Pharmacological Targeting of GLUT1 to Control Autoreactive T Cell Responses


Abstract
:1. Introduction
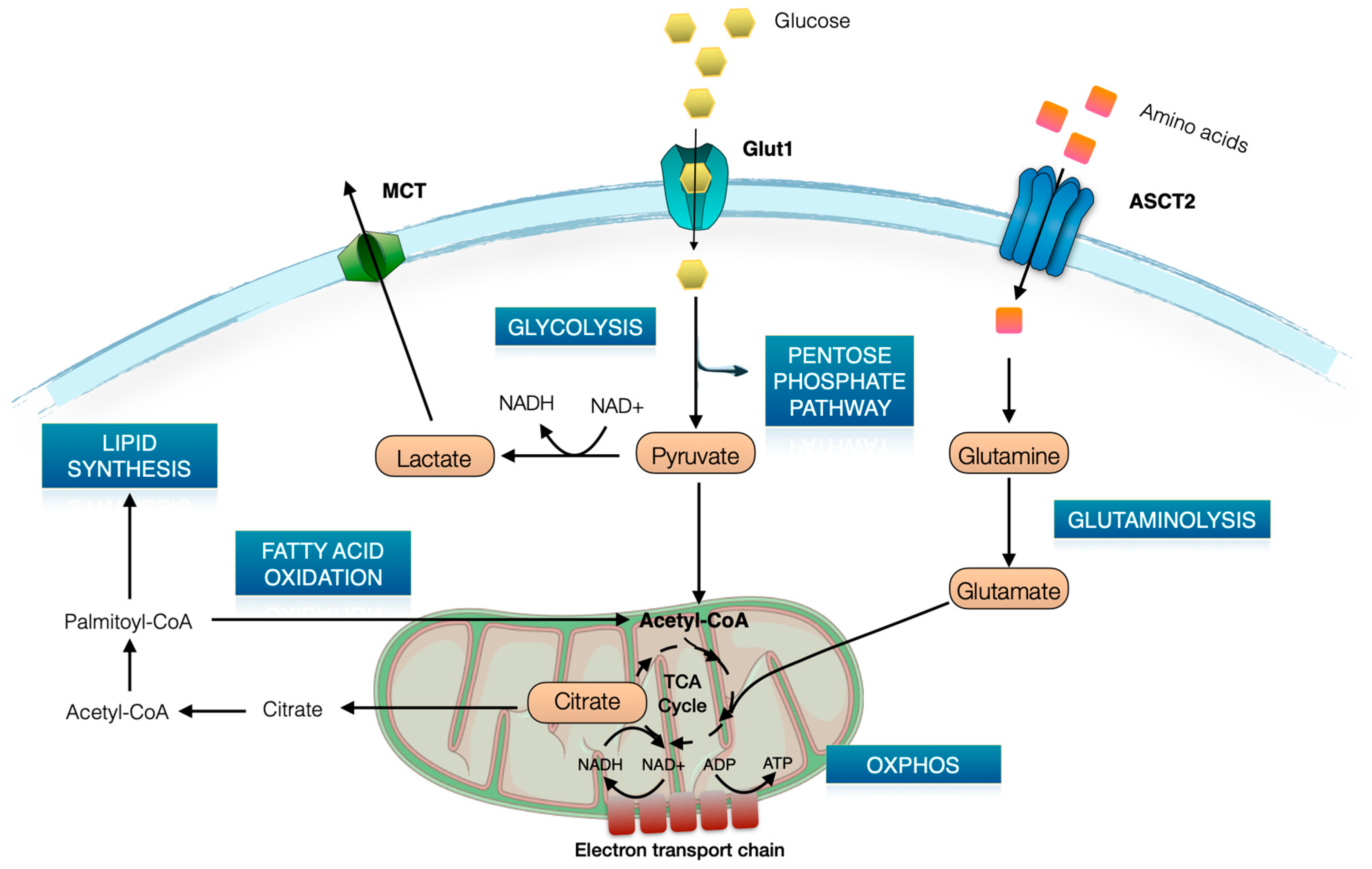
2. Brief Overview of T Cell Metabolism
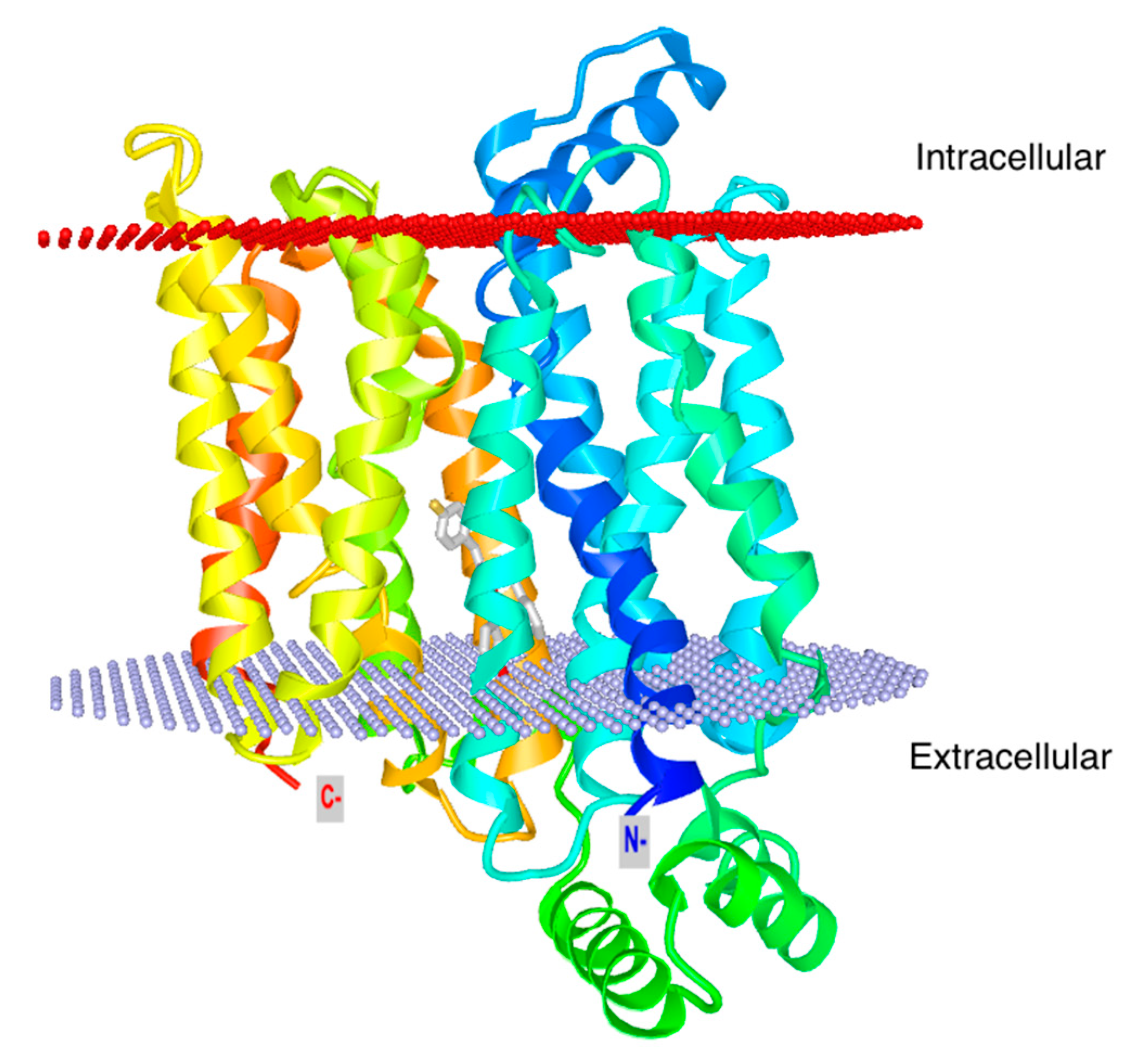
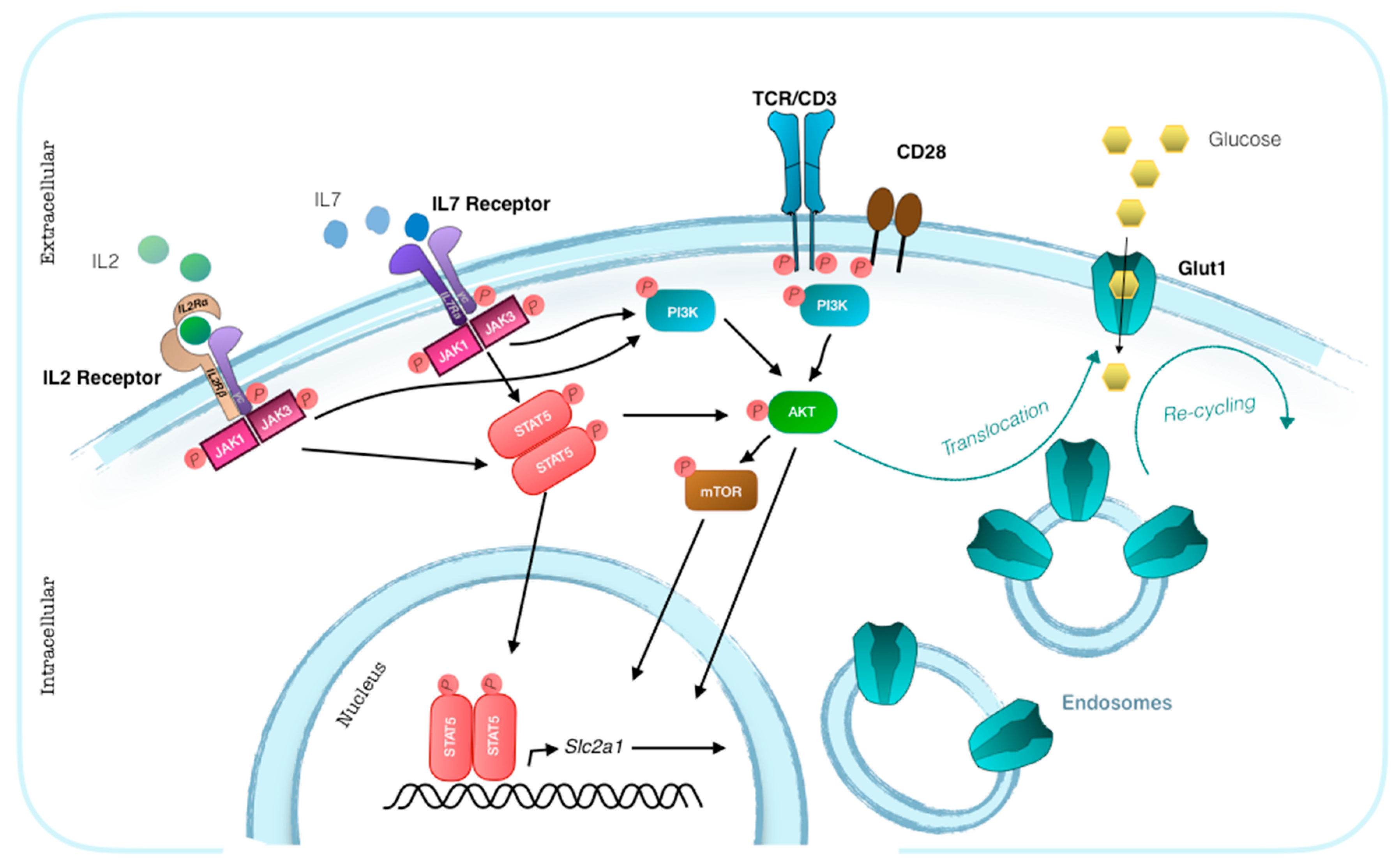
3. Glut1 Expression, Usage, and Blockade in T Cells
4. Metabolic Modulators in the Treatment of Autoimmunity
5. Novel drugs targeting Glut1
6. Potential off-Target and Side Effects of Pharmacological Glut1 Blockade
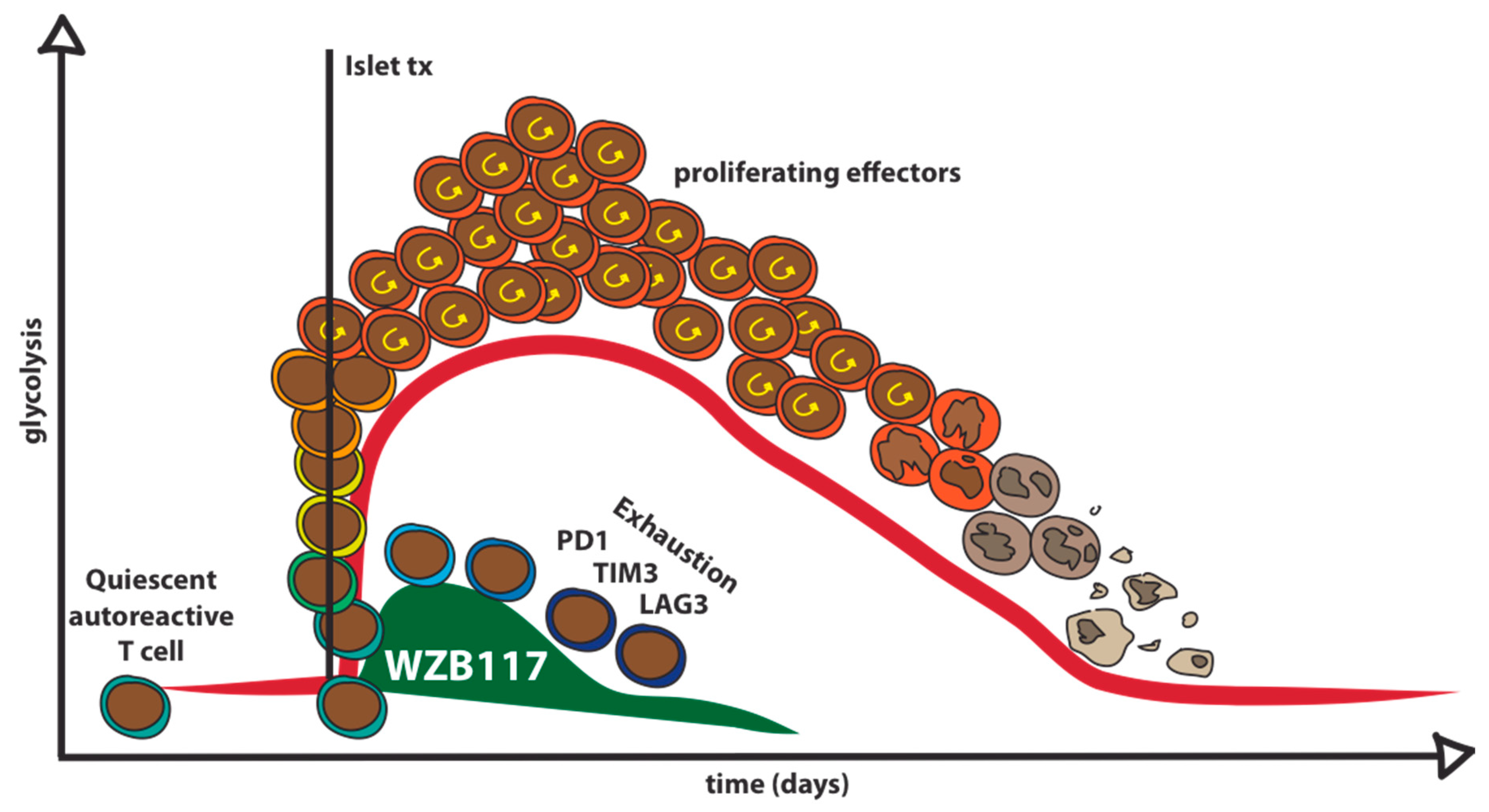
7. Pharmacological Glut1 Blockade to Target Autoreactive T Cells in Type 1 Diabetes
8. Feasibility and Potential Roadblocks
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| T1D | Type 1 Diabetes |
| OXPHOS | Oxidative Phosphorylation |
| FAO | Fatty Acid Oxidation |
| TCA | Trycarboxylic Acid |
| Tscm | Stem Cell Memory T Cells |
| 2DG | 2-deoxy-D-glucose |
| SLE | Systemic Lupus Erythematosus |
| EAE | Experimental Autoimmune Encephalomyelitis |
| VHL | Von Hippel-Lindau |
| BBB | Blood Brain Barrier |
| CNS | Central Nervous System |
| GLUT1-DS | GLUT1 deficiency syndrome |
References
- Babaya, N.; Nakayama, M.; Eisenbarth, G.S. The stages of type 1A diabetes. Ann. N. Y. Acad. Sci. 2005, 1051, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Kent, S.C.; Chen, Y.; Bregoli, L.; Clemmings, S.M.; Kenyon, N.S.; Ricordi, C.; Hering, B.J.; Hafler, D.A. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature 2005, 435, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.T.; Dotta, F.; Amirian, N.; Campbell, P.D.; Kay, T.W.H.; Atkinson, M.A.; Roep, B.O.; von Herrath, M.G. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J. Exp. Med. 2012, 209, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.L.; Peakman, M. Peptide immunotherapy for type 1 diabetes-clinical advances. Front. Immunol. 2018, 9, 1–5. [Google Scholar] [CrossRef]
- Jacobsen, L.M.; Newby, B.N.; Perry, D.J.; Posgai, A.L.; Haller, M.J.; Brusko, T.M. Immune Mechanisms and Pathways Targeted in Type 1 Diabetes. Curr. Diab. Rep. 2018, 18, 90. [Google Scholar] [CrossRef]
