Dec1 Deficiency Suppresses Cardiac Perivascular Fibrosis Induced by Transverse Aortic Constriction

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
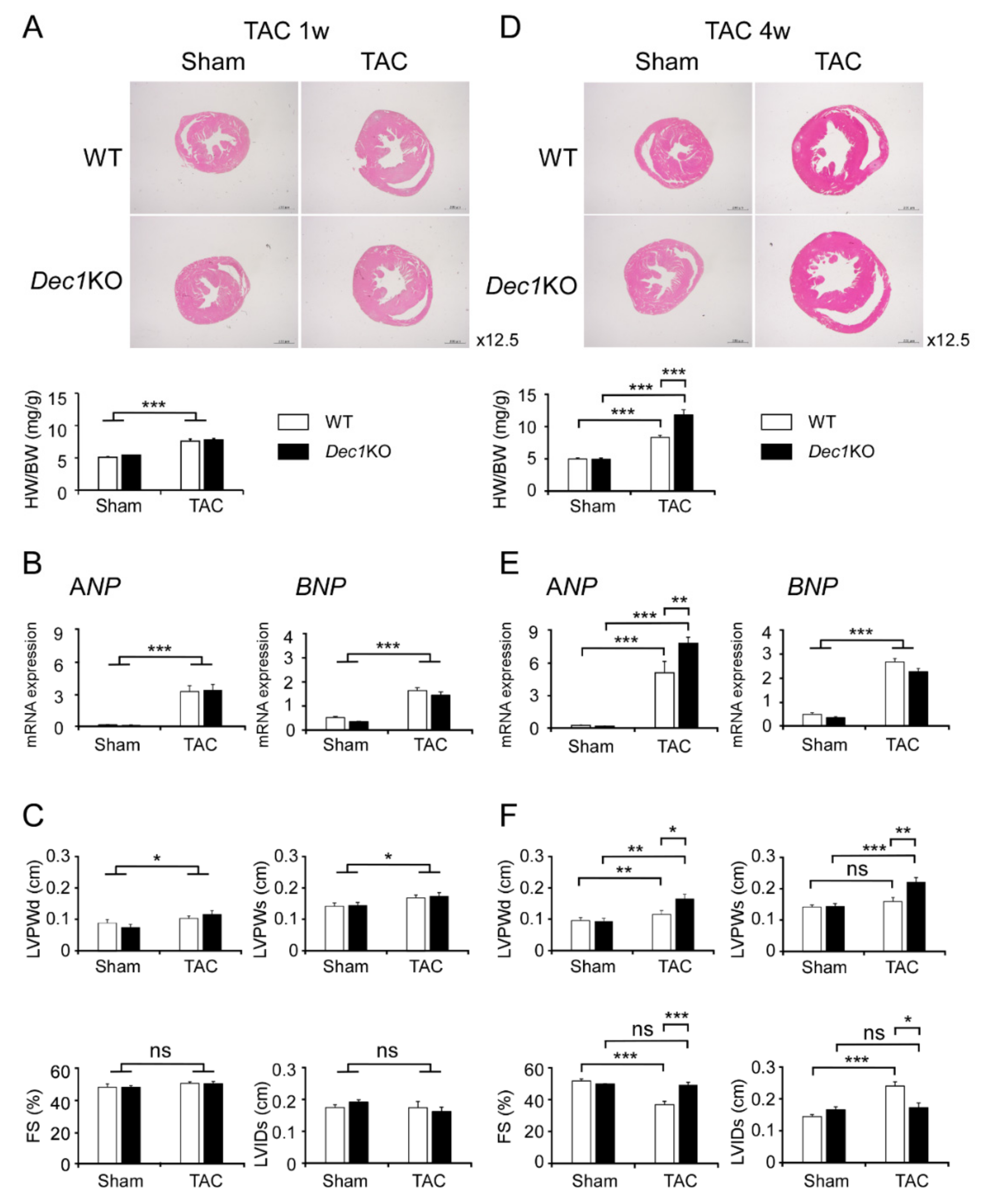
2.1. Dec1 Deficiency Protects Cardiac Function in Pressure Overload-Induced Cardiac Hypertrophy
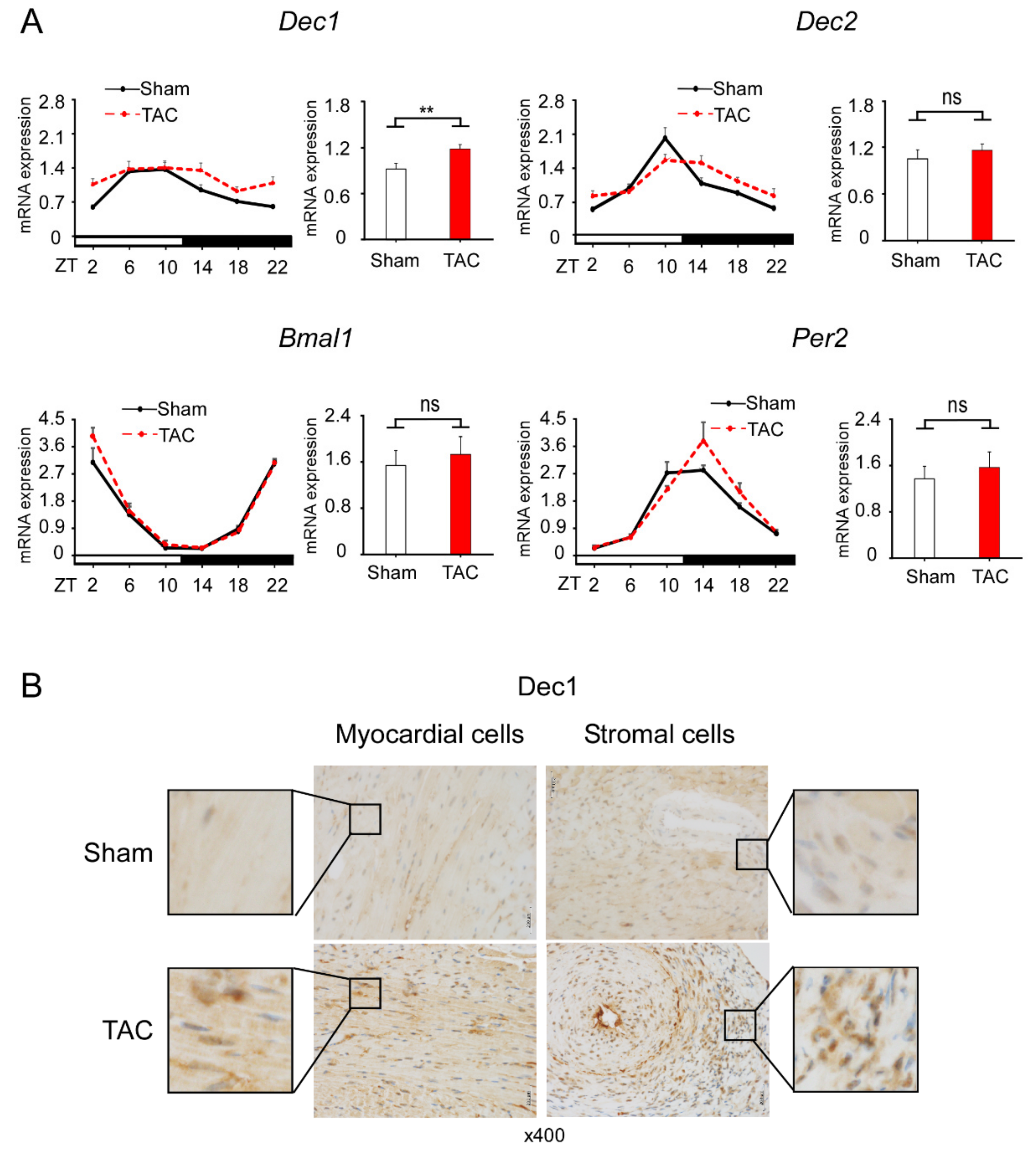
2.2. TAC Increases Dec1 Expression
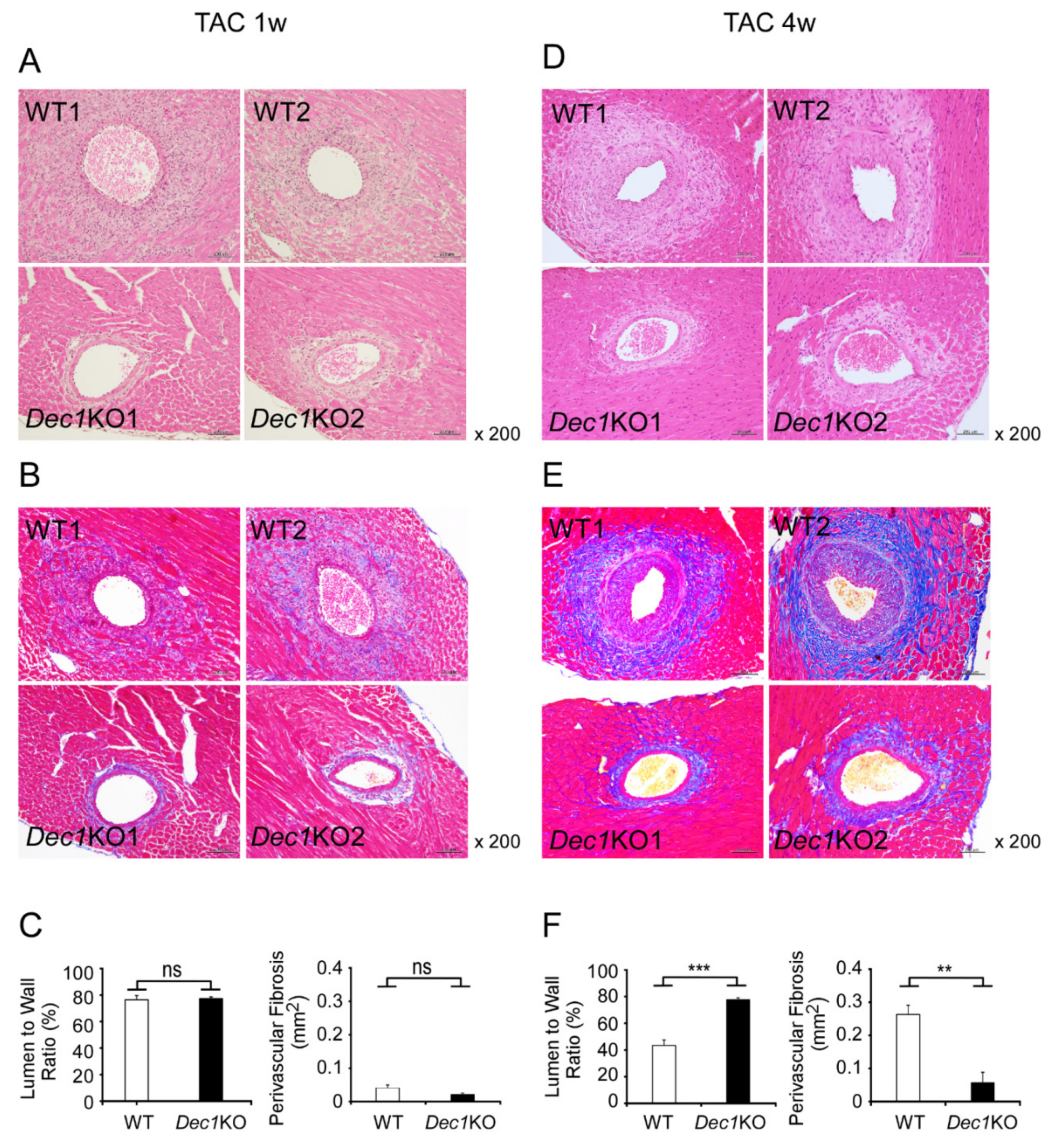
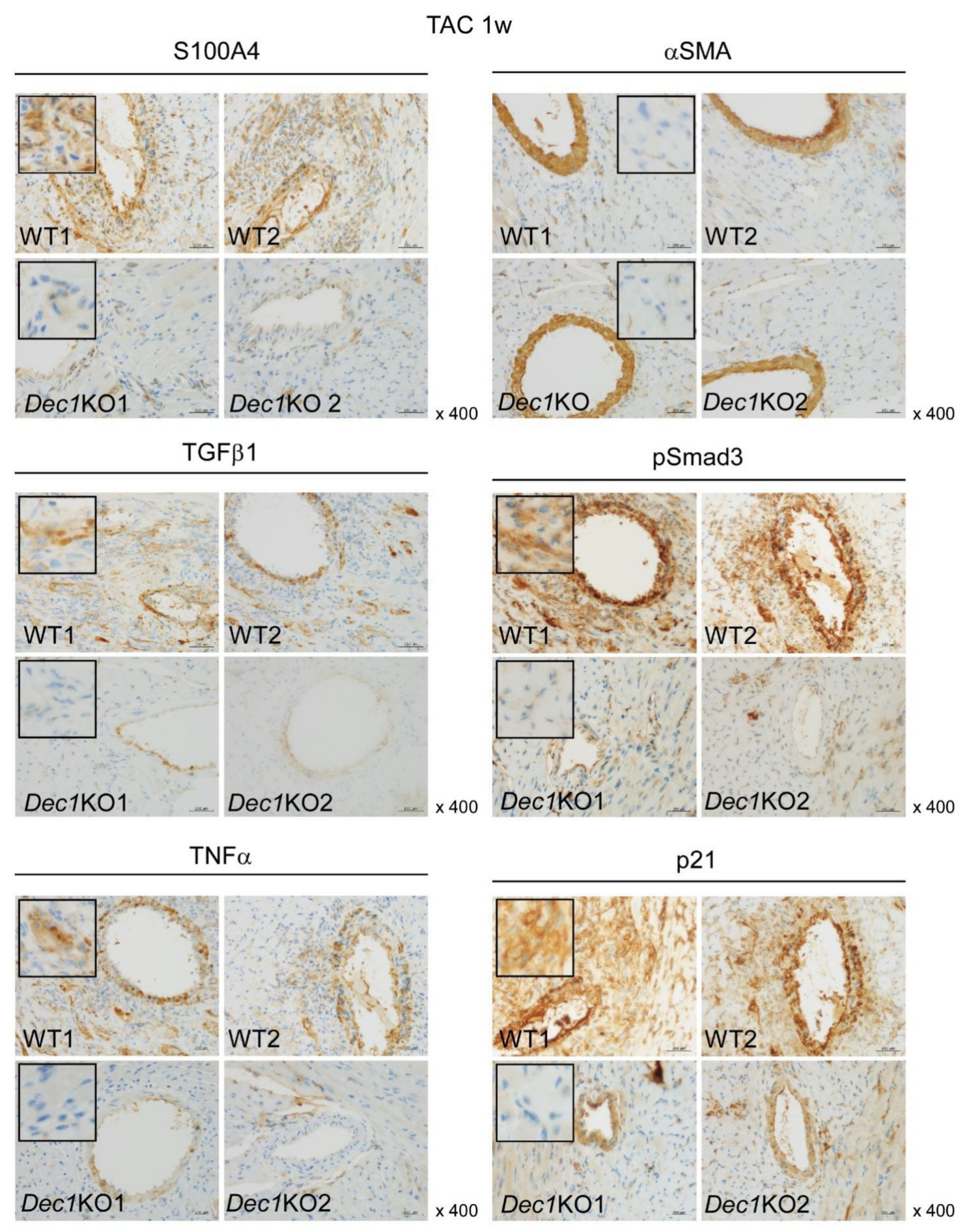
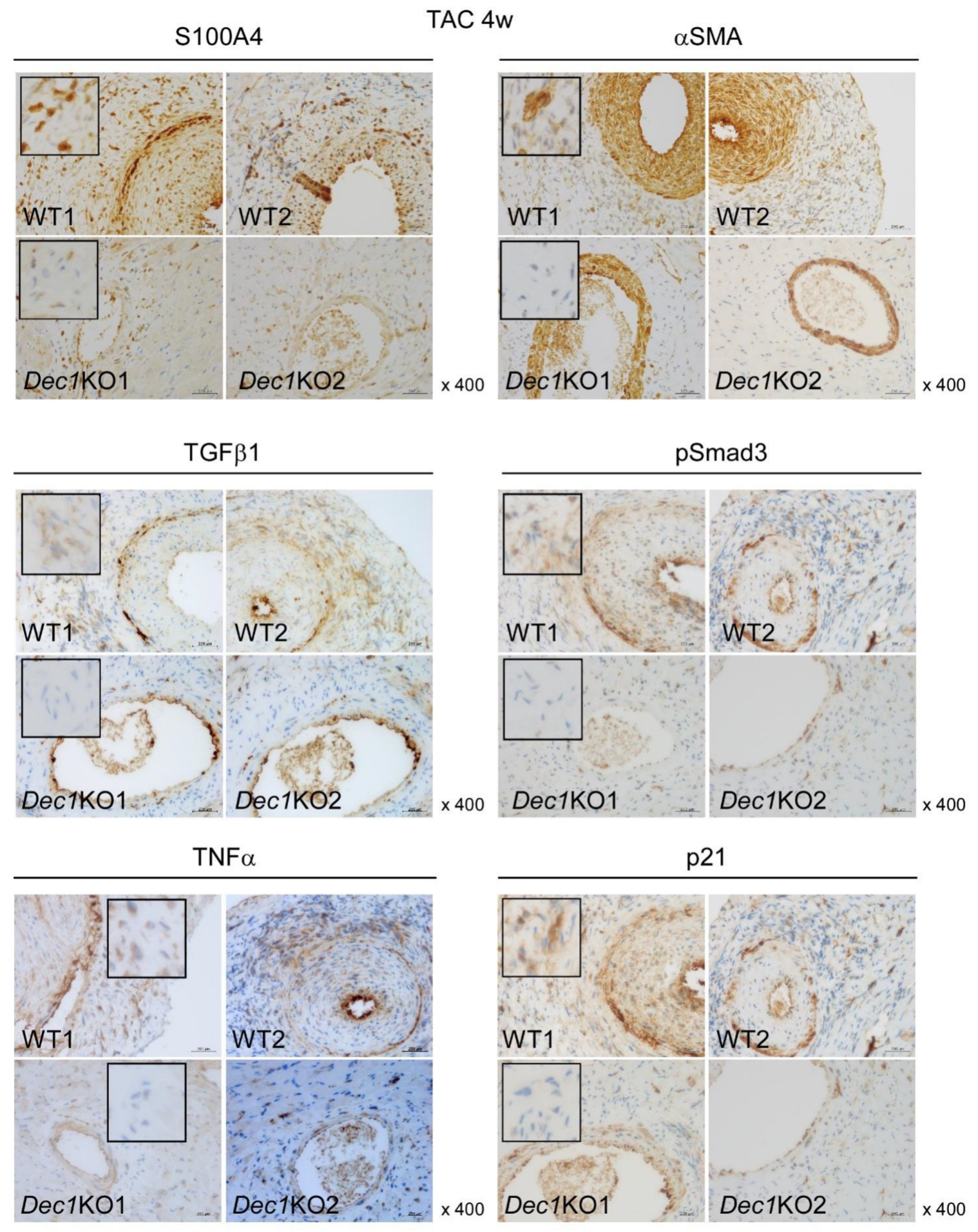
2.3. Dec1 Deficiency Suppresses TAC-Induced Cardiac Perivascular Fibrosis
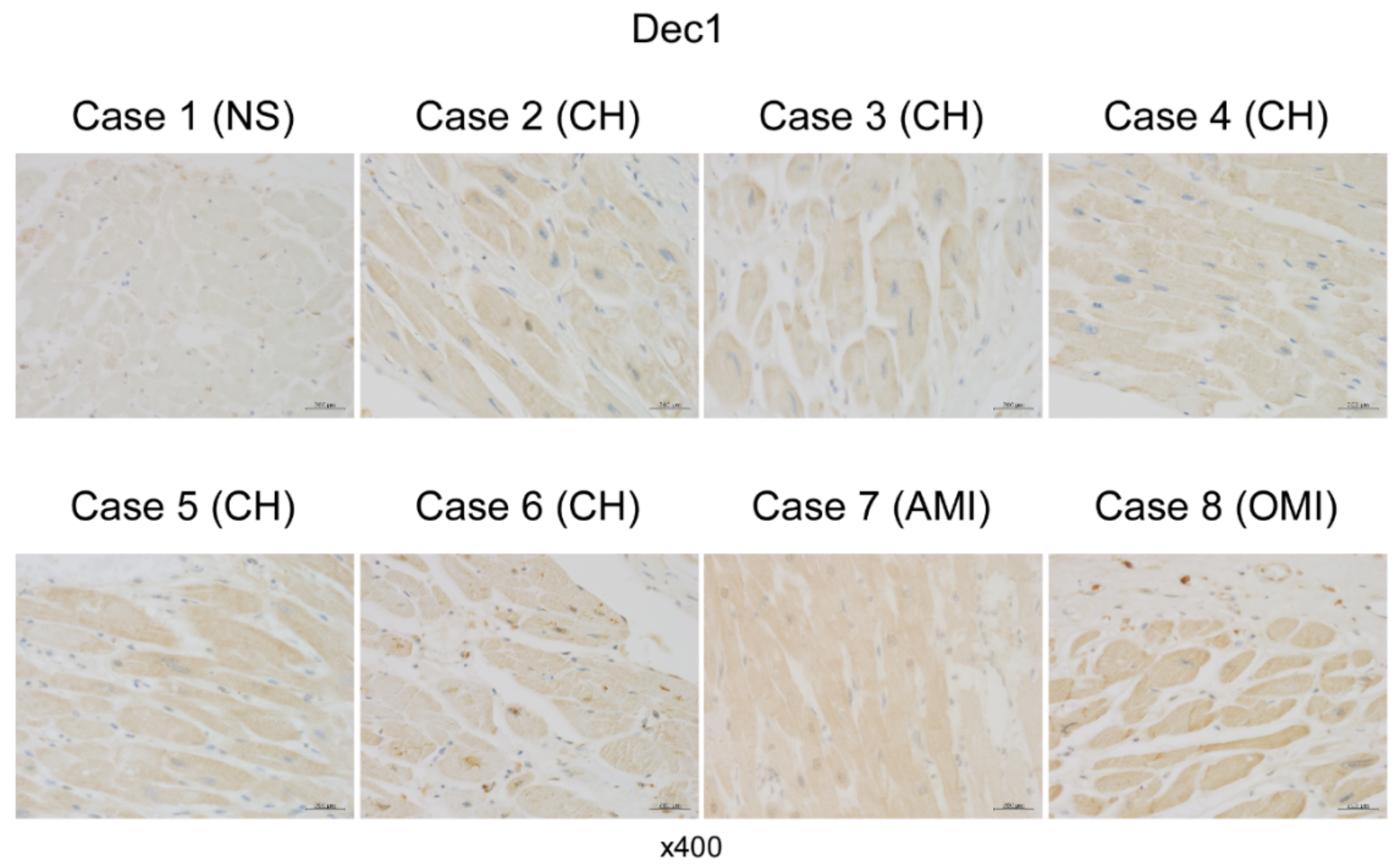
2.4. Dec1 Expression Is Increased in Human Cardiac Hypertrophy and Myocardial Infarction
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Ethics Approval and Consent to Participate
4.3. Transverse Aortic Constriction (TAC)
4.4. Transthoracic Echocardiography
4.5. Tissue Preparation
4.6. H&E and MT Staining
4.7. Immunohistochemistry
4.8. Real-Time PCR
4.9. Antibodies
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Dec1 | Differentiated embryonic chondrocyte gene 1 |
| Dec2 | Differentiated embryonic chondrocyte gene 2 |
| TAC | Transverse aortic constriction |
| H&E | Hematoxylin and eosin |
| MT | Masson’s trichrome |
| ANP | Atrial natriuretic peptide |
| BNP | B-type natriuretic peptide |
| FS | Fractional shortening |
| LVPWd | Left ventricular posterior wall thickness at the end of diastole |
| LVPWs | Left ventricular posterior wall thickness at the end of systole |
