Deleterious Variants in WNT10A, EDAR, and EDA Causing Isolated and Syndromic Tooth Agenesis: A Structural Perspective from Molecular Dynamics Simulations

 ,
, 
 , , , , and add
Show full author list
, , , , and add
Show full author list
Abstract
:1. Introduction
2. Results
2.1. Clinical Findings
2.2. Variant Screening and Pathogenicity
2.3. Protein Structure and Stability Prediction
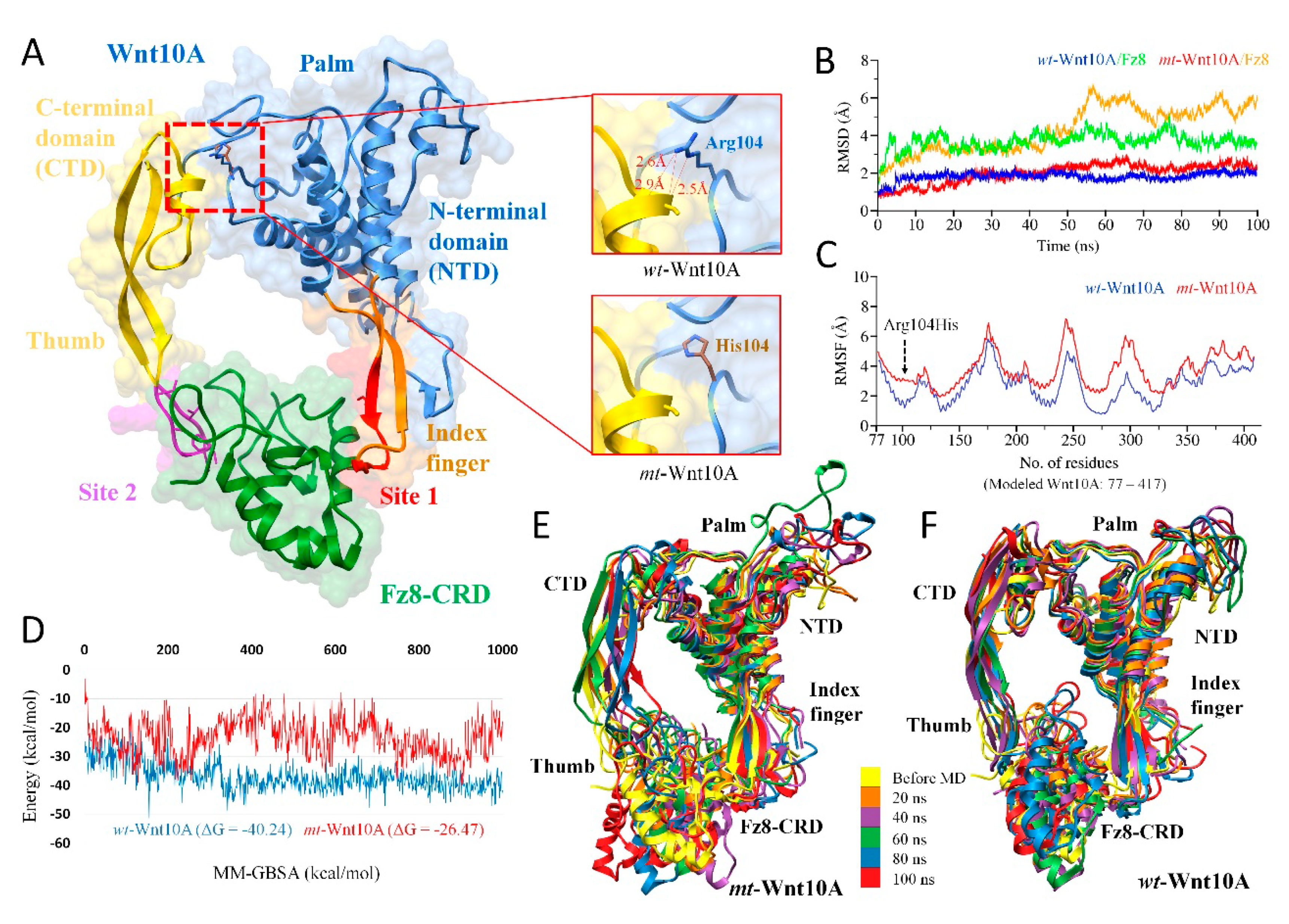
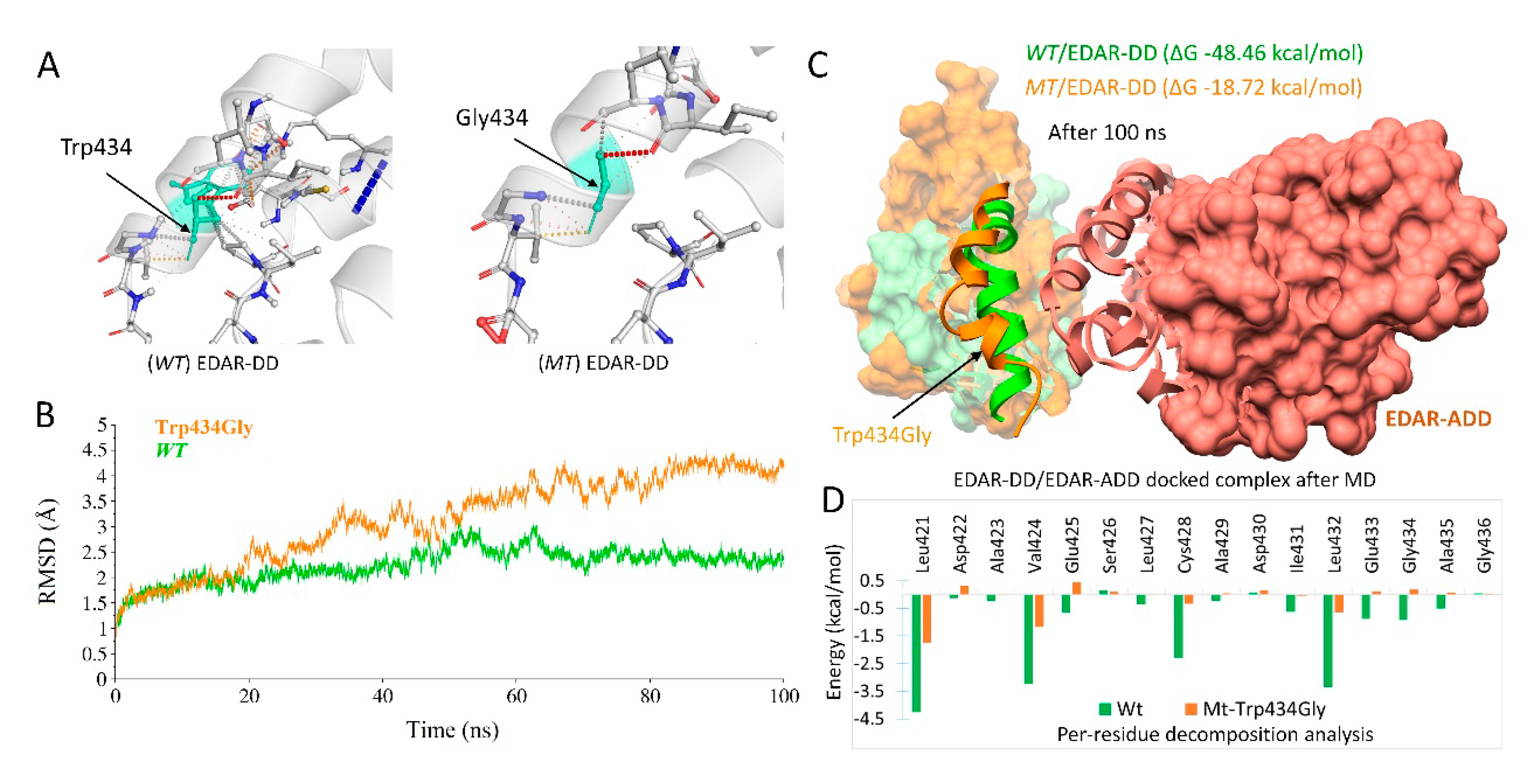
2.4. MD Simulations and Interpretations of Novel Missense Variants
2.4.1. WNT10A Missense Variant (c.311G>A; p.Arg104His)
2.4.2. EDAR Missense Variant (c.1300T>G; p.Trp434Gly)
3. Discussion
4. Material and Methods
4.1. Project Approval, Recruitment of Patients and DNA Extraction
4.2. Exome Sequencing
4.3. Sanger Sequencing
4.4. Pathogenicity Context of the Missense Variants
4.5. Molecular Modeling and Protein Stability Predictions
4.6. MD Simulations and Binding Free Energy Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pinheiro, M.; Freire-Maia, N. Ectodermal dysplasias: A clinical classification and a causal review. Am. J. Med. Genet. 1994, 53, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Chishti, M.S.; Muhammad, D.; Haider, M.; Ahmad, W. A novel missense mutation in MSX1 underlies autosomal recessive oligodontia with associated dental anomalies in Pakistani families. J. Hum. Genet. 2006, 51, 872–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cluzeau, C.; Hadj-Rabia, S.; Jambou, M.; Mansour, S.; Guigue, P.; Masmoudi, S.; Bal, E.; Chassaing, N.; Vincent, M.C.; Viot, G. Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum. Mutat. 2011, 32, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Issa, Y.A.; Kamal, L.; Rayyan, A.A.; Dweik, D.; Pierce, S.; Lee, M.K.; King, M.-C.; Walsh, T.; Kanaan, M. Mutation of KREMEN1, a modulator of Wnt signaling, is responsible for ectodermal dysplasia including oligodontia in Palestinian families. Eur. J. Hum. Genet. 2016, 24, 1430. [Google Scholar] [CrossRef] [PubMed]
- Massink, M.P.; Créton, M.A.; Spanevello, F.; Fennis, W.M.; Cune, M.S.; Savelberg, S.M.; Nijman, I.J.; Maurice, M.M.; van den Boogaard, M.-J.H.; van Haaften, G. Loss-of-function mutations in the WNT co-receptor LRP6 cause autosomal-dominant oligodontia. Am. J. Hum. Genet. 2015, 97, 621–626. [Google Scholar] [CrossRef]
