Replication Stress at Telomeric and Mitochondrial DNA: Common Origins and Consequences on Ageing


Abstract
:1. Introduction: Senescence and Ageing
2. The Contribution of Telomeres to Senescence and Ageing
2.1. Replicative Senescence or Replication Stress-Induced Senescence
2.2. Different Sources of Replication Stress at Telomeres
2.3. Oxidative Stress at Telomeres: Origin and Regulation
3. The Origin of the Mitochondrial Metabolic Compromise during Ageing
4. mtDNA and telDNA Maintenance: The Same Causes Produce the Same Effects
4.1. D-loops
4.2. Epigenetic Regulation
4.3. G-Quadruplex
4.4. RNA:DNA Hybrids
4.5. Torsion and Supercoiling
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5hmC | 5-hydroxymethylcytosine |
| adPEO | autosomal dominant progressive external ophthalmoplegia |
| ALT | alternative lengthening of telomeres |
| CDKI | CDKI cyclin-dependent kinase inhibitor |
| DDR | DNA damage repair |
| D-loop | displacement loop |
| DNMT | DNA methyltransferases |
| ECT | electron chain transport |
| G4 | G-quadruplex |
| HHS | Hoyeraal-Hreidarsson syndrome |
| HSP | heavy strand promoter |
| LSP | light strand promoter |
| MMF | mitochondrial metabolic failure |
| mtDNA | mitochondrial DNA |
| mt-lncRNA | long-non coding mtRNA |
| mt-lncRNA | long-non coding mtRNA |
| MTS | mitochondrial targeting signal |
| nDNA | nuclear DNA |
| OH | heavy strand origin of replication |
| OL | ligth strand origin of replication |
| ORC | origin replication complex |
| RdRP | RNA-dependent RNA Polymerase |
| RITOLS | ribonucleotide incorporation throughout the lagging strand |
| ROS | reactive oxygen species |
| SASP | senescence-associated secretory phenotype |
| telDNA | telomeric DNA |
| TET | ten-eleven-translocation |
| T-loop | telomeric loop |
References
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular Senescence in Aging Primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.-M.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Gui, D.Y.; Heiden, M.G.V. Altered metabolite levels in cancer: Implications for tumour biology and cancer therapy. Nat. Rev. Cancer 2016, 16, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Martínez, P.; Blasco, M.A. Telomere-driven diseases and telomere-targeting therapies. J. Cell Biol. 2017, 216, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, K.L.; Chang, S.; Lee, H.W.; Blasco, M.A.; Gottlieb, G.J.; Greider, C.; DePinho, R.A. Longevity, Stress Response, and Cancer in Aging Telomerase-Deficient Mice. Cell 1999, 96, 701–712. [Google Scholar] [CrossRef] [Green Version]
- Jaskelioff, M.; Muller, F.L.; Paik, J.H.; Thomas, E.; Jiang, S.; Adams, A.C.; Sahin, E.; Kost-Alimova, M.; Protopopov, A.; Cadiñanos, J.; et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 2011, 469, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.; Blasco, M.A. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat. Rev. Cancer 2011, 11, 161–176. [Google Scholar] [CrossRef]
- Arndt, G.M.; MacKenzie, K.L. New prospects for targeting telomerase beyond the telomere. Nat. Rev. Cancer 2016, 16, 508–524. [Google Scholar] [CrossRef] [PubMed]
- Savage, S.A. Beginning at the ends: Telomeres and human disease. F1000 Res. 2018, 7, 1–15. [Google Scholar] [CrossRef]
- De Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Verdun, R.E.; Karlseder, J. Replication and protection of telomeres. Nature 2007, 447, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-Y.; Redon, S.; Lingner, J. The human CST complex is a terminator of telomerase activity. Nature 2012, 488, 540–544. [Google Scholar] [CrossRef]
- Feng, X.; Hsu, S.-J.; Bhattacharjee, A.; Wang, Y.; Diao, J.; Price, C.M. CTC1-STN1 terminates telomerase while STN1-TEN1 enables C-strand synthesis during telomere replication in colon cancer cells. Nat. Commun. 2018, 9, 2827. [Google Scholar] [CrossRef] [Green Version]
- Glousker, G.; Touzot, F.; Revy, P.; Tzfati, Y.; Savage, S.A. Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Br. J. Haematol. 2015, 170, 457–471. [Google Scholar] [CrossRef]
- Simon, A.J.; Lev, A.; Zhang, Y.; Weiss, B.; Rylova, A.; Eyal, E.; Kol, N.; Barel, O.; Cesarkas, K.; Soudack, M.; et al. Mutations in STN1 cause Coats plus syndrome and are associated with genomic and telomere defects. J. Exp. Med. 2016, 213, 1429–1440. [Google Scholar] [CrossRef]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beauséjour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nature 2012, 14, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Parkinson, G.N.; Lee, M.P.H.; Neidle, S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 2002, 417, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Azzalin, C.M.; Reichenbach, P.; Khoriauli, L.; Giulotto, E.; Lingner, J. Telomeric Repeat Containing RNA and RNA Surveillance Factors at Mammalian Chromosome Ends. Science 2007, 318, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Doksani, Y.; Wu, J.Y.; De Lange, T.; Zhuang, X. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell 2013, 155, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; De Lange, T. Mammalian Telomeres End in a Large Duplex Loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Vannier, J.-B.; Sandhu, S.; Petalcorin, M.I.; Wu, X.; Nabi, Z.; Ding, H.; Boulton, S.J. RTEL1 Is a Replisome-Associated Helicase That Promotes Telomere and Genome-Wide Replication. Science 2013, 342, 239–242. [Google Scholar] [CrossRef]