| HW/BW | Heart weight/body weight |
References
- Kuwahara, F.; Kai, H.; Tokuda, K.; Takeya, M.; Takeshita, A.; Egashira, K.; Imaizumi, T. Hypertensive myocardial fibrosis and diastolic dysfunction: Another model of inflammation. Hypertension 2004, 43, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Suetomi, T.; Willeford, A.; Brand, C.S.; Cho, Y.; Ross, R.S.; Miyamoto, S.; Brown, J.H. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca2+/Calmodulin-dependent protein Kinase II δ signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation 2018, 138, 2530–2544. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Wang, G.; Liu, W.; Ding, W.; Dong, M.; Zheng, N.; Ye, H.; Liu, J. Hypoxia-induced mitogenic factor promotes cardiac hypertrophy via calcium-dependent and hypoxia-inducible factor-1α mechanisms. Hypertension 2018, 72, 331–342. [Google Scholar] [CrossRef]
- Sano, M.; Minamino, T.; Toko, H.; Miyauchi, H.; Orimo, M.; Qin, Y.; Akazawa, H.; Tateno, K.; Kayama, Y.; Harada, M.; et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007, 446, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Lee, K.; Li, N.; Corbett, D.; Mendoza, L.; Frangogiannis, N.G. Characterization of Inflammatory and fibrotic response in a mousse model of cardiac pressure overload. Histochem. Cell Biol. 2009, 131, 471–481. [Google Scholar]