- Bone, R.N.; Evans-Molina, C. Combination Immunotherapy for Type 1 Diabetes. Curr. Diab. Rep. 2017, 17, 1–19. [Google Scholar] [CrossRef]
- Von Herrath, M.; Peakman, M.; Roep, B. Progress in immune-based therapies for type 1 diabetes. Clin. Exp. Immunol. 2013, 172, 186–202. [Google Scholar] [CrossRef]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-H.; Pearce, E.L. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat. Immunol. 2016, 17, 364–368. [Google Scholar] [CrossRef] [Green Version]
- Bettencourt, I.A.; Powell, J.D. Targeting Metabolism as a Novel Therapeutic Approach to Autoimmunity, Inflammation, and Transplantation. J. Immunol. 2017, 198, 999–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Benny, J.; Hale, L.P.; et al. Cell Activation and Effector Function. Cell Metab. 2015, 20, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.F. Oxidative phosphorylation: Regulation and role in cellular and tissue metabolism. J. Physiol. 2017, 595, 7023–7038. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.C.; O’Neill, L.A.J. A role for the krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation. Front. Immunol. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Chapman, N.M.; Boothby, M.R.; Chi, H. Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st Century. Am. J. Physiol.-Endocrinol. Metab. 2010, 298, 141–145. [Google Scholar] [CrossRef]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal structure of the human glucose transporter GLUT1. Nature 2014, 510, 121–125. [Google Scholar] [CrossRef]
- Filigheddu, N.; Gnocchi, V.F.; Coscia, M.; Cappelli, M.; Porporato, P.E.; Taulli, R.; di Pietro, S.M.; Falcon-Perez, J.M.; Tenza, D.; Setty, S.R.G.; et al. Ghrelin and Des-Acyl Ghrelin Promote Differentiation and Fusion of C2C12 Skeletal Muscle Cells. Mol. Biol. Cell 2007, 18, 986–994. [Google Scholar]