- Leman, A.R.; Dheekollu, J.; Deng, Z.; Lee, S.W.; Das, M.M.; Lieberman, P.M.; Noguchi, E. Timeless preserves telomere length by promoting efficient DNA replication through human telomeres. Cell Cycle 2012, 11, 2337–2347. [Google Scholar] [CrossRef] [Green Version]
- Vannier, J.-B.; Pavicic-Kaltenbrunner, V.; Petalcorin, M.I.; Ding, H.; Boulton, S.J. RTEL1 Dismantles T Loops and Counteracts Telomeric G4-DNA to Maintain Telomere Integrity. Cell 2012, 149, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Opresko, P.L.; Mason, P.A.; Podell, E.R.; Lei, M.; Hickson, I.D.; Cech, T.R.; Bohr, V.A.; Opresko, P. POT1 Stimulates RecQ Helicases WRN and BLM to Unwind Telomeric DNA Substrates. J. Biol. Chem. 2005, 280, 32069–32080. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, M.; Kibe, T.; Kabir, S.; De Lange, T. TRF1 negotiates TTAGGG repeat-associated replication problems by recruiting the BLM helicase and the TPP1/POT1 repressor of ATR signaling. Genome Res. 2014, 28, 2477–2491. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yang, J.; Wild, A.T.; Wu, W.H.; Shah, R.; Danussi, C.; Riggins, G.J.; Kannan, K.; Sulman, E.P.; Chan, T.A.; et al. G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat. Commun. 2019, 10, 943. [Google Scholar] [CrossRef]
- Lin, W.; Sampathi, S.; Dai, H.; Liu, C.; Zhou, M.; Hu, J.; Huang, Q.; Campbell, J.; Shin-Ya, K.; Zheng, L.; et al. Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 2013, 32, 1425–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.T.; Voon, H.P.J.; Xella, B.; Scott, C.; Clynes, D.; Babbs, C.; Ayyub, H.; Kerry, J.; Sharpe, J.A.; Sloane-Stanley, J.A.; et al. The chromatin remodelling factor ATRX suppresses R-loops in transcribed telomeric repeats. EMBO Rep. 2017, 18, 914–928. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Lee, Y.; Wischnewski, H.; Brun, C.M.; Schwarz, T.; Azzalin, C.M. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 2014, 5, 5220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teasley, D.C.; Parajuli, S.; Nguyen, M.; Moore, H.R.; Alspach, E.; Lock, Y.J.; Honaker, Y.; Saharia, A.; Piwnica-Worms, H.; Stewart, S.A. Flap Endonuclease 1 Limits Telomere Fragility on the Leading Strand*. J. Biol. Chem. 2015, 290, 15133–15145. [Google Scholar] [CrossRef] [PubMed]
- Germe, T.; Miller, K.; Cooper, J.P. A non-canonical function of topoisomerase II in disentangling dysfunctional telomeres. EMBO J. 2009, 28, 2803–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, M.H.; Tsai, C.H.; Lin, C.C.; Li, T.K.; Hung, T.W.; Chang, L.T.; Hsin, L.W.; Teng, S.C. Topoisomerase II inhibition suppresses the proliferation of telomerase-negative cancers. Cell. Mol. Life Sci. CMLS 2015, 72, 1825–1837. [Google Scholar] [CrossRef]
- Ye, J.; Lenain, C.; Bauwens, S.; Rizzo, A.; Saint-Léger, A.; Poulet, A.; Benarroch, D.; Magdinier, F.; Morere, J.; Amiard, S.; et al. TRF2 and apollo cooperate with topoisomerase 2alpha to protect human telomeres from replicative damage. Cell 2010, 142, 230–242. [Google Scholar] [CrossRef]
- Sarek, G.; Vannier, J.B.; Panier, S.; Petrini, J.H.; Boulton, S.J. TRF2 recruits RTEL1 to telomeres in S phase to promote t-loop unwinding. Mol. Cell 2015, 57, 622–635. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Rossi, M.L.; Singh, D.K.; Dunn, C.; Ramamoorthy, M.; Croteau, D.L.; Liu, Y.; Bohr, V.A. RECQL4, the Protein Mutated in Rothmund-Thomson Syndrome, Functions in Telomere Maintenance. J. Biol. Chem. 2012, 287, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Higa, M.; Kushiyama, T.; Kurashige, S.; Kohmon, D.; Enokitani, K.; Iwahori, S.; Sugimoto, N.; Yoshida, K.; Fujita, M. TRF2 recruits ORC through TRFH domain dimerization. Biochim. Biophys. Acta BBA Bioenerg. 2017, 1864, 191–201. [Google Scholar] [CrossRef]
- Loayza, D.; De Lange, T. POT1 as a terminal transducer of TRF1 telomere length control. Nature 2003, 423, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Zaug, A.J.; Podell, E.R.; Cech, T.R. Human POT1 disrupts telomeric G-quadruplexes allowing telomerase extension in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 10864–10869. [Google Scholar] [CrossRef] [PubMed]
- Martínez, P.; Thanasoula, M.; Muñoz, P.; Liao, C.; Tejera, A.; McNees, C.; Flores, J.M.; Fernandez-Capetillo, O.; Tarsounas, M.; Blasco, M.A. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genome Res. 2009, 23, 2060–2075. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A.; Kosiyatrakul, S.T.; Hockemeyer, D.; Macrae, S.L.; Karlseder, J.; Schildkraut, C.L.; De Lange, T. Mammalian Telomeres Resemble Fragile Sites and Require TRF1 for Efficient Replication. J. End End Test. 2009, 138, 90–103. [Google Scholar] [CrossRef]
- D’alcontres, M.S.; Palacios, A.; Mejias, D.; Blasco, M.A.; Palacios, J.A. TopoIIα prevents telomere fragility and formation of ultra thin DNA bridges during mitosis through TRF1-dependent binding to telomeres. Cell Cycle 2014, 13, 1463–1481. [Google Scholar]