- Schirone, L.; Forte, M.; Palmerio, S.; Yee, D.; Nocella, C.; Angelini, F.; Pagano, F.; Schiavon, S.; Bordin, A.; Carrizzo, A.; et al. A review of the molecular mechanisms underlying the development and progression of cardiac remodeling. Oxid. Med. Cell Longev. 2017, 1–16. [Google Scholar] [CrossRef]
- King, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Suthahar, N.; Meijers, W.C.; Silljé, H.H.W.; Boer, R.A.D. From inflammation to fibrosis-molecular and cellular mechanisms of myocardial tissue remodelling and perspectives on differential treatment on opportunities. Curr. Heart Fail. Rep. 2017, 14, 235–250. [Google Scholar] [CrossRef]
- Tamaki, Y.; Iwanaga, Y.; Niizuma, S.; Kawashima, T.; Kato, T.; Inuzuka, Y.; Horie, T.; Morooka, H.; Takase, T.; Akahashi, Y.; et al. Metastasis-associated protein, S100A4 mediates cardiac fibrosis potentially through the modulation of p53 in cardiac fibroblasts. J. Mod. Cell Cardiol. 2013, 57, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Koitabashi, N.; Danner, T.; Zaiman, A.L.; Pinto, Y.M.; Rowell, J.; Mankowski, J.; Zhang, D.; Nakamura, T.; Takimoto, E.; Kass, D.A. Pivotal role of cardiomyocyte TGF-β signaling in the murine pathological response to sustained pressure overload. J. Clin. Invest. 2011, 121, 2301–2312. [Google Scholar] [CrossRef]
- Schneider, M.; Hansen, J.L.; Sheikh, S.P. S100A4: A common mediator of epithelial-mesenchymal transition, fibrosis and regeneration in diseases. J. Mol. Med. 2008, 86, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-messenchymal transition