Pathophysiology of Calcium Mediated Ventricular Arrhythmias and Novel Therapeutic Options with Focus on Gene Therapy



Abstract
:1. The Role of Calcium in Action Potential Generation
1.1. The Role of β-Adrenergic Receptor Stimulation
1.2. Cardiac Mitochondrial Ca2+ (mCa2+) Handling
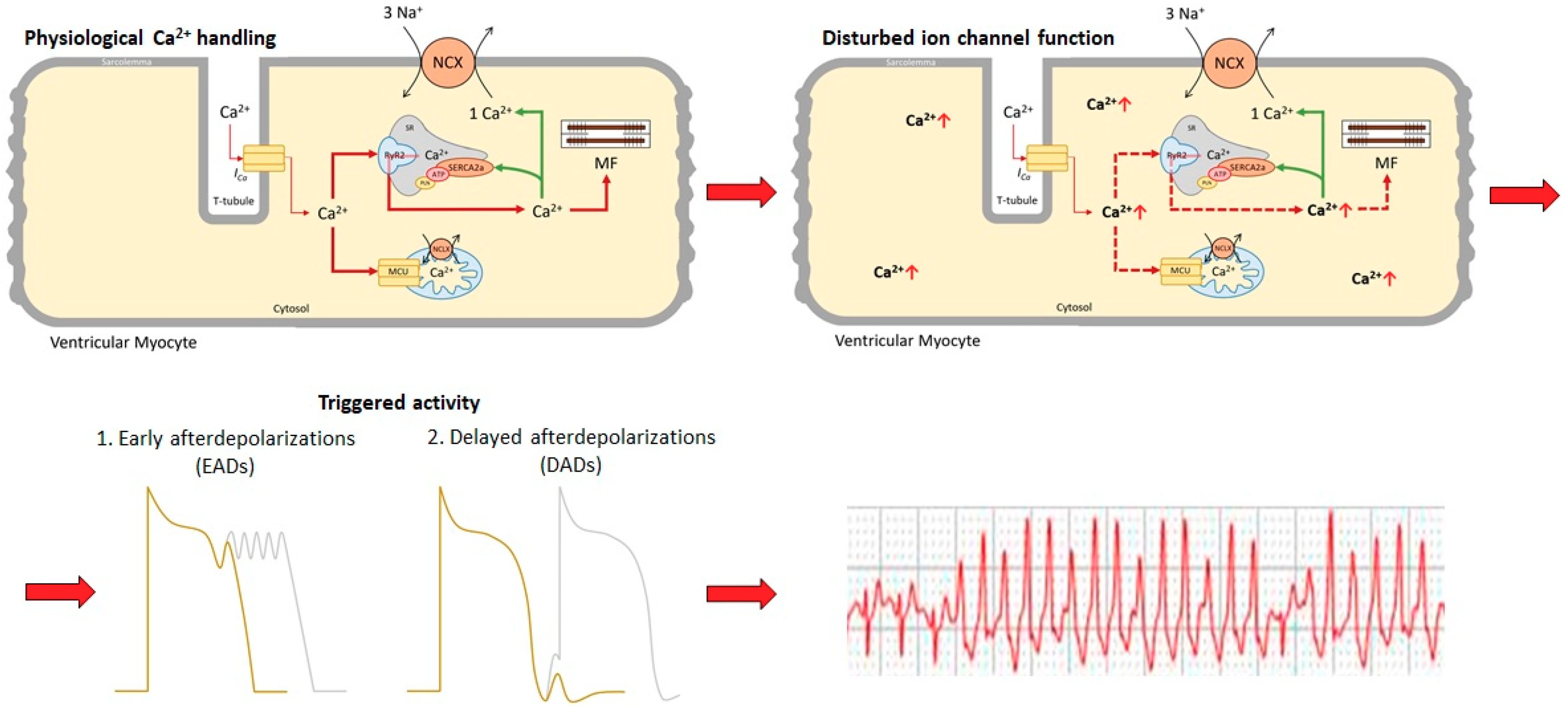
2. Pathophysiology of Calcium Mediated Ventricular Arrhythmia
2.1. Spontaneous Ca2+ Release Events (SCRs)
2.2. Action Potential Prolongation and Afterdepolarizations
2.3. The Role of Mitochondria in Ca2+ Handling of Diseased Heart
3. Novel Therapeutic Options for Calcium Mediated Ventricular Arrhythmias
3.1. Gene Therapy Targeting LTCC
3.2. SERCA2a Gene Therapy
3.3. Gene Therapy Targeting RyR2 Complex
3.4. Cardiac NCX Constituting a Target for Gene Therapy?
3.5. Mitochondrial Ca2+ Channels—Possible Targets for Gene Therapy
4. New Targets for Ca2+ Mediated Ventricular Arrhythmias
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| AD | Adenovirus |
| AIP | Autocamitide-2-related inhibitory peptide |
| AP | Action potential |
| APD | Action potential duration |
| ATP | Adenosine triphosphate |
| β-AR | β-adrenergic receptor |
| Ca2+ | Calcium |
| CGP | CGP317157 |
| CaM | Calmodulin |
| CaMKII | Calcium/calmodulin-dependent protein kinase type II |
| CASQ | Calsequestrin |
| CAST | Cardiac Arrhythmia Suppression Trial |
| CICR | Calcium-induced calcium-release |
| CM | Cardiomyocyte |
| CPVT | Catecholaminergic polymorphic ventricular tachycardia |
| CRAC | Ca2+ release-activated Ca2+ |
| CRISPR/Cas9 | Clustered regularly interspaced short palindromic repeats with caspase 9 |
| DAD | Delayed afterdepolarizations |
| DN | Dominant negative |
| DNA | Deoxyribonucleic acid |
| DR | Downregulation |
| EAD | Early afterdepolarizations |
| EMRE | Essential mitochondrial calcium uniporter regulator |
| GFP | Green fluorescent protein |
| HF | Heart failure |
| I-1c | Inhibitor of protein phosphatase 1 |
| ICa-L | L-type calcium current |
| ICD | Implantable cardioverter defibrillator |
| IMAC | Inner membrane anion channel |
| iPSC | Induced pluripotent stem cell |
| I/R | Ischemia reperfusion |
| Iti | Transient inward current |
| JPH2 | Junctophilin 2 |
| kb | Kilobases |
| kg | Kilograms |
| LTCC | L-type calcium channel |
| LV | Left ventricle |
| MCU | Mitochondrial calcium uniporter |
| MICU1/2 | Mitochondrial calcium uptake 1 and 2 |
| mNCX | Mitochondrial sodium/calcium exchanger |
| Na+ | Sodium |
| NAb | Neutralizing antibodies |
| NCX | Sodium/calcium exchanger |
| NCLX | Mitochondrial sodium/calcium exchanger |
| NKA | Sodium/potassium-ATPase |
| O2− | Superoxide anions |
| OE | Overexpression |
| ORAI | Calcium release-activated calcium channel protein |
| OS | Oxidative stress |
| PAH | Pulmonary arterial hypertension |
| PKA | Protein kinase A |
| PLN | Phospholamban |
| PTP | Permeability transition pore |