- Yue, H.; Liang, J.; Yang, K.; Hua, B.; Bian, Z. Functional analysis of a novel missense mutation in AXIN 2 associated with non-syndromic tooth agenesis. Eur. J. Oral Sci. 2016, 124, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhao, X.; Hou, F.; Sun, Y.; Wu, J.; Ma, T.; Zhang, X. A novel PAX9 mutation found in a Chinese patient with hypodontia via whole exome sequencing. Oral Dis. 2019, 25, 234–241. [Google Scholar] [CrossRef]
- Bibi, N.; Ahmad, S.; Ahmad, W.; Naeem, M. Molecular genetic analysis of consanguineous Pakistani families with autosomal recessive hypohidrotic ectodermal dysplasia. Australas. J. Dermatol. 2011, 52, 37–42. [Google Scholar] [CrossRef]
- Reed, W.; Lopez, D.; Landing, B. Clinical spectrum of anhidrotic ectodermal dysplasia. Arch. Dermatol. 1970, 102, 134. [Google Scholar] [CrossRef]
- Clarke, A. Hypohidrotic ectodermal dysplasia. J. Med Genet. 1987, 24, 659. [Google Scholar] [CrossRef]
- Viljoen, D.L.; Winship, W.S.; Opitz, J.M.; Reynolds, J.F. A new form of hypohidrotic ectodermal dysplasia. Am. J. Med. Genet. 1988, 31, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, M.; Ideriha, M.; Chautard-Freire-Maia, E.; Freire-Maia, N.; Primo-Parmo, S. Christ-Siemens-Touraine syndrome. Investigations on two large Brazilian kindreds with a new estimate of the manifestation rate among carriers. Hum. Genet. 1981, 57, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Elomaa, O.; Pulkkinen, K.; Hannelius, U.; Mikkola, M.; Saarialho-Kere, U.; Kere, J. Ectodysplasin is released by proteolytic shedding and binds to the EDAR protein. Hum. Mol. Genet. 2001, 10, 953–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppinen, P.; Pispa, J.; Laurikkala, J.; Thesleff, I.; Mikkola, M. Signaling and subcellular localization of the TNF receptor Edar. Exp. Cell Res. 2001, 269, 180–192. [Google Scholar] [CrossRef]
- Kumar, A.; Eby, M.T.; Sinha, S.; Jasmin, A.; Chaudhary, P.M. The ectodermal dysplasia receptor activates the nuclear factor-κB, JNK, and cell death pathways and binds to ectodysplasin A. J. Biol. Chem. 2001, 276, 2668–2677. [Google Scholar] [CrossRef]
- Yan, M.; Wang, L.-C.; Hymowitz, S.G.; Schilbach, S.; Lee, J.; Goddard, A.; de Vos, A.M.; Gao, W.-Q.; Dixit, V.M. Two-amino acid molecular switch in an epithelial morphogen that regulates binding to two distinct receptors. Science 2000, 290, 523–527. [Google Scholar] [CrossRef]
- Bal, E.; Baala, L.; Cluzeau, C.; El Kerch, F.; Ouldim, K.; Hadj-Rabia, S.; Bodemer, C.; Munnich, A.; Courtois, G.; Sefiani, A. Autosomal dominant anhidrotic ectodermal dysplasias at the EDARADD locus. Hum. Mutat. 2007, 28, 703–709. [Google Scholar] [CrossRef]