- Jacobs, S.R.; Herman, C.E.; MacIver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose Uptake Is Limiting in T Cell Activation and Requires CD28-Mediated Akt-Dependent and Independent Pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, R.; Jung, C.Y.; Lee, T.P.; Liu, H.; Mookerjee, B.K. Changes in glucose transport and transporter isoforms during the activation of human peripheral blood lymphocytes by phytohemagglutinin. J. Immunol. 1994, 152, 2660–2668. [Google Scholar] [PubMed]
- Wofford, J.A.; Wieman, H.L.; Jacobs, S.R.; Zhao, Y.; Rathmell, J.C. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood 2008, 111, 2101–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, M.J.; Mahajan, V.S.; Trajman, L.; Lauffenburger, D.A.; Chen, J. Perspectives on the quantitative immunobiology of the IL-7 signaling network. Cell. Mol. Immunol. 2009, 5, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.; Cantarelli, E.; Bordignon, C.; Canu, A.; Citro, A.; Annoni, A.; Piemonti, L.; Monti, P. Detection and characterization of CD8+ autoreactive memory Stem T cells in patients with type 1 diabetes. Diabetes 2018, 67, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Qiu, J.; Sullivan, D.O.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van, G.J.W.; et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cellular 2016, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Siska, P.J.; van der Windt, G.J.W.; Kishton, R.J.; Cohen, S.; Eisner, W.; MacIver, N.J.; Kater, A.P.; Weinberg, J.B.; Rathmell, J.C. Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J. Immunol. 2016, 197, 2532–2540. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef]
- Patel, C.H.; Powell, J.D. Targeting T cell metabolism to regulate T cell activation, differentiation and function in disease. Curr. Opin. Immunol. 2017, 46, 82–88. [Google Scholar] [CrossRef]