| RIRR | Reactive oxygen species-induced reactive oxygen species-release |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| Ru360 | Ruthenium 360 |
| RuR | Ruthenium red |
| RyR2 | Ryanodine receptor type 2 |
| S100A1 | S100 calcium-binding protein A1 |
| SCD | Sudden cardiac death |
| SCR | Spontaneous calcium release events |
| SERCA2a | Sarcoplasmic reticulum calcium-ATPase type-2a |
| siRNA | Short-interfering ribonucleic-acid |
| SOC | Store-operated Ca2+ entry channels |
| SR | Sarcoplasmic reticulum |
| STIM1 | Stromal interaction molecule 1 |
| SUMO-1 | Small ubiquitin-like modifier type 1 |
| TAC | Transverse aortic constriction |
| UCP | Uncoupling protein |
| VF | Ventricular fibrillation |
| VT | Ventricular tachycardia |
| wt | Wild-type |
References
- Schocken, D.D.; Arrieta, M.I.; Leaverton, P.E.; Ross, E.A. Prevalence and mortality rate of congestive heart failure in the United States. J. Am. Coll. Cardiol. 1992, 20, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, C.; Bloise, R.; Monteforte, N.; Priori, S.G. Sudden cardiac death and genetic ion channelopathies: Long QT, Brugada, short QT, catecholaminergic polymorphic ventricular tachycardia, and idiopathic ventricular fibrillation. Circulation 2012, 125, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Jalife, J. Is TGF-β1 (Transforming Growth Factor-β1) an Enabler of Myofibroblast-Cardiomyocyte Cross Talk? Circ. Arrhythm. Electrophysiol. 2017, 10, e005289. [Google Scholar] [CrossRef] [PubMed]
- Estes, N.A.M.; Weinstock, J.; Wang, P.J.; Homoud, M.K.; Link, M.S. Use of antiarrhythmics and implantable cardioverter-defibrillators in congestive heart failure. Am. J. Cardiol. 2003, 91, 45D–52D. [Google Scholar] [CrossRef]
- Echt, D.S.; Liebson, P.R.; Mitchell, L.B.; Peters, R.W.; Obias-Manno, D.; Barker, A.H.; Arensberg, D.; Baker, A.; Friedman, L.; Greene, H.L. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N. Engl. J. Med. 1991, 324, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Schram, G.; Pourrier, M.; Melnyk, P.; Nattel, S. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ. Res. 2002, 90, 939–950. [Google Scholar] [CrossRef]
- Akar, F.G.; Wu, R.C.; Juang, G.J.; Tian, Y.; Burysek, M.; Disilvestre, D.; Xiong, W.; Armoundas, A.A.; Tomaselli, G.F. Molecular mechanisms underlying K+ current downregulation in canine tachycardia-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2887–H2896. [Google Scholar] [CrossRef]
- Jin, H.; Chemaly, E.R.; Lee, A.; Kho, C.; Hadri, L.; Hajjar, R.J.; Akar, F.G. Mechanoelectrical remodeling and arrhythmias during progression of hypertrophy. FASEB J. 2010, 24, 451–463. [Google Scholar] [CrossRef]
- Yano, M.; Ikeda, Y.; Matsuzaki, M. Altered intracellular Ca2+ handling in heart failure. J. Clin. Investig. 2005, 115, 556–564. [Google Scholar] [CrossRef]