- Headon, D.J.; Emmal, S.A.; Ferguson, B.M.; Tucker, A.S.; Justice, M.J.; Sharpe, P.T.; Zonana, J.; Overbeek, P.A. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 414, 913. [Google Scholar] [CrossRef]
- Tariq, M.; Wasif, N.; Ayub, M.; Ahmad, W. A novel 4-bp insertion mutation in EDA1 gene in a Pakistani family with X-linked hypohidrotic ectodermal dysplasia. Eur. J. Dermatol. 2007, 17, 209–212. [Google Scholar]
- Van Der Hout, A.H.; Oudesluijs, G.G.; Venema, A.; Verheij, J.B.; Mol, B.G.; Rump, P.; Brunner, H.G.; Vos, Y.J.; Van Essen, A.J. Mutation screening of the Ectodysplasin-A receptor gene EDAR in hypohidrotic ectodermal dysplasia. Eur. J. Hum. Genet. 2008, 16, 673–679. [Google Scholar] [CrossRef] [Green Version]
- Wasif, N.; Tariq, M.; Ali, G.; Hassan, M.J.; Ahmad, W. A Novel Splice Site Mutation in the EDAR Gene Underlies Autosomal Recessive Hypohidrotic Ectodermal Dysplasia in a Pakistani Family. Pediatric Dermatol. 2010, 27, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Adaimy, L.; Chouery, E.; Mégarbané, H.; Mroueh, S.; Delague, V.; Nicolas, E.; Belguith, H.; de Mazancourt, P.; Mégarbané, A. Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: The odonto-onycho-dermal dysplasia. Am. J. Hum. Genet. 2007, 81, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Arzoo, P.S.; Klar, J.; Bergendal, B.; Norderyd, J.; Dahl, N. WNT10A mutations account for ¼ of population-based isolated oligodontia and show phenotypic correlations. Am. J. Med. Genet. Part A 2014, 164, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Bohring, A.; Stamm, T.; Spaich, C.; Haase, C.; Spree, K.; Hehr, U.; Hoffmann, M.; Ledig, S.; Sel, S.; Wieacker, P. WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am. J. Hum. Genet. 2009, 85, 97–105. [Google Scholar] [CrossRef]
- Nagy, N.; Wedgeworth, E.; Hamada, T.; White, J.M.; Hashimoto, T.; McGrath, J.A. Schöpf-Schulz-Passarge syndrome resulting from a homozygous nonsense mutation in WNT10A. J. Dermatol. Sci. 2010, 58, 220–222. [Google Scholar] [CrossRef]
- Van den Boogaard, M.-J.; Créton, M.; Bronkhorst, Y.; van der Hout, A.; Hennekam, E.; Lindhout, D.; Cune, M.; van Amstel, H.K.P. Mutations in WNT10A are present in more than half of isolated hypodontia cases. J. Med. Genet. 2012, 49, 327–331. [Google Scholar] [CrossRef]