- Cham, C.M.; Driessens, G.; O’Keefe, J.P.; Gajewski, T.F. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur. J. Immunol. 2008, 38, 2438–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Invest. 2013, 123, 4479–4488. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Choi, S.S.-C.; Xu, Z.; Perry, D.J.; Seay, H.; Croker, B.P.; Sobel, E.S.; Brusko, T.M.; Morel, L. Normalization of CD4+ T cell metabolism reverses lupus. Sci. Transl. Med. 2015, 7, 274ra18. [Google Scholar] [CrossRef] [PubMed]
- Abboud, G.; Choi, S.C.; Kanda, N.; Zeumer-Spataro, L.; Roopenian, D.C.; Morel, L. Inhibition of glycolysis reduces disease severity in an autoimmune model of rheumatoid arthritis. Front. Immunol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Tian, T.; Gao, J.; Liu, X.; Hou, H.; Cao, R.; Li, B.; Quan, M.; Guo, L. Metformin ameliorates the development of experimental autoimmune encephalomyelitis by regulating T helper 17 and regulatory T cells in mice. J. Neuroimmunol. 2016, 292, 58–67. [Google Scholar] [CrossRef]
- Garyu, J.W.; Uduman, M.; Stewart, A.; Rui, J.; Deng, S.; Shenson, J.; Staron, M.M.; Kaech, S.M.; Kleinstein, S.H.; Herold, K.C. Characterization of Diabetogenic CD8+ T Cells. J. Biol. Chem. 2016, 291, 11230–11240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- 37 Granchi, C.; Fortunato, S.; Minutolo, F. Anticancer agents interacting with membrane glucose transporters. Medchemcomm 2016, 7, 1716–1729. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.T.; Wu, J.; Solow-Cordero, D.E.; et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70. [Google Scholar] [CrossRef]
- Williamson, M.K.; Coombes, N.; Juszczak, F.; Athanasopoulos, M.; Khan, M.B.; Eykyn, T.R.; Srenathan, U.; Taams, L.S.; Zeidler, J.D.; Da Poian, A.T.; et al. Upregulation of glucose uptake and hexokinase activity of primary human CD4+ T cells in response to infection with HIV-1. Viruses 2018, 10, 114. [Google Scholar] [CrossRef]
- Pingitore, A.; Ruz-Maldonado, I.; Liu, B.; Huang, G.C.; Choudhary, P.; Persaud, S.J. Dynamic Profiling of Insulin Secretion and ATP Generation in Isolated Human and Mouse Islets Reveals Differential Glucose Sensitivity. Cell. Physiol. Biochem. 2017, 44, 1352–1359. [Google Scholar] [CrossRef] [Green Version]