contributes to cardiac fibrosis. Nat Med 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Andrew, L. Getting to the heart of the matter: New insights into cardiac fibrosis. Circ. Res. 2015, 116, 1269–1276. [Google Scholar]
- Tomcik, M.; Palumbo-Zerr, K.; Zerr, P.; Avouac, J.; Dees, C.; Sumova, B.; Distler, A.; Beyer, C.; Cerezo, L.A.; Becvar, R.; et al. S100A4 amplifies TGF-β-induced fibroblast activation in systemic sclerosis. Ann. Rheum. Dis. 2014, 74, 1748–1755. [Google Scholar] [CrossRef]
- Hu, B.; Wu, Z.; Phan, S.H. Smad3 mediates transforming growth factor-β-induced α-smooth muscle actin expression. Am. J. Respir. Cel. Mol. Biol. 2003, 29, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L.; Tyner, A.L. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer. Ther. 2002, 1, 639–649. [Google Scholar] [PubMed]
- Sato, F.; Kohsaka, A.; Takahashi, K.; Otao, S.; Kitada, Y.; Iwasaki, Y.; Muragaki, Y. Smad3 and Bmal1 regulate p21 and S100A4 expression in myocardial stromal fibroblast via TNF-α. Histochem. Cell Biol. 2017, 148, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Sriramula, S.; Francis, J. Tumor necrosis factor-alpha is essential for angiotensin II-induced ventricular remodeling: Role for oxidative stress. Plos ONE 2015, 10, e0138372. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.R.; Chung, A.C.K.; Yang, F.; Deng, C.; Lau, C.P.; Tse, H.F.; Lan, H.Y. Smad3 mediates cardiac inflammation and fibrosis in angiotensin II-induced hypertensive cardiac remodeling. Hypertension 2010, 55, 1165–1171. [Google Scholar] [CrossRef] [PubMed]