- Van Geel, M.; Gattas, M.; Kesler, Y.; Tong, P.; Yan, H.; Tran, K.; Steijlen, P.; Murrell, D.; Van Steensel, M. Phenotypic variability associated with WNT10A nonsense mutations. Br. J. Dermatol. 2010, 162, 1403–1406. [Google Scholar] [CrossRef]
- Parveen, A.; Mirza, M.U.; Vanmeert, M.; Akhtar, J.; Bashir, H.; Khan, S.; Shehzad, S.; Froeyen, M.; Ahmed, W.; Ansar, M. A novel pathogenic missense variant in CNNM4 underlying Jalili syndrome: Insights from molecular dynamics simulations. Mol. Genet. Genom. Med. 2019, 7, 902. [Google Scholar] [CrossRef]
- Saeed, S.; Bonnefond, A.; Tamanini, F.; Mirza, M.U.; Manzoor, J.; Janjua, Q.M.; Din, S.M.; Gaitan, J.; Milochau, A.; Durand, E. Loss-of-function mutations in ADCY3 cause monogenic severe obesity. Nat. Genet. 2018, 50, 175. [Google Scholar] [CrossRef]
- Sun, Y.Z.; Chen, X.B.; Wang, R.R.; Li, W.Y.; Ma, Y. Exploring the effect of N308D mutation on protein tyrosine phosphatase-2 cause gain-of-function activity by a molecular dynamics study. J. Cell. Biochem. 2019, 120, 5949–5961. [Google Scholar] [CrossRef]
- Pirolli, D.; Sciandra, F.; Bozzi, M.; Giardina, B.; Brancaccio, A.; De Rosa, M.C. Insights from molecular dynamics simulations: Structural basis for the V567D mutation-induced instability of zebrafish alpha-dystroglycan and comparison with the murine model. PLoS ONE 2014, 9, e103866. [Google Scholar] [CrossRef] [PubMed]
- Durrani, F.G.; Gul, R.; Mirza, M.U.; Kaderbhai, N.N.; Froeyen, M.; Saleem, M. Mutagenesis of DsbAss is Crucial for the Signal Recognition Particle Mechanism in Escherichia coli: Insights from Molecular Dynamics Simulations. Biomolecules 2019, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Daggett, V.; Levitt, M. Realistic simulations of native-protein dynamics in solution and beyond. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 353–380. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Eastwood, M.P.; Bank, J.A.; Jumper, J.M.; Salmon, J.K.; Shan, Y. Atomic-level characterization of the structural dynamics of proteins. Science 2010, 330, 341–346. [Google Scholar] [CrossRef]
- Janda, C.Y.; Waghray, D.; Levin, A.M.; Thomas, C.; Garcia, K.C. Structural basis of Wnt recognition by Frizzled. Science 2012, 337, 59–64. [Google Scholar] [CrossRef]
- Frappier, V.; Chartier, M.; Najmanovich, R.J. ENCoM server: Exploring protein conformational space and the effect of mutations on protein function and stability. Nucleic Acids Res. 2015, 43, W395–W400. [Google Scholar] [CrossRef]