- Takai, K.K.; Hooper, S.; Blackwood, S.; Gandhi, R.; de Lange, T. In vivo stoichiometry of shelterin components. J. Biol. Chem. 2010, 285, 1457–1467. [Google Scholar] [CrossRef]
- Sugimoto, M. A cascade leading to premature aging phenotypes including abnormal tumor profiles in Werner syndrome (review). Int. J. Mol. Med. 2014, 33, 247–253. [Google Scholar] [CrossRef]
- Lu, L.; Jin, W.; Wang, L.L. Aging in Rothmund-Thomson syndrome and related RECQL4 genetic disorders. Ageing Res. Rev. 2017, 33, 30–35. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Sanderson, S.L.; Simon, A.K. In aged primary T cells, mitochondrial stress contributes to telomere attrition measured by a novel imaging flow cytometry assay. Aging Cell 2017, 16, 1234–1243. [Google Scholar] [CrossRef]
- Liu, L.; Trimarchi, J.R.; Smith, P.J.S.; Keefe, D.L. Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell 2002, 1, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef] [PubMed]
- Oexle, K.; Zwirner, A. Advanced Telomere Shortening in Respiratory Chain Disorders. Hum. Mol. Genet. 1997, 6, 905–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.C.; Min, R.; Ji, J.J.; Zhang, S.; Tong, A.L.; Xu, J.P.; Li, Z.Y.; Zhang, H.B.; Li, Y.X. Analysis of association among clinical features and shorter leukocyte telomere length in mitochondrial diabetes with m.3243A>G mitochondrial DNA mutation. BMC Med. Genet. 2015, 16, 92. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Kawanishi, S. Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett. 1999, 453, 365–368. [Google Scholar] [CrossRef] [Green Version]
- Petersen, S.; Saretzki, G.; Von Zglinicki, T. Preferential Accumulation of Single-Stranded Regions in Telomeres of Human Fibroblasts. Exp. Cell Res. 1998, 239, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T.; Pilger, R.; Sitte, N. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic. Biol. Med. 2000, 28, 64–74. [Google Scholar] [CrossRef]
- Opresko, P.L.; Fan, J.; Danzy, S.; Wilson, D.M.; Bohr, V.A. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 2005, 33, 1230–1239. [Google Scholar] [CrossRef]
- Aeby, E.; Ahmed, W.; Redon, S.; Simanis, V.; Lingner, J. Peroxiredoxin 1 Protects Telomeres from Oxidative Damage and Preserves Telomeric DNA for Extension by Telomerase. Cell Rep. 2016, 17, 3107–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uziel, O.; Reshef, H.; Ravid, A.; Fabian, I.; Halperin, D.; Ram, R.; Bakhanashvili, M.; Nordenberg, J.; Lahav, M. Oxidative stress causes telomere damage in Fanconi anaemia cells—A possible predisposition for malignant transformation. Br. J. Haematol. 2008, 142, 82–93. [Google Scholar] [CrossRef]
- Fouquerel, E.; Barnes, R.P.; Uttam, S.; Watkins, S.C.; Bruchez, M.P.; Opresko, P.L. Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Mol. Cell 2019, 75, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Van Der Reest, J.; Lilla, S.; Zheng, L.; Zanivan, S.; Gottlieb, E. Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat. Commun. 2018, 9, 1581. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Lingner, J. PRDX1 and MTH1 cooperate to prevent ROS-mediated inhibition of telomerase. Genes Dev. 2018, 32, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Spilsbury, A.; Miwa, S.; Attems, J.; Saretzki, G. The role of telomerase protein TERT in Alzheimer’s disease and in tau-related pathology in vitro. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 1659–1674. [Google Scholar] [CrossRef] [PubMed]
- Eitan, E.; Braverman, C.; Tichon, A.; Gitler, D.; Hutchison, E.R.; Mattson, M.P.; Priel, E.; Braverman, C. Excitotoxic and Radiation Stress Increase TERT Levels in the Mitochondria and Cytosol of Cerebellar Purkinje Neurons. Cerebellum 2016, 15, 509–517. [Google Scholar] [CrossRef]
- Zhuang, X.Y.; Yao, Y.G. Mitochondrial dysfunction and nuclear-mitochondrial shuttling of TERT are involved in cell proliferation arrest induced by G-quadruplex ligands. FEBS Lett. 2013, 587, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Piciocchi, M.; Cardin, R.; Cillo, U.; Vitale, A.; Cappon, A.; Mescoli, C.; Guido, M.; Rugge, M.; Burra, P.; Floreani, A.; et al. Differential timing of oxidative DNA damage and telomere shortening in hepatitis C and B virus–related liver carcinogenesis. Transl. Res. 2016, 168, 122–133. [Google Scholar] [CrossRef]
- Campelo, R.; Díaz Lozano, I.; Figarella, K.; Osuna, A.; Ramírez, J.L. Leishmania major telomerase TERT protein has a nuclear/mitochondrial eclipsed distribution that is affected by oxidative stress. Infect. Immun. 2015, 83, 57–66. [Google Scholar] [CrossRef]
- Miwa, S.; Czapiewski, R.; Wan, T.; Bell, A.; Hill, K.N.; Von Zglinicki, T.; Saretzki, G. Decreased mTOR signalling reduces mitochondrial ROS in brain via accumulation of the telomerase protein TERT within mitochondria. Aging 2016, 8, 2551–2564. [Google Scholar] [CrossRef]
- Haendeler, J.; Drose, S.; Buchner, N.; Jakob, S.; Altschmied, J.; Goy, C.; Spyridopoulos, I.; Zeiher, A.M.; Brandt, U.; Dimmeler, S.; et al. Mitochondrial Telomerase Reverse Transcriptase Binds to and Protects Mitochondrial DNA and Function From Damage. Arter. Thromb. Vasc. Biol. 2009, 29, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Haendeler, J.; Hoffmann, J.; Brandes, R.P.; Zeiher, A.M.; Dimmeler, S. Hydrogen Peroxide Triggers Nuclear Export of Telomerase Reverse Transcriptase Via Src Kinase Family-Dependent Phosphorylation of Tyrosine 707; F1000 (Faculty of 1000 Ltd): London, UK, 2003; Volume 23, pp. 4598–4610. [Google Scholar]