- Ojelabi, O.A.; Lloyd, K.P.; Simon, A.H.; De Zutter, J.K.; Carruthers, A. WZB117 (2-fluoro-6-(m-hydroxybenzoyloxy) Phenyl m-Hydroxybenzoate) inhibits GLUT1-mediated sugar transport by binding reversibly at the exofacial sugar binding site. J. Biol. Chem. 2016, 291, 26762–26772. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J.; Ito, D.; Rees, M.G.; Seashore-Ludlow, B.; Puyang, X.; Ramos, A.H.; Cheah, J.H.; Clemons, P.A.; Warmuth, M.; Zhu, P.; et al. NAMPT is the cellular target of STF-31-like small-molecule probes. ACS Chem. Biol. 2014, 9, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Siebeneicher, H.; Cleve, A.; Rehwinkel, H.; Neuhaus, R.; Heisler, I.; Müller, T.; Bauser, M.; Buchmann, B. Identification and Optimization of the First Highly Selective GLUT1 Inhibitor BAY-876. Chem. Med. Chem. 2016, 11, 2261–2271. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, W.; Idowu, M.O.; Oh, U.; Wang, X.Y.; Temkin, S.M.; Fang, X. Ovarian cancer relies on glucose transporter 1 to fuel glycolysis and growth: Anti-tumor activity of BAY-876. Cancers 2019, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Helgerson, A.L.; Carruthers, A. Equilibrium ligand binding to the human erythrocyte sugar transporter. Evidence for two sugar-binding sites per carrier. J. Biol. Chem. 1987, 262, 5464–5475. [Google Scholar] [PubMed]
- Sebastian, A.; Harris, S.; Ottaway, J.; Todd, K.; Morris, R. Improved mineral balance and skeletal metabolism in postmenopausal women treated with potassium bicarbonate. N. Engl. J. Med. 1994, 330, 1776–1781. [Google Scholar] [CrossRef] [PubMed]
- De Giorgis, V.; Veggiotti, P. GLUT1 Deficiency syndrome 2013: Current state of the art. Seizure 2013, 22, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.S.; Akman, C.; Hinton, V.J.; Engelstad, K.; De Vivo, D.C. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr. Neurol. Neurosci. Rep. 2013, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Leen, W.G.; Taher, M.; Verbeek, M.M.; Kamsteeg, E.J.; Van De Warrenburg, B.P.; Willemsen, M.A. GLUT1 deficiency syndrome into adulthood: A follow-up study. J. Neurol. 