- Hsieh, J.-C.; Rattner, A.; Smallwood, P.M.; Nathans, J. Biochemical characterization of Wnt-frizzled interactions using a soluble, biologically active vertebrate Wnt protein. Proc. Natl. Acad. Sci. USA 1999, 96, 3546–3551. [Google Scholar] [CrossRef]
- Wang, J.; Shackleford, G.M. Murine Wnt10a and Wnt10b: Cloning and expression in developing limbs, face and skin of embryos and in adults. Oncogene 1996, 13, 1537–1544. [Google Scholar]
- Andl, T.; Reddy, S.T.; Gaddapara, T.; Millar, S.E. WNT signals are required for the initiation of hair follicle development. Dev. Cell 2002, 2, 643–653. [Google Scholar] [CrossRef]
- Järvinen, E.; Birchmeier, W.; Taketo, M.M.; Jernvall, J.; Thesleff, I. Continuous tooth generation in mouse is induced by activated epithelial Wnt/β-catenin signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 18627–18632. [Google Scholar] [CrossRef]
- Liu, F.; Chu, E.Y.; Watt, B.; Zhang, Y.; Gallant, N.M.; Andl, T.; Yang, S.H.; Lu, M.-M.; Piccolo, S.; Schmidt-Ullrich, R. Wnt/β-catenin signaling directs multiple stages of tooth morphogenesis. Dev. Biol. 2008, 313, 210–224. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Kulozik, A.E. A perfect message: RNA surveillance and nonsense-mediated decay. Cell 1999, 96, 307–310. [Google Scholar] [CrossRef]
- Chassaing, N.; Bourthoumieu, S.; Cossee, M.; Calvas, P.; Vincent, M.C. Mutations in EDAR account for one-quarter of non-ED1-related hypohidrotic ectodermal dysplasia. Hum. Mutat. 2006, 27, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, Y.; Wajid, M.; Weiser, J.; Kraemer, L.; Ishii, Y.; Lombillo, V.; Bale, S.; Christiano, A. Identification of mutations in the EDA and EDAR genes in Pakistani families with hypohidrotic ectodermal dysplasia. Clin. Genet. 2009, 75, 582–584. [Google Scholar] [PubMed]
- Gohlke, H.; Kuhn, L.A.; Case, D.A. Change in protein flexibility upon complex formation: Analysis of Ras-Raf using molecular dynamics and a molecular framework approach. PROTEINS Struct. Funct. Bioinform. 2004, 56, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Goethe, M.; Fita, I.; Rubi, J.M. Vibrational entropy of a protein: Large differences between distinct conformations. J. Chem. Theory Comput. 2014, 11, 351–359. [Google Scholar] [CrossRef]
- Ghosh, T.; Garde, S.; García, A.E. Role of backbone hydration and salt-bridge formation in stability of α-helix in solution. Biophys. J. 2003, 85, 3187–3193. [Google Scholar] [CrossRef]