- Betzenhauser, M.J.; Pitt, G.S.; Antzelevitch, C. Calcium Channel Mutations in Cardiac Arrhythmia Syndromes. Curr. Mol. Pharmacol. 2015, 8, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Györke, I.; Hester, N.; Jones, L.R.; Györke, S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys. J. 2004, 86, 2121–2128. [Google Scholar] [CrossRef]
- Kotta, M.-C.; Sala, L.; Ghidoni, A.; Badone, B.; Ronchi, C.; Parati, G.; Zaza, A.; Crotti, L. Calmodulinopathy: A Novel, Life-Threatening Clinical Entity Affecting the Young. Front. Cardiovasc. Med. 2018, 5, 175. [Google Scholar] [CrossRef] [PubMed]
- Michels, G.; Khan, I.F.; Endres-Becker, J.; Rottlaender, D.; Herzig, S.; Ruhparwar, A.; Wahlers, T.; Hoppe, U.C. Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels. Circulation 2009, 119, 2435–2443. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Liu, X.; Wang, W. Regulation of metabolism in individual mitochondria during excitation-contraction coupling. J. Mol. Cell. Cardiol. 2014, 76, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Trimarco, B.; Iaccarino, G.; Santulli, G. New Insights in Cardiac Calcium Handling and Excitation-Contraction Coupling. Adv. Exp. Med. Biol. 2018, 1067, 373–385. [Google Scholar]
- Santulli, G.; Lewis, D.; des Georges, A.; Marks, A.R.; Frank, J. Ryanodine Receptor Structure and Function in Health and Disease. Subcell. Biochem. 2018, 87, 329–352. [Google Scholar] [Green Version]
- Santulli, G.; Nakashima, R.; Yuan, Q.; Marks, A.R. Intracellular calcium release channels: An update. J. Physiol. 2017, 595, 3041–3051. [Google Scholar] [CrossRef]
- Fabiato, A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983, 245, C1–C14. [Google Scholar] [CrossRef]
- Stern, M.D.; Cheng, H. Putting out the fire: What terminates calcium-induced calcium release in cardiac muscle? Cell Calcium 2004, 35, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Bassani, J.W.; Bassani, R.A.; Bers, D.M. Relaxation in rabbit and rat cardiac cells: Species-dependent differences in cellular mechanisms. J. Physiol. 1994, 476, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Viatchenko-Karpinski, S.; Valdivia, H.H.; Escobar, A.L.; Györke, S. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ. Res. 2002, 91, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, K.S.; Bers, D.M. Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J. Physiol. 2004, 556, 463–480. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Orchard, C.H. Sarcoplasmic reticulum Ca2+ content, L-type Ca2+ current and the Ca2+ transient in rat myocytes during beta-adrenergic stimulation. J. Physiol. 1997, 505 Pt 2, 385–402. [Google Scholar] [CrossRef]
- Grimm, M.; Brown, J.H. Beta-adrenergic receptor signaling in the heart: Role of CaMKII. J. Mol. Cell. Cardiol. 2010, 48, 322–330. [Google Scholar] [CrossRef]
- Reiken, S.; Gaburjakova, M.; Guatimosim, S.; Gomez, A.M.; D’Armiento, J.; Burkhoff, D.; Wang, J.; Vassort, G.; Lederer, W.J.; Marks, A.R. Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. J. Biol. Chem. 2003, 278, 444–453. [Google Scholar] [CrossRef]