2014, 261, 589–599. [Google Scholar] [CrossRef]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; Dipaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase i dose-escalation trial of 2-deoxy-d-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 523–530. [Google Scholar] [CrossRef] [PubMed]
- De Vos, A.; Heimberg, H.; Quartier, E.; Huypens, P.; Bouwens, L.; Pipeleers, D.; Schuit, F. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J. Clin. Invest. 1995, 96, 2489–2495. [Google Scholar] [CrossRef] [PubMed]
- Kover, K.; Tong, P.Y.; Watkins, D.; Clements, M.; Stehno-Bittel, L.; Novikova, L.; Bittel, D.; Kibiryeva, N.; Stuhlsatz, J.; Yan, Y.; et al. Expression and Regulation of Nampt in Human Islets. PLoS ONE 2013, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Heimberg, H.; De Vos, A.; Pipeleers, D.; Thorens, B.; Schuit, F. Differences in glucose transporter gene expression between rat pancreatic α- and β-cells are correlated to differences in glucose transport but not in glucose utilization. J. Biol. Chem. 1995, 270, 8971–8977. [Google Scholar] [CrossRef] [PubMed]
- Heninger, A.-K.; Monti, P.; Wilhelm, C.; Schwaiger, P.; Kuehn, D.; Ziegler, A.-G.; Bonifacio, E. Activation of Islet Autoreactive Naive T Cells in Infants Is Influenced by Homeostatic Mechanisms and Antigen-Presenting Capacity. Diabetes 2013, 62, 2059–2066. [Google Scholar] [CrossRef] [PubMed]
- Monti, P.; Scirpoli, M.; Rigamonti, A.; Mayr, A.; Jaeger, A.; Bonfanti, R.; Chiumello, G.; Ziegler, A.G.; Bonifacio, E. Evidence for in vivo primed and expanded autoreactive T cells as a specific feature of patients with type 1 diabetes. J. Immunol. 2007, 179, 5785–5792. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.M.J.; Pokrywczynska, M.; Ricordi, C. Clinical pancreatic islet transplantation. Nat. Rev. Endocrinol. 2017, 13, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Gruessner, R.W.G.; Gruessner, A.C. The current state of pancreas transplantation. Nat. Rev. Endocrinol. 2013, 9, 555–562. [Google Scholar] [CrossRef]