- Dokainish, H.; Teo, K.; Zhu, J.; Roy, A.; Alhabib, K.F.; Elsayed, A.; Palileo-Villaneuva, L.; Lopez-Jaramillo, P.; Karaye, K.; Yusoff, K.; et al. Global mortality variations in patients with heart failure: Results from the international congestive heart failure (INTER-CHF) prospective cohort study. Lancet Glo. Health 2017, 5, 665–672. [Google Scholar] [CrossRef]
- Sato, F.; Bhawal, U.K.; Yoshimura, T.; Muragaki, Y. Dec1 and Dec2 crosstalk between circadian rhythm and tumor progression. J. Cancer 2016, 7, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Honma, S.; Kawamoto, T.; Takagi, Y.; Fujimoto, K.; Sato, F.; Noshiro, M.; Kato, Y.; Honma, K. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature 2002, 419, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Otsuka, T.; Kohsaka, A.; Le, H.T.; Bhawal, U.K.; Muragaki, Y. Smad3 suppress epithelial cell migration and proliferation via the clock gene Dec1, which negatively regulates the expression of clock genes Dec2 and Per1. Am. J. Patho. 2019, 189, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Suzuki, M.; Kim, I.S.; Kobayashi, R.; Hamada, N.; Sato, F.; Bhawal, U.K. Transcription factor Dec1 is required for maximal experimentally induced periodontal inflammation. J. Periodont Res. 2018, 53, 883–893. [Google Scholar] [CrossRef]