- Santos, J.H.; Meyer, J.N.; Skorvaga, M.; Annab, L.A.; Van Houten, B. Mitochondrial hTERT exacerbates free-radical-mediated mtDNA damage. Aging Cell 2004, 3, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.K.; Reyes, A.; Green, P.; Caron, M.J.; Bonini, M.G.; Gordon, D.M.; Holt, I.J.; Santos, J.H. Human telomerase acts as a hTR-independent reverse transcriptase in mitochondria. Nucleic Acids Res. 2012, 40, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Singhapol, C.; Pal, D.; Czapiewski, R.; Porika, M.; Nelson, G.; Saretzki, G.C. Mitochondrial Telomerase Protects Cancer Cells from Nuclear DNA Damage and Apoptosis. PLoS ONE 2013, 8, e52989. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Passos, J.F.; Birket, M.J.; Beckmann, T.; Brings, S.; Peters, H.; Birch-Machin, M.A.; Von Zglinicki, T.; Saretzki, G. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J. Cell Sci. 2008, 121, 1046–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, P.D.; Sharma, N.K.; Santos, J.H. Telomerase Impinges on the Cellular Response to Oxidative Stress Through Mitochondrial ROS-Mediated Regulation of Autophagy. Int. J. Mol. Sci. 2019, 20, 1509. [Google Scholar] [CrossRef]
- Maida, Y.; Yasukawa, M.; Masutomi, K. De Novo RNA Synthesis by RNA-Dependent RNA Polymerase Activity of Telomerase Reverse Transcriptase. Mol. Cell. Biol. 2016, 36, 1248–1259. [Google Scholar] [CrossRef]
- Maida, Y.; Yasukawa, M.; Furuuchi, M.; Lassmann, T.; Possemato, R.; Okamoto, N.; Kasim, V.; Hayashizaki, Y.; Hahn, W.C.; Masutomi, K. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature 2009, 461, 230–235. [Google Scholar] [CrossRef]
- De Jesus, B.B.; Vera, E.; Schneeberger, K.; Tejera, A.M.; Ayuso, E.; Bosch, F.; Blasco, M.A. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol. Med. 2012, 4, 691–704. [Google Scholar] [CrossRef]
- Vera, E.; De Jesus, B.B.; Foronda, M.; Flores, J.M.; Blasco, M.A. Telomerase Reverse Transcriptase Synergizes with Calorie Restriction to Increase Health Span and Extend Mouse Longevity. PLoS ONE 2013, 8, e53760. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, P.; Zheng, Q.; Gao, G.; Yuan, J.; Wang, P.; Huang, J.; Xie, L.; Lu, X.; Tong, T.; et al. Mitochondrial Trafficking and Processing of Telomerase RNA TERC. Cell Rep. 2018, 24, 2589–2595. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.Y.; Zhang, Y.; Zhang, Q.; Li, H.; Luo, Z.; Fang, H.; Kim, S.H.; Qin, L.; Yotnda, P.; Xu, J.; et al. Mitochondrial localization of telomeric protein TIN2 links telomere regulation to metabolic control. Mol. Cell 2012, 47, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Li, F.; He, Q.; Deng, T.; Xu, J.; Jin, F.; Coarfa, C.; Putluri, N.; Liu, D.; Songyang, Z. Systematic analysis of human telomeric dysfunction using inducible telosome/shelterin CRISPR/Cas9 knockout cells. Cell Discov. 2017, 3, 17034. [Google Scholar] [CrossRef] [PubMed]
- Angrisani, A.; Turano, M.; Paparo, L.; Di Mauro, C.; Furia, M. A new human dyskerin isoform with cytoplasmic localization. Biochim. Biophys. Acta (BBA) Gen. Subj. 2011, 1810, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Angrisani, A.; Matrone, N.; Belli, V.; Vicidomini, R.; Di Maio, N.; Turano, M.; Scialò, F.; Netti, P.A.; Porcellini, A.; Furia, M. A functional connection between dyskerin and energy metabolism. Redox Biol. 2018, 14, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Vasileiou, P.V.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Passos, J.F. Mitochondria: Are they causal players in cellular senescence? Biochim. Biophys. Acta 2015, 1847, 1373–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halsne, R.; Esbensen, Y.; Wang, W.; Scheffler, K.; Suganthan, R.; Bjørås, M.; Eide, L. Lack of the DNA glycosylases MYH and OGG1 in the cancer prone double mutant mouse does not increase mitochondrial DNA mutagenesis. DNA Repair 2012, 11, 278–285. [Google Scholar] [CrossRef]
- Kauppila, J.H.K.; Bonekamp, N.A.; Mourier, A.; Isokallio, M.A.; Just, A.; Kauppila, T.E.S.; Stewart, J.B.; Larsson, N.G. Base-excision repair deficiency alone or combined with increased oxidative stress does not increase mtDNA point mutations in mice. Nucleic Acids Res. 2018, 46, 6642–6669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, S.R.; Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Ultra-Sensitive Sequencing Reveals an Age-Related Increase in Somatic Mitochondrial Mutations That Are Inconsistent with Oxidative Damage. PLoS Genet. 2013, 9, e1003794. [Google Scholar] [CrossRef]
- Zheng, W.; Khrapko, K.; Coller, H.A.; Thilly, W.G.; Copeland, W.C. Origins of human mitochondrial point mutations as DNA polymerase gamma-mediated errors. Mutat. Res. 2006, 599, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.F.; Millet, A.R.; Tigano, M.; Dubois, S.M.; Crimmins, H.; Babin, L.; Charpentier, M.; Piganeau, M.; Brunet, E.; Sfeir, A. Single-Molecule Analysis of mtDNA Replication Uncovers the Basis of the Common Deletion. Mol. Cell 2017, 65, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Beal, M.F.; Wallace, D.C. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Pang, C.Y.; Hsu, H.S.; Wei, Y.H. Differential accumulations of 4977 bp deletion in mitochondrial DNA of various tissues in human ageing. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1994, 1226, 37–43. [Google Scholar] [CrossRef]