- Vila, J.A.; Ripoll, D.R.; Scheraga, H. Physical reasons for the unusual α-helix stabilization afforded by charged or neutral polar residues in alanine-rich peptides. Proc. Natl. Acad. Sci. USA 2000, 97, 13075–13079. [Google Scholar] [CrossRef]
- Garcia, A.E.; Sanbonmatsu, K.Y. α-Helical stabilization by side chain shielding of backbone hydrogen bonds. Proc. Natl. Acad. Sci. USA 2002, 99, 2782–2787. [Google Scholar] [CrossRef]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into protein–protein binding by binding free energy calculation and free energy decomposition for the Ras–Raf and Ras–RalGDS complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]
- Vincent, M.C.; Biancalana, V.; Ginisty, D.; Mandel, J.L.; Calvas, P. Mutational spectrum of the ED1 gene in X-linked hypohidrotic ectodermal dysplasia. Eur. J. Hum. Genet. 2001, 9, 355. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Zhang, Z.; Brady, J.R.; Schilbach, S.; Fairbrother, W.J.; Dixit, V.M. Identification of a novel death domain-containing adaptor molecule for ectodysplasin-A receptor that is mutated in crinkled mice. Curr. Biol. 2002, 12, 409–413. [Google Scholar] [CrossRef]
- Masui, Y.; Farooq, M.; Sato, N.; Fujimoto, A.; Fujikawa, H.; Ito, M.; Shimomura, Y. A missense mutation in the death domain of EDAR abolishes the interaction with EDARADD and underlies hypohidrotic ectodermal dysplasia. Dermatology 2011, 223, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, Y.; Sato, N.; Miyashita, A.; Hashimoto, T.; Ito, M.; Kuwano, R. A rare case of hypohidrotic ectodermal dysplasia caused by compound heterozygous mutations in the EDAR gene. J. Investig. Dermatol. 2004, 123, 649–655. [Google Scholar] [CrossRef]
- Rasool, M.; Schuster, J.; Aslam, M.; Tariq, M.; Ahmad, I.; Ali, A.; Entesarian, M.; Dahl, N.; Baig, S.M. A novel missense mutation in the EDA gene associated with X-linked recessive isolated hypodontia. J. Hum. Genet. 2008, 53, 894. [Google Scholar] [CrossRef] [PubMed]
- Shahid, M. Single nucleotide polymorphism (SNPs) in the genes associated with tooth agenesis. Eur. Exp. Biol. 2017, 7, 17. [Google Scholar] [CrossRef]
- Kurban, M.; Michailidis, E.; Wajid, M.; Shimomura, Y.; Christiano, A.M. A common founder mutation in the EDA-A1 gene in X-linked hypodontia. Dermatology 2010, 221, 243–247. [Google Scholar] [CrossRef]