- Matthis, J.; Nepom, G.T. T cell autoreactivity in the transplant milieu. Am. J. Transplant. 2012, 12, 1674–1681. [Google Scholar] [CrossRef]
- Pugliese, A.; Reijonen, H.K.; Nepom, J.; Burke, G.W. Recurrence of autoimmunity in pancreas transplant patients: research update. Diabetes Manag. 2011, 1, 229–238. [Google Scholar] [CrossRef]
- Piemonti, L.; Everly, M.J.; Maffi, P.; Scavini, M.; Poli, F.; Nano, R.; Cardillo, M.; Melzi, R.; Mercalli, A.; Sordi, V.; et al. Alloantibody and autoantibody monitoring predicts islet transplantation outcome in human type 1 diabetes. Diabetes 2013, 62, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Monti, P.; Scirpoli, M.; Maffi, P.; Ghidoli, N.; De Taddeo, F.; Bertuzzi, F.; Piemonti, L.; Falcone, M.; Secchi, A.; Bonifacio, E. Islet transplantation in patients with autoimmune diabetes induces homeostatic cytokines that expand autoreactive memory T cells. J. Clin. Invest. 2008, 118, 1806–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, D.C.; Kopetskie, H.A.; Sayre, P.H.; Alejandro, R.; Cagliero, E.; Shapiro, A.M.J.; Goldstein, J.S.; Desmarais, M.R.; Booher, S.; Bianchine, P.J. Long-Term Follow-Up of the Edmonton Protocol of Islet Transplantation in the United States. Am. J. Transplant. 2016, 16, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Monti, P.; Piemonti, L. Homeostatic T cell proliferation after islet transplantation. Clin. Dev. Immunol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Monti, P.; Brigatti, C.; Heninger, A.K.; Scirpoli, M.; Bonifacio, E. Disengaging the IL-2 receptor with daclizumab enhances IL-7-mediated proliferation of CD4(+) and CD8(+) T cells. Am. J. Transplant 2009, 9, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Potter, K.J.; Westwell-Roper, C.Y.; Klimek-Abercrombie, A.M.; Warnock, G.L.; Verchere, C.B. Death and dysfunction of transplanted β-cells: Lessons learned from type 2 diabetes? Diabetes 2014, 63, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Wherrett, D.K.; Bundy, B.; Becker, D.J.; Dimeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Marks, J.B.; et al. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: A randomised double-blind trial. Lancet 2011, 378, 319–327. [Google Scholar] [CrossRef]
- Bolla, A.M.; Caretto, A.; Laurenzi, A.; Scavini, M.; Piemonti, L. Low-carb and ketogenic diets in type 1 and type 2 diabetes. Nutrients 2019, 11, 962. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Structure | MW | IC50 (µ) | Characteristics | Human Cell Target (ref) |
|---|---|---|---|---|---|
| STF-31 |  | 423.53 | 1 | Low solubility | Renal cancer RCC4 [38] CD4+ T cells [39] Beta cells [40] |
| WZB-117 |  | 368.31 | 0.5 | High solubility | Multiple cancer cell lines [42] Autoreactive CD8+ T cells [25] |
| BAY 876 |  | 496.42 | 0.002 | Highly selective Orally bioavailable | Colon cancer DLD1 [44] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Dedda, C.; Vignali, D.; Piemonti, L.; Monti, P. Pharmacological Targeting of GLUT1 to Control Autoreactive T Cell Responses. Int. J. Mol. Sci. 2019, 20, 4962. https://doi.org/10.3390/ijms20194962
Di Dedda C, Vignali D, Piemonti L, Monti P. Pharmacological Targeting of GLUT1 to Control Autoreactive T Cell Responses. International Journal of Molecular Sciences. 2019; 20(19):4962. https://doi.org/10.3390/ijms20194962
Chicago/Turabian StyleDi Dedda, Carla, Debora Vignali, Lorenzo Piemonti, and Paolo Monti. 2019. "Pharmacological Targeting of GLUT1 to Control Autoreactive T Cell Responses" International Journal of Molecular Sciences 20, no. 19: 4962. https://doi.org/10.3390/ijms20194962
APA StyleDi Dedda, C., Vignali, D., Piemonti, L., & Monti, P. (2019). Pharmacological Targeting of GLUT1 to Control Autoreactive T Cell Responses. International Journal of Molecular Sciences, 20(19), 4962. https://doi.org/10.3390/ijms20194962