- Inoue, K.; Nakada, K.; Ogura, A.; Isobe, K.; Goto, Y.I.; Nonaka, I.; Hayashi, J.I. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat. Genet. 2000, 26, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Nakada, K.; Inoue, K.; Ono, T.; Isobe, K.; Ogura, A.; Goto, Y.-I.; Nonaka, I.; Hayashi, J.-I. Inter-mitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat. Med. 2001, 7, 934–940. [Google Scholar] [CrossRef]
- Persson, Ö.; Muthukumar, Y.; Basu, S.; Jenninger, L.; Uhler, J.P.; Berglund, A.-K.; McFarland, R.; Taylor, R.W.; Gustafsson, C.M.; Larsson, E.; et al. Copy-choice recombination during mitochondrial L-strand synthesis causes DNA deletions. Nat. Commun. 2019, 10, 759. [Google Scholar] [CrossRef]
- Rahman, S.; Copeland, W.C. POLG-related disorders and their neurological manifestations. Nat. Rev. Neurol. 2019, 15, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, T.J.; Minczuk, M. In D-loop: 40 years of mitochondrial 7S DNA. Exp. Gerontol. 2014, 56, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.-G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef]
- Chi, Z.; Nie, L.; Peng, Z.; Yang, Q.; Yang, K.; Tao, J.; Mi, Y.; Fang, X.; Balajee, A.S.; Zhao, Y. RecQL4 cytoplasmic localization: Implications in mitochondrial DNA oxidative damage repair. Int. J. Biochem. Cell Biol. 2012, 44, 1942–1951. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-T.; Xu, X.; Alontaga, A.Y.; Chen, Y.; Liu, Y. Impaired p32 regulation caused by the lymphoma-prone RECQ4 mutation drives mitochondrial dysfunction. Cell Rep. 2014, 7, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Saxena, S.; Kaushal, A.; Nagaraju, G. RAD51C/XRCC3 Facilitates Mitochondrial DNA Replication and Maintains Integrity of the Mitochondrial Genome. Mol. Cell. Biol. 2018, 38, e00489. [Google Scholar] [CrossRef] [PubMed]
- Herbers, E.; Kekäläinen, N.J.; Hangas, A.; Pohjoismäki, J.L.; Goffart, S. Tissue specific differences in mitochondrial DNA maintenance and expression. Mitochondrion 2018, 44, 85–92. [Google Scholar] [CrossRef]
- Pohjoismäki, J.L.O.; Goffart, S.; Tyynismaa, H.; Willcox, S.; Ide, T.; Kang, D.; Suomalainen, A.; Karhunen, P.J.; Griffith, J.D.; Holt, I.J.; et al. Human heart mitochondrial DNA is organized in complex catenated networks containing abundant four-way junctions and replication forks. J. Biol. Chem. 2009, 284, 21446–21457. [Google Scholar] [CrossRef] [PubMed]
- Kraytsberg, Y. Recombination of Human Mitochondrial DNA. Science 2004, 304, 981. [Google Scholar] [CrossRef] [PubMed]
- Sobinoff, A.P.; Pickett, H.A. Alternative Lengthening of Telomeres: DNA Repair Pathways Converge. Trends Genet. 2017, 33, 921–932. [Google Scholar] [CrossRef]
- Ngo, H.B.; Lovely, G.A.; Phillips, R.; Chan, D.C. Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat. Commun. 2014, 5, 3077. [Google Scholar] [CrossRef] [PubMed]
- Benarroch-Popivker, D.; Pisano, S.; Mendez-Bermudez, A.; Lototska, L.; Kaur, P.; Bauwens, S.; Djerbi, N.; Latrick, C.M.; Fraisier, V.; Pei, B.; et al. TRF2-Mediated Control of Telomere DNA Topology as a Mechanism for Chromosome-End Protection. Mol. Cell 2016, 61, 274–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potts, P.R.; Yu, H. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat. Struct. Mol. Biol. 2007, 14, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [PubMed]
- García-Expósito, L.; Bournique, E.; Bergoglio, V.; Bose, A.; Barroso-González, J.; Zhang, S.; Roncaioli, J.L.; Lee, M.; Wallace, C.T.; Watkins, S.C.; et al. Proteomic Profiling Reveals a Specific Role for Translesion DNA Polymerase η in the Alternative Lengthening of Telomeres. Cell Rep. 2016, 17, 1858–1871. [Google Scholar] [CrossRef] [PubMed]
- Tadi, S.K.; Sebastian, R.; Dahal, S.; Babu, R.K.; Choudhary, B.; Raghavan, S.C. Microhomology-mediated end joining is the principal mediator of double-strand break repair during mitochondrial DNA lesions. Mol. Biol. Cell 2016, 27, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Stroik, S.; Kurtz, K.; Hendrickson, E.A. CtIP is essential for telomere replication. Nucleic Acids Res. 2019, 47, 8927–8940. [Google Scholar] [CrossRef] [Green Version]
- Rivera, T.; Haggblom, C.; Cosconati, S.; Karlseder, J. A balance between elongation and trimming regulates telomere stability in stem cells. Nat. Struct. Mol. Biol. 2017, 24, 30–39. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Arnoult, N.; Lackner, D.H.; Oganesian, L.; Haggblom, C.; Corpet, A.; Almouzni, G.; Karlseder, J. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol. 2014, 21, 167–174. [Google Scholar] [CrossRef]