- Schneider, P.; Street, S.L.; Gaide, O.; Hertig, S.; Tardivel, A.; Tschopp, J.; Runkel, L.; Alevizopoulos, K.; Ferguson, B.M.; Zonana, J. Mutations leading to X-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-A. J. Biol. Chem. 2001, 276, 18819–18827. [Google Scholar] [CrossRef]
- Wang, J.; Ha, W.-W.; Wang, W.; Tang, H.-Y.; Tang, X.-F.; Zheng, X.-D.; Zhu, J.; Yin, X.-Y.; Yang, S.; Zhang, X.-J. One mutation of the ED1 gene in a chinese han family with X-linked hypohidrotic ectodermal dysplasia. Ann. Dermatol. 2014, 26, 111–113. [Google Scholar] [CrossRef]
- Salas-Alanis, J.C.; Wozniak, E.; Mein, C.A.; Duran Mckinster, C.C.; Ocampo-Candiani, J.; Kelsell, D.P.; Hua, R.; Garza-Rodriguez, M.L.; Choate, K.A.; Barrera Saldaña, H.A. Mutations in EDA and EDAR genes in a large Mexican Hispanic cohort with hypohidrotic ectodermal dysplasia. Ann. Dermatol. 2015, 27, 474–477. [Google Scholar] [CrossRef]
- Bayés, M.; Hartung, A.J.; Ezer, S.; Pispa, J.; Thesleff, I.; Srivastava, A.K.; Kere, J. The anhidrotic ectodermal dysplasia gene (EDA) undergoes alternative splicing and encodes ectodysplasin-A with deletion mutations in collagenous repeats. Hum. Mol. Genet. 1998, 7, 1661–1669. [Google Scholar] [CrossRef]
- Ullah, I.; Kakar, N.; Schrauwen, I.; Hussain, S.; Chakchouk, I.; Liaqat, K.; Acharya, A.; Wasif, N.; Santos-Cortez, R.L.P.; Khan, S. Variants in KIAA0825 underlie autosomal recessive postaxial polydactyly. Hum. Genet. 2019, 138, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Rossi, I.; Casadio, R. A three-state prediction of single point mutations on protein stability changes. BMC Bioinform. 2008, 9, S6. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255. [Google Scholar] [CrossRef] [PubMed]
- May, P.; Woldt, E.; Matz, R.L.; Boucher, P. The LDL receptor-related protein (LRP) family: An old family of proteins with new physiological functions. Ann. Med. 2007, 39, 219–228. [Google Scholar] [CrossRef]
- Shen, G.; Ke, J.; Wang, Z.; Cheng, Z.; Gu, X.; Wei, Y.; Melcher, K.; Xu, H.E.; Xu, W. Structural basis of the Norrin-Frizzled 4 interaction. Cell Res. 2015, 25, 1078. [Google Scholar] [CrossRef]
- Mikkola, M.L.; Thesleff, I. Ectodysplasin signaling in development. Cytokine Growth Factor Rev. 2003, 14, 211–224. [Google Scholar] [CrossRef]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.; Cheatham Iii, T.; Darden, T.; Duke, R.; Gohlke, H. Amber 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2010, 51, 69–82. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models Using SWISS-MODEL | Molprobity | ProSA z-Score | |||
