FoxP3 and generate adaptive CD4+CD25+ regulatory cells. J. Immunol. 2006, 176, 3321–3329. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Subudhi, S.K.; Blando, J.; Scutti, J.; Vence, L.; Wargo, J.; Allison, J.P.; Ribas, A.; Sharma, P. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3(+) Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res. 2019, 25, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Allison, J.P. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 1996, 183, 2533–2540. [Google Scholar] [CrossRef]
- Jain, N.; Nguyen, H.; Chambers, C.; Kang, J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc. Natl. Acad. Sci. USA 2010, 107, 1524–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedline, R.H.; Brown, D.S.; Nguyen, H.; Kornfeld, H.; Lee, J.; Zhang, Y.; Appleby, M.; Der, S.D.; Kang, J.; Chambers, C.A. CD4+ regulatory T cells require CTLA-4 for the maintenance of systemic tolerance. J. Exp. Med. 2009, 206, 421–434. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.-J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Bhattacharya, P.; Prabhakar, B.S. A comprehensive review on the role of co-signaling receptors and Treg homeostasis in autoimmunity and tumor immunity. J. Autoimmun. 2018, 95, 77–99. [Google Scholar] [CrossRef]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef]
- Sheppard, K.A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Bennett, F.; Luxenberg, D.; Ling, V.; Wang, I.M.; Marquette, K.; Lowe, D.; Khan, N.; Veldman, G.; Jacobs, K.A.; Valge-Archer, V.E.; et al. Program death-1 engagement upon TCR activation has distinct effects on costimulation and cytokine-driven proliferation: Attenuation of ICOS, IL-4, and IL-21, but not CD28, IL-7, and IL-15 responses. J. Immunol. 2003, 170, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Kato, R.; Yamasaki, M.; Urakawa, S.; Nishida, K.; Makino, T.; Morimoto-Okazawa, A.; Kawashima, A.; Iwahori, K.; Suzuki, S.; Ueda, R.; et al. Increased Tim-3(+) T cells in PBMCs during nivolumab therapy correlate with responses and prognosis of advanced esophageal squamous cell carcinoma patients. Cancer Immunol. Immunother. 2018, 67, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther. 2018, 11, 7005–7009. [Google Scholar] [CrossRef]
- Brummelman, J.; Mazza, E.M.C.; Alvisi, G.; Colombo, F.S.; Grilli, A.; Mikulak, J.; Mavilio, D.; Alloisio, M.; Ferrari, F.; Lopci, E.; et al. High-dimensional single cell analysis identifies stem-like cytotoxic CD8(+) T cells infiltrating human tumors. J. Exp. Med. 2018, 215, 2520–2535. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Akerley, W.; Borghaei, H.; Chang, A.C.; Cheney, R.T.; Chirieac, L.R.; D’Amico, T.A.; Demmy, T.L.; Ganti, A.K.P.; Govindan, R.; et al. Non-small cell lung cancer. J. Natl. Compr. Canc. Netw. 2012, 10, 1236–1271. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013, 20, 456–464. [Google Scholar] [CrossRef]
- Phillips, J.D.; Knab, L.M.; Blatner, N.R.; Haghi, L.; DeCamp, M.M.; Meyerson, S.L.; Heiferman, M.J.; Heiferman, J.R.; Gounari, F.; Bentrem, D.J.; et al. Preferential expansion of pro-inflammatory Tregs in human non-small cell lung cancer. Cancer Immunol. Immunother. 2015, 64, 1185–1191. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Contribution of regulatory T cells to cancer: A review. J. Cell. Physiol. 2019, 234, 7983–7993. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Prognostic Biomarker | Current State of Development |
|---|---|
| PD-L1 expression | FDA-approved and fully implemented in clinical practice |
| Tumor mutational burden | Under investigation |
| Differential agretopicity index | Under investigation |
| STK11 mutations | Under investigation |
| High levels of tumor infiltrating lymphocytes (CD4+, CD8+, CD8+/CD4+ ratio) | Under investigation |
| Kynurenine/tryptophan ratios | Under investigation |
| Quinolinic acid concentrations | Under investigation |
| Gut microbiome | Under investigation |
| ICI Treatment | Pivotal Trial | Setting | Target Population | FDA Approval |
|---|---|---|---|---|
| Nivolumab monotherapy versus docetaxel | CheckMate017 | II line after chemotherapy failure | Stage III-B or IV Squamous NSCLC | March 2015 |
| Nivolumab monotherapy versus docetaxel | CheckMate057 | II line after chemotherapy failure | Stage III-B or IV Nonsquamous NSCLC | October 2015 |
| Pembrolizumab monotherapy versus platinum- based chemotherapy | KEYNOTE-024 | I line (PD-L1 ≥ 50%) | Stage IV Nonsquamous and squamous NSCLC | October 2016 |
| Pembrolizumab monotherapy versus docetaxel | KEYNOTE-010 | II line after chemotherapy failure (PD-L1 ≥ 1%) | Nonsquamous and squamous NSCLC | October 2016 |
| Atezolizumab monotherapy | OAK | II line after chemotherapy failure | Stage III-B or IV Nonsquamous and squamous NSCLC | October 2016 |
| Durvalumab monotherapy versus placebo | PACIFIC | Durvalumab after chemoradiotherapy | Stage III unresectable Nonsquamous and squamous NSCLC | February 2018 |
| Pembrolizumab + cis/carboplatin + pemetrexed | KEYNOTE-189 | I line | Nonsquamous NSCLC | August 2018 |
| Pembrolizumab + paclitaxel/nab-paclitaxel + carboplatin | KEYNOTE-407 | I line | Stage IV Squamous NSCLC | October 2018 |
| Atezolizumab + carboplatin + paclitaxel + bevacizumab | IMpower 150 | I line | Stage IV or recurrent metastatic Nonsquamous NSCLC | December 2018 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianco, A.; Perrotta, F.; Barra, G.; Malapelle, U.; Rocco, D.; De Palma, R. Prognostic Factors and Biomarkers of Responses to Immune Checkpoint Inhibitors in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 4931. https://doi.org/10.3390/ijms20194931
Bianco A, Perrotta F, Barra G, Malapelle U, Rocco D, De Palma R. Prognostic Factors and Biomarkers of Responses to Immune Checkpoint Inhibitors in Lung Cancer. International Journal of Molecular Sciences. 2019; 20(19):4931. https://doi.org/10.3390/ijms20194931
Chicago/Turabian StyleBianco, Andrea, Fabio Perrotta, Giusi Barra, Umberto Malapelle, Danilo Rocco, and Raffaele De Palma. 2019. "Prognostic Factors and Biomarkers of Responses to Immune Checkpoint Inhibitors in Lung Cancer" International Journal of Molecular Sciences 20, no. 19: 4931. https://doi.org/10.3390/ijms20194931
APA StyleBianco, A., Perrotta, F., Barra, G., Malapelle, U., Rocco, D., & De Palma, R. (2019). Prognostic Factors and Biomarkers of Responses to Immune Checkpoint Inhibitors in Lung Cancer. International Journal of Molecular Sciences, 20(19), 4931. https://doi.org/10.3390/ijms20194931