- Tomaska, L.; Nosek, J.; Kar, A.; Willcox, S.; Griffith, J.D. A New View of the T-Loop Junction: Implications for Self-Primed Telomere Extension, Expansion of Disease-Related Nucleotide Repeat Blocks, and Telomere Evolution. Front. Genet. 2019, 10, 792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisnovsky, S.; Jean, S.R.; Liyanage, S.; Schimmer, A.; Kelley, S.O.; Kelley, S. Mitochondrial DNA repair and replication proteins revealed by targeted chemical probes. Nat. Methods 2016, 12, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Du, Q.; Chen, L.; Fu, G.; Li, S.; Fu, L.; Zhang, X.; Ma, C.; Bin, C. CpG methylation patterns of human mitochondrial DNA. Sci. Rep. 2016, 6, 23421. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.K.; Mangalhara, K.C.; Prakasam, G.; Bamezai, R.N.K. DNA Methyltransferase1 (DNMT1) Isoform3 methylates mitochondrial genome and modulates its biology. Sci. Rep. 2017, 7, 1525. [Google Scholar] [CrossRef] [PubMed]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, M.; Gertz, B.; Chestnut, B.A.; Martin, L.J. Mitochondrial DNMT3A and DNA methylation in skeletal muscle and CNS of transgenic mouse models of ALS. Front. Cell. Neurosci. 2013, 7, 279. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Du, Z.; Pian, L.; Li, T.; Wen, X.; Li, W.; Kim, S.J.; Xiao, J.; Cohen, P.; Cui, J.; et al. Mitochondrial DNA Hypomethylation Is a Biomarker Associated with Induced Senescence in Human Fetal Heart Mesenchymal Stem Cells. Stem Cells Int. 2017, 2017, 1764549. [Google Scholar] [CrossRef] [PubMed]
- Blanch, M.; Mosquera, J.L.; Ansoleaga, B.; Ferrer, I.; Barrachina, M. Altered Mitochondrial DNA Methylation Pattern in Alzheimer Disease–Related Pathology and in Parkinson Disease. Am. J. Pathol. 2016, 186, 385–397. [Google Scholar] [CrossRef]
- Gonzalo, S.; Jaco, I.; Fraga, M.F.; Chen, T.; Li, E.; Esteller, M.; Blasco, M.A. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nature 2006, 8, 416–424. [Google Scholar] [CrossRef]
- Dan, J.; Rousseau, P.; Hardikar, S.; Veland, N.; Wong, J.; Autexier, C.; Chen, T. Zscan4 Inhibits Maintenance DNA Methylation to Facilitate Telomere Elongation in Mouse Embryonic Stem Cells. Cell Rep. 2017, 20, 1936–1949. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Sengupta, S.; Scaria, V. Hydroxymethyl cytosine marks in the human mitochondrial genome are dynamic in nature. Mitochondrion 2016, 27, 25–31. [Google Scholar] [CrossRef]
- Chen, H.; Dzitoyeva, S.; Manev, H. Effect of valproic acid on mitochondrial epigenetics. Eur. J. Pharmacol. 2012, 690, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Guo, R.; Wang, H.; Ye, X.; Zhou, Z.; Dan, J.; Wang, H.; Gong, P.; Deng, W.; Yin, Y.; et al. Tet Enzymes Regulate Telomere Maintenance and Chromosomal Stability of Mouse ESCs. Cell Rep. 2016, 15, 1809–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novakovic, B.; Napier, C.E.; Vryer, R.; Dimitriadis, E.; Manuelpillai, U.; Sharkey, A.; Craig, J.M.; Reddel, R.R.; Saffery, R. DNA methylation mediated up-regulation of TERRA non-coding RNA is coincident with elongated telomeres in the human placenta. Mol. Hum. Reprod. 2016, 22, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Li, B.; Duan, S. The Alteration of Subtelomeric DNA Methylation in Aging-Related Diseases. Front. Genet. 2018, 9, 697. [Google Scholar] [CrossRef] [PubMed]
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res. Rev. 2017, 33, 89–104. [Google Scholar] [CrossRef] [PubMed]
- King, G.A.; Shabestari, M.H.; Taris, K.K.H.; Pandey, A.K.; Venkatesh, S.; Thilagavathi, J.; Singh, K.; Koppisetti, R.K.; Temiakov, D.; Roos, W.H.; et al. Acetylation and phosphorylation of human TFAM regulate TFAM–DNA interactions via contrasting mechanisms. Nucleic Acids Res. 2018, 46, 3633–3642. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, S.; Liu, X.; Chen, Y.; Deng, H. SIRT3 Overexpression Inhibits Growth of Kidney Tumor Cells and Enhances Mitochondrial Biogenesis. J. Proteome Res. 2018, 17, 3143–3152. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Vigilanza, P.; Baldelli, S.; Pagliei, B.; Rotilio, G.; Ciriolo, M.R. Peroxisome proliferator-activated receptor gamma co-activator 1alpha (PGC-1alpha) and sirtuin 1 (SIRT1) reside in mitochondria: Possible direct function in mitochondrial biogenesis. J. Biol. Chem. 2010, 285, 21590–21599. [Google Scholar] [CrossRef] [PubMed]
- Tvardovskiy, A.; Schwämmle, V.; Kempf, S.J.; Rogowska-Wrzesinska, A.; Jensen, O.N. Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Res. 2017, 45, 9272–9289. [Google Scholar] [CrossRef] [PubMed]
- Piazzesi, A.; Papić, D.; Bertan, F.; Salomoni, P.; Nicotera, P.; Bano, D. Replication-Independent Histone Variant H3.3 Controls Animal Lifespan through the Regulation of Pro-longevity Transcriptional Programs. Cell Rep. 2016, 17, 987–996. [Google Scholar] [CrossRef] [Green Version]