- Wehrens, X.H.T.; Lehnart, S.E.; Reiken, S.R.; Marks, A.R. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 2004, 94, e61–e70. [Google Scholar] [CrossRef]
- Kwong, J.Q. The mitochondrial calcium uniporter in the heart: Energetics and beyond. J. Physiol. 2017, 595, 3743–3751. [Google Scholar] [CrossRef]
- Ramesh, V.; Sharma, V.K.; Sheu, S.S.; Franzini-Armstrong, C. Structural proximity of mitochondria to calcium release units in rat ventricular myocardium may suggest a role in Ca2+ sequestration. Ann. N. Y. Acad. Sci. 1998, 853, 341–344. [Google Scholar] [CrossRef]
- Weiss, R.G.; Gerstenblith, G.; Bottomley, P.A. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc. Natl. Acad. Sci. USA 2005, 102, 808–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Maack, C.; Cortassa, S.; Aon, M.A.; Ganesan, A.N.; Liu, T.; O’Rourke, B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 2006, 99, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Litsky, M.L.; Pfeiffer, D.R. Regulation of the mitochondrial Ca2+ uniporter by external adenine nucleotides: The uniporter behaves like a gated channel which is regulated by nucleotides and divalent cations. Biochemistry 1997, 36, 7071–7080. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Mallilankaraman, K.; Doonan, P.; Cárdenas, C.; Chandramoorthy, H.C.; Müller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgó, J.; Birnbaum, M.J.; et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 2013, 8, e55785. [Google Scholar] [CrossRef]
- Ahuja, M.; Muallem, S. The gatekeepers of mitochondrial calcium influx: MICU1 and MICU2. EMBO Rep. 2014, 15, 205–206. [Google Scholar] [CrossRef] [PubMed]
- Waldeck-Weiermair, M.; Malli, R.; Parichatikanond, W.; Gottschalk, B.; Madreiter-Sokolowski, C.T.; Klec, C.; Rost, R.; Graier, W.F. Rearrangement of MICU1 multimers for activation of MCU is solely controlled by cytosolic Ca2+. Sci. Rep. 2015, 5, 15602. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, U.C. Mitochondrial calcium channels. FEBS Lett. 2010, 584, 1975–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondarenko, A.I.; Parichatikanond, W.; Madreiter, C.T.; Rost, R.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. UCP2 modulates single-channel properties of a MCU-dependent Ca2+ inward current in mitochondria. Pflügers Arch.-Eur. J. Physiol. 2015, 467, 2509–2518. [Google Scholar] [CrossRef]
- Motloch, L.J.; Larbig, R.; Gebing, T.; Reda, S.; Schwaiger, A.; Leitner, J.; Wolny, M.; Eckardt, L.; Hoppe, U.C. By Regulating Mitochondrial Ca2+-Uptake UCP2 Modulates Intracellular Ca2+. PLoS ONE 2016, 11, e0148359. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Parker, N.; Buckingham, J.A.; Vidal-Puig, A.; Halestrap, A.P.; Gunter, T.E.; Nicholls, D.G.; Bernardi, P.; Lemasters, J.J.