- Noshiro, M.; Kawamoto, T.; Nakashima, A.; Ozaki, N.; Ueno, T.; Saeki, M.; Honda, K.; Fujimoto, K.; Kato, Y. Deficiency of the basic helix-loop-helix transcription factor Dec1 prevents obesity induced by a high-fat diet in mice. Genes Cells 2018, 23, 658–669. [Google Scholar] [CrossRef]
- Nakashima, A.; Kawamoto, T.; Noshiro, M.; Ueno, T.; Doi, S.; Honda, K.; Maruhashi, T.; Noma, K.; Honma, S.; Masaki, T.; et al. Dec1 and CLOCK regulate Na+/K+-ATPase β1 subunit expression and blood pressure. Hypertension 2018, 72, 746–754. [Google Scholar] [CrossRef]
- Wu, Y.; Sato, F.; Yamada, T.; Bhawal, U.K.; Kawamoto, T.; Fujimoto, K.; Noshiro, M.; Seino, H.; Morohashi, S.; Hakamada, K.; et al. The BHLH transcription factor DEC1 plays an important role in the epithelial-mesenchymal transition of pancreatic cancer. Int. J. Oncol. 2012, 41, 1337–1346. [Google Scholar] [CrossRef] [Green Version]
- Kohsaka, A.; Das, P.; Hashimoto, I.; Nakao, T.; Deguchi, Y.; Gouraud, S.S.; Waki, H.; Muragaki, Y.; Maeda, M. The circadian clock maintains cardiac function by regulating mitochondrial metabolism in mice. PLoS ONE 2014, 9, e112811. [Google Scholar] [CrossRef]
- Alibhai, F.J.; LaMarre, J.; Reitz, C.J.; Tsimakouridze, E.V.; Kroetsch, J.T.; Bolz, S.S.; Shulman, A.; Steinberg, S.; Burris, T.P.; Oudit, G.Y.; et al. Disrupting the key circadian regulator CLOCK leads to age-dependent cardiovascular disease. J. Mol. Cell Cardiol. 2017, 105, 24–37. [Google Scholar] [CrossRef] [Green Version]