- Tennen, R.I.; Chua, K.F. Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem. Sci. 2011, 36, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacios, J.A.; Herranz, D.; De Bonis, M.L.; Velasco, S.; Serrano, M.; Blasco, M.A. SIRT1 contributes to telomere maintenance and augments global homologous recombination. J. Cell Biol. 2010, 191, 1299–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Bi, X.; Czarny-Ratajczak, M.; Dai, J.; Welsh, D.A.; Myers, L.; Welsch, M.A.; Cherry, K.E.; Arnold, J.; Poon, L.W.; et al. Telomere maintenance genes SIRT1 and XRCC6 impact age-related decline in telomere length but only SIRT1 is associated with human longevity. Biogerontology 2012, 13, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Cluett, T.J.; Akman, G.; Reyes, A.; Kazak, L.; Mitchell, A.; Wood, S.R.; Spinazzola, A.; Spelbrink, J.N.; Holt, I.J. Transcript availability dictates the balance between strand-asynchronous and strand-coupled mitochondrial DNA replication. Nucleic Acids Res. 2018, 46, 10771–10781. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, T.; Kang, D. An overview of mammalian mitochondrial DNA replication mechanisms. J. Biochem. 2018, 164, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Sommers, J.A.; Zhou, J.; Kaplan, D.L.; Spelbrink, J.N.; Mergny, J.L.; Brosh, R.M. DNA Sequences Proximal to Human Mitochondrial DNA Deletion Breakpoints Prevalent in Human Disease Form G-quadruplexes, a Class of DNA Structures Inefficiently Unwound by the Mitochondrial Replicative Twinkle Helicase. J. Biol. Chem. 2014, 289, 29975–29993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falabella, M.; Kolesar, J.E.; Wallace, C.; De Jesus, D.; Sun, L.; Taguchi, Y.V.; Wang, C.; Wang, T.; Xiang, I.M.; Alder, J.K.; et al. G-quadruplex dynamics contribute to regulation of mitochondrial gene expression. Sci. Rep. 2019, 9, 5605. [Google Scholar] [CrossRef]
- Fusté, J.M.; Shi, Y.; Wanrooij, S.; Zhu, X.; Jemt, E.; Persson, Ö.; Sabouri, N.; Gustafsson, C.M.; Falkenberg, M. In Vivo Occupancy of Mitochondrial Single-Stranded DNA Binding Protein Supports the Strand Displacement Mode of DNA Replication. PLoS Genet. 2014, 10, e1004832. [Google Scholar]
- Yang, Q.; Zhang, R.; Horikawa, I.; Fujita, K.; Afshar, Y.; Kokko, A.; Laiho, P.; Aaltonen, L.A.; Harris, C.C. Functional Diversity of Human Protection of Telomeres 1 Isoforms in Telomere Protection and Cellular Senescence. Cancer Res. 2007, 67, 11677–11686. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.; Bandaria, J.N.; Qureshi, M.H.; Yildiz, A.; Balci, H. G-quadruplex formation in telomeres enhances POT1/TPP1 protection against RPA binding. Proc. Natl. Acad. Sci. USA 2014, 111, 2990–2995. [Google Scholar] [CrossRef] [Green Version]
- Flynn, R.L.; Centore, R.C.; O’Sullivan, R.J.; Rai, R.; Tse, A.; Songyang, Z.; Chang, S.; Karlseder, J.; Zou, L. TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 2011, 471, 532–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, J.; Lin, Y.F.; Xu, K.; Lee, K.J.; Wang, D.; Chen, B.P. DNA-PKcs phosphorylates hnRNP-A1 to facilitate the RPA-to-POT1 switch and telomere capping after replication. Nucleic Acids Res. 2015, 43, 5971–5983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazak, L.; Reyes, A.; Duncan, A.L.; Rorbach, J.; Wood, S.R.; Brea-Calvo, G.; Gammage, P.A.; Robinson, A.J.; Minczuk, M.; Holt, I.J. Alternative translation initiation augments the human mitochondrial proteome. Nucleic Acids Res. 2013, 41, 2354–2369. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.L.; Rossi, M.L.; Canugovi, C.; Tian, J.; Sykora, P.; Ramamoorthy, M.; Wang, Z.M.; Singh, D.K.; Akbari, M.; Kasiviswanathan, R.; et al. RECQL4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell 2012, 11, 456–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshima, J.; Kato, H.; Maezawa, Y.; Yokote, K. RECQ helicase disease and related progeroid syndromes: RECQ2018 meeting. Mech. Ageing Dev. 2018, 173, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Sparks, M.A.; Singh, S.P.; Burgers, P.M.; Galletto, R. Complementary roles of Pif1 helicase and single stranded DNA binding proteins in stimulating DNA replication through G-quadruplexes. Nucleic Acids Res. 2019, 47, 8595–8605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duxin, J.P.; Dao, B.; Martinsson, P.; Rajala, N.; Guittat, L.; Campbell, J.L.; Spelbrink, J.N.; Stewart, S.A. Human Dna2 Is a Nuclear and Mitochondrial DNA Maintenance Protein. Mol. Cell. Biol. 2009, 29, 4274–4282. [Google Scholar] [CrossRef] [Green Version]
- Yasukawa, T.; Reyes, A.; Cluett, T.J.; Yang, M.-Y.; Bowmaker, M.; Jacobs, H.T.; Holt, I.J. Replication of vertebrate mitochondrial DNA entails transient ribonucleotide incorporation throughout the lagging strand. EMBO J. 2006, 25, 5358–5371. [Google Scholar] [CrossRef]
- Akman, G.; Desai, R.; Bailey, L.J.; Yasukawa, T.; Rosa, I.D.; Durigon, R.; Holmes, J.B.; Moss, C.F.; Mennuni, M.; Houlden, H.; et al. Pathological ribonuclease H1 causes R-loop depletion and aberrant DNA segregation in mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, E4276–E4285. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Holmes, J.B.; Cerritelli, S.M.; Sakhuja, K.; Minczuk, M.; Holt, I.J.; Crouch, R.J. An Upstream Open Reading Frame and the Context of the Two AUG Codons Affect the Abundance of Mitochondrial and Nuclear RNase H1. Mol. Cell. Biol. 2010, 30, 5123–5134. [Google Scholar] [CrossRef] [Green Version]
- Kazak, L.; Reyes, A.; He, J.; Wood, S.R.; Brea-Calvo, G.; Holen, T.T.; Holt, I.J. A Cryptic Targeting Signal Creates a Mitochondrial FEN1 Isoform with Tailed R-Loop Binding Properties. PLoS ONE 2013, 8, e62340. [Google Scholar] [CrossRef] [PubMed]