|---|---|---|---|---|---|
| Molprobity Score | Ram.Fav (%) | Ram.Out (%) | Rot.Out (%) | ||
| WNT10A-wt | 1.45 | 91.3 | 2.81 | 2 | −6.82 |
| EDAR-wt | 1.98 | 92.94 | 2.35 | 2.6 | −6.34 |
| EDARADD | 1.82 | 90.91 | 3.21 | 2.84 | −6.02 |
| Mutated Models | DUET | ENCoM | |
|---|---|---|---|
| Consensus Prediction from mCSM and SDM (ΔΔG kcal.mol−1) | Vibrational Entropy Energy (ΔΔS vib kcal.mol−1.K−1) | Thermal Stability (ΔΔG kcal.mol−1) | |
| WNT10A-mt (Arg104His) | −1.431 (destabilizing) | 0.523 (increase in flexibility) | −0.348 (destabilizing) |
| EDAR-mt (Trp434Gly) | −2.762 (destabilizing) | 1.661 (increase in flexibility) | −1.329 (destabilizing) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parveen, A.; Khan, S.A.; Mirza, M.U.; Bashir, H.; Arshad, F.; Iqbal, M.; Ahmad, W.; Wahab, A.; Fiaz, A.; Naz, S.; et al. Deleterious Variants in WNT10A, EDAR, and EDA Causing Isolated and Syndromic Tooth Agenesis: A Structural Perspective from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2019, 20, 5282. https://doi.org/10.3390/ijms20215282
Parveen A, Khan SA, Mirza MU, Bashir H, Arshad F, Iqbal M, Ahmad W, Wahab A, Fiaz A, Naz S, et al. Deleterious Variants in WNT10A, EDAR, and EDA Causing Isolated and Syndromic Tooth Agenesis: A Structural Perspective from Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2019; 20(21):5282. https://doi.org/10.3390/ijms20215282
Chicago/Turabian StyleParveen, Asia, Sher Alam Khan, Muhammad Usman Mirza, Hina Bashir, Fatima Arshad, Maria Iqbal, Waseem Ahmad, Ahsan Wahab, Amal Fiaz, Sidra Naz, and et al. 2019. "Deleterious Variants in WNT10A, EDAR, and EDA Causing Isolated and Syndromic Tooth Agenesis: A Structural Perspective from Molecular Dynamics Simulations" International Journal of Molecular Sciences 20, no. 21: 5282. https://doi.org/10.3390/ijms20215282
APA StyleParveen, A., Khan, S. A., Mirza, M. U., Bashir, H., Arshad, F., Iqbal, M., Ahmad, W., Wahab, A., Fiaz, A., Naz, S., Ashraf, F., Mobeen, T., Aziz, S., Ahmed, S. S., Muhammad, N., Hassib, N. F., Mostafa, M. I., Gaboon, N. E., Gul, R., ... Wasif, N. (2019). Deleterious Variants in WNT10A, EDAR, and EDA Causing Isolated and Syndromic Tooth Agenesis: A Structural Perspective from Molecular Dynamics Simulations. International Journal of Molecular Sciences, 20(21), 5282. https://doi.org/10.3390/ijms20215282