- Virag, J.A.I.; Dries, J.L.; Easton, P.R.; Friesland, A.M.; DeAntonio, J.H.; Chintalgattu, V.; Cozzi, E.; Lehmann, B.D.; Ding, J.M.; Lust, R.M. Attenuation of myocardial injury in mice with functional deletion of the circadian rhythm gene mPer2. Am. J. Physiol. Heart Cir. Physiol. 2010, 298, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Bhawal, U.K.; Kawamoto, T.; Fujimoto, K.; Imaizumi, T.; Imanaka, T.; Kondo, J.; Koyanagi, S.; Noshiro, M.; Yoshida, H.; et al. Basic-helix-loop-helix (bHLH) transcription factor DEC2 negatively regulates vascular endothelial growth factor expression. Genes Cells 2008, 13, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sato, F.; Kawamoto, T.; Fujimoto, K.; Morohashi, S.; Akasaka, H.; Kondon, J.; Wu, Y.; Noshiro, M.; Kato, Y.; et al. Anti-apoptotic effect of the basic helix-loop-helix (bHLH) transcription factor DEC2 in human breast cancer cells. Genes Cells 2010, 15, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Makishima, M.; Bhawal, U.K. Differentiated embryo chondrocyte 1 (DEC1) is a novel negative regulator of hepatic fibroblast growth factor 21 (FGF21) in aging mice. Biochem. Bioph. Res. Co. 2016, 469, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.; Kawamoto, T.; Honda, K.K.; Ueshima, T.; Noshiro, M.; Iwata, T.; Fujimoto, K.; Kubo, H.; Honma, S.; Yorioka, N.; et al. Dec1 modulates the circadian phase of clock gene expression. Mol. Cell Biol. 2008, 28, 4080–4092. [Google Scholar] [CrossRef] [PubMed]