- Saharia, A.; Teasley, D.C.; Duxin, J.P.; Dao, B.; Chiappinelli, K.B.; Stewart, S.A. FEN1 Ensures Telomere Stability by Facilitating Replication Fork Re-initiation. J. Biol. Chem. 2010, 285, 27057–27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Gómez, S.; Reyes, A.; Martínez-Jiménez, M.I.; Chocrón, E.S.; Mouron, S.; Terrados, G.; Powell, C.; Salido, E.; Méndez, J.; Holt, I.J.; et al. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Šviković, S.; Crisp, A.; Tan-Wong, S.M.; Guilliam, T.A.; Doherty, A.J.; Proudfoot, N.J.; Guilbaud, G.; Sale, J.E. R-loop formation during S phase is restricted by PrimPol-mediated repriming. EMBO J. 2019, 38, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, D.; Jozwiakowski, S.K.; Romanello, M.; Guilbaud, G.; Guilliam, T.A.; Bailey, L.J.; Sale, J.E.; Doherty, A.J. PrimPol Is Required for Replicative Tolerance of G Quadruplexes in Vertebrate Cells. Mol. Cell 2016, 61, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Hangas, A.; Aasumets, K.; Kekäläinen, N.J.; Paloheinä, M.; Pohjoismäki, J.L.; Gerhold, J.M.; Goffart, S. Ciprofloxacin impairs mitochondrial DNA replication initiation through inhibition of Topoisomerase 2. Nucleic Acids Res. 2018, 46, 9625–9636. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, T.J.; Nadalutti, C.A.; Motori, E.; Sommerville, E.W.; Gorman, G.S.; Basu, S.; Hoberg, E.; Turnbull, D.M.; Chinnery, P.F.; Larsson, N.-G.; et al. Topoisomerase 3α Is Required for Decatenation and Segregation of Human mtDNA. Mol. Cell 2018, 69, 9–23. [Google Scholar] [CrossRef]
- Sobinoff, A.P.; Allen, J.A.; Neumann, A.A.; Yang, S.F.; Walsh, M.E.; Henson, J.D.; Reddel, R.R.; Pickett, H.A. BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J. 2017, 36, 2907–2919. [Google Scholar] [CrossRef]
- Martin, C.-A.; Sarlós, K.; Logan, C.V.; Thakur, R.S.; Parry, D.A.; Bizard, A.H.; Leitch, A.; Cleal, L.; Ali, N.S.; Al-Owain, M.A.; et al. Mutations in TOP3A Cause a Bloom Syndrome-like Disorder. Am. J. Hum. Genet. 2018, 103, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Rosa, I.D.; Huang, S.Y.N.; Agama, K.; Khiati, S.; Zhang, H.; Pommier, Y. Mapping Topoisomerase Sites in Mitochondrial DNA with a Poisonous Mitochondrial Topoisomerase I (Top1mt). J. Biol. Chem. 2014, 289, 18595–18602. [Google Scholar] [CrossRef] [Green Version]
- Yadav, P.; Harcy, V.; Argueso, J.L.; Dominska, M.; Jinks-Robertson, S.; Kim, N. Topoisomerase I Plays a Critical Role in Suppressing Genome Instability at a Highly Transcribed G-Quadruplex-Forming Sequence. PLoS Genet. 2014, 10, e1004839. [Google Scholar] [CrossRef] [PubMed]
- Lionaki, E.; Gkikas, I.; Tavernarakis, N. Differential Protein Distribution between the Nucleus and Mitochondria: Implications in Aging. Front. Genet. 2016, 7, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
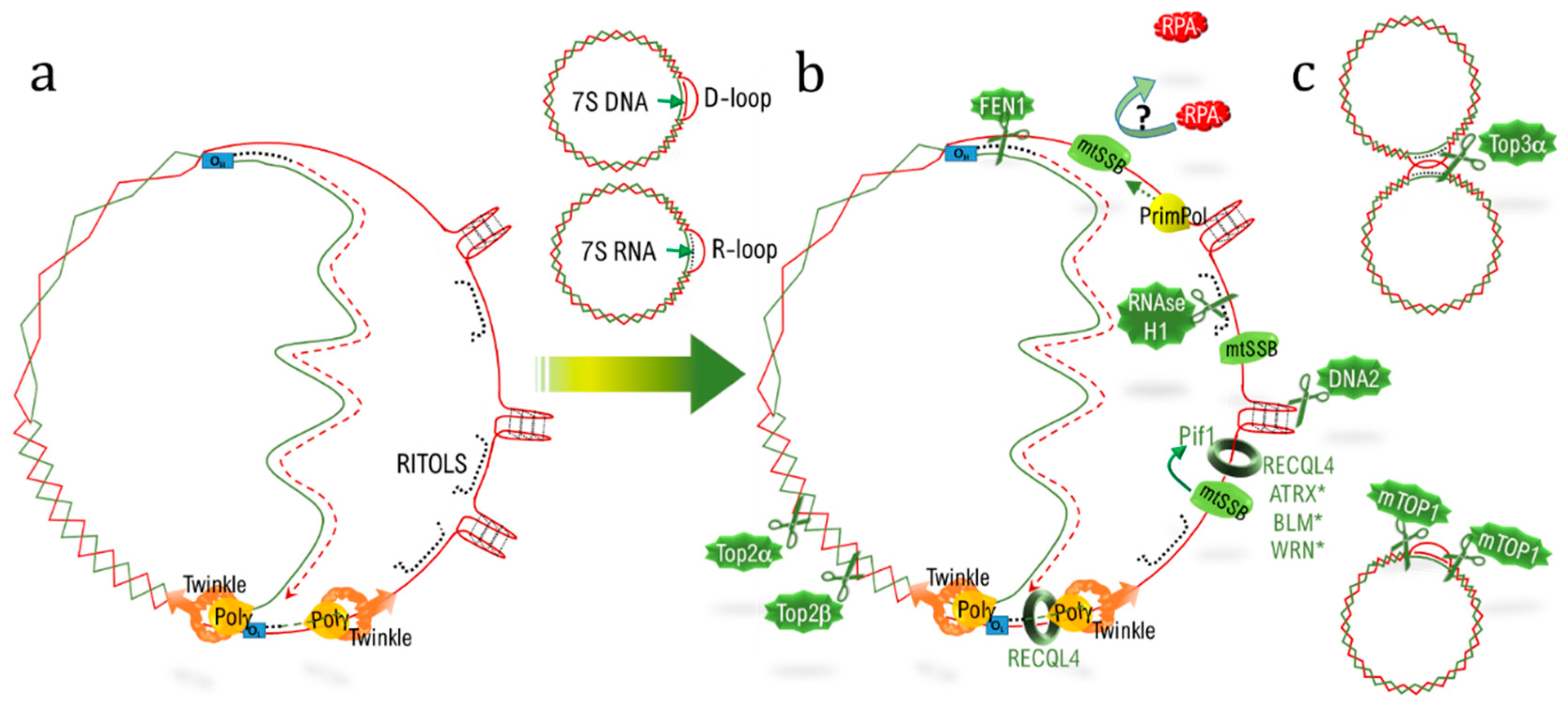{kind=link}
| Mechanisms | Features/Actors | mtDNA | telDNA |
|---|---|---|---|
| Structuration | D-Loop | D-loop (NCR) | D-loop (T-loop) |
| G rich sequences | H strand, G4 +++ | G strand, G4 +++ | |
| Curvature | TFAM [112] | TRF2 [113] | |
| RNA:DNA hybrid | R-loop (7SRNA), RITOLS § | R-loop(TERRA) § | |
| Specialized telomeric factors | TERT, TERC, TIN2 § | Telomerase complex, shelterin complex § | |
| Chromatin organization | Specific proteins | TFAM § | H3.3 § |
| Epigenetic modulators | DNMT1, 3b § | DNMT1/3A/3B § | |
| TET1, TET2 § SIRT1/3/4/5 § | TET1, TET 2, TET3 § SIRT1, SIRT6 § | ||
| Structuration | SMC6 * | SMC6 [114] | |
| Replication | Polymerase | POLG §, POLQ * | POLD, POLE §, POLQ [115] |
| (Translesional) | REV3L * | Pol η [116] | |
| ssDNA binding proteins | mtSSB §, RPA3 * | POT1, RPA § | |
| RNAse | RNASe H1 § | RNAse H1 § | |
| Torsion/supercoiling | TOP1mt, TOP2α, TOP2β § | TOP1 #, TOP2α, TOP2β § | |
| G4 unwinding, G4 clivage | Pif1, ATRX *, WRN *, BLM *, DNA2 | Pif1#, ATRX, WRN, BLM, DNA2 | |
| Primase | PrimPol | PrimPol # | |
| D-loop dissociation | RECQL4/Twinkle # | RECQL4/WRN | |
| Homologous recombination | Homology search/maturation | ctIP [117], RAD51 [107], MRE11 [117] XRCC3 [107] | ctIP [118], RAD51, MRE11 XRCC3 [119] |
| Endonucleases | EXO1 *, GEN1 * | EXO1 [120], GEN1 [121] | |
| MUS81 * | MUS81 [121] | ||
| Flap-exonuclease | FEN1, DNA2 § | FEN1, DNA2 § | |
| dJH resolutions | TOP3A § | TOP3A § |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Billard, P.; Poncet, D.A. Replication Stress at Telomeric and Mitochondrial DNA: Common Origins and Consequences on Ageing. Int. J. Mol. Sci. 2019, 20, 4959. https://doi.org/10.3390/ijms20194959
Billard P, Poncet DA. Replication Stress at Telomeric and Mitochondrial DNA: Common Origins and Consequences on Ageing. International Journal of Molecular Sciences. 2019; 20(19):4959. https://doi.org/10.3390/ijms20194959
Chicago/Turabian StyleBillard, Pauline, and Delphine A Poncet. 2019. "Replication Stress at Telomeric and Mitochondrial DNA: Common Origins and Consequences on Ageing" International Journal of Molecular Sciences 20, no. 19: 4959. https://doi.org/10.3390/ijms20194959
APA StyleBillard, P., & Poncet, D. A. (2019). Replication Stress at Telomeric and Mitochondrial DNA: Common Origins and Consequences on Ageing. International Journal of Molecular Sciences, 20(19), 4959. https://doi.org/10.3390/ijms20194959