- Nakao, T.; Kohsaka, A.; Otsuka, T.; Thein, Z.L.; Le, H.T.; Waki, H.; Gouraud, S.S.; Ihara, H.; Nakanishi, M.; Sato, F.; et al. Impact of heart-specific disruption of circadian clock on systemic glucose metabolism in mice. Chronobiol. Int. 2018, 35, 499–510. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, H.T.; Sato, F.; Kohsaka, A.; Bhawal, U.K.; Nakao, T.; Muragaki, Y.; Nakata, M. Dec1 Deficiency Suppresses Cardiac Perivascular Fibrosis Induced by Transverse Aortic Constriction. Int. J. Mol. Sci. 2019, 20, 4967. https://doi.org/10.3390/ijms20194967
Le HT, Sato F, Kohsaka A, Bhawal UK, Nakao T, Muragaki Y, Nakata M. Dec1 Deficiency Suppresses Cardiac Perivascular Fibrosis Induced by Transverse Aortic Constriction. International Journal of Molecular Sciences. 2019; 20(19):4967. https://doi.org/10.3390/ijms20194967
Chicago/Turabian StyleLe, Hue Thi, Fuyuki Sato, Akira Kohsaka, Ujjal K. Bhawal, Tomomi Nakao, Yasuteru Muragaki, and Masanori Nakata. 2019. "Dec1 Deficiency Suppresses Cardiac Perivascular Fibrosis Induced by Transverse Aortic Constriction" International Journal of Molecular Sciences 20, no. 19: 4967. https://doi.org/10.3390/ijms20194967
APA StyleLe, H. T., Sato, F., Kohsaka, A., Bhawal, U. K., Nakao, T., Muragaki, Y., & Nakata, M. (2019). Dec1 Deficiency Suppresses Cardiac Perivascular Fibrosis Induced by Transverse Aortic Constriction. International Journal of Molecular Sciences, 20(19), 4967. https://doi.org/10.3390/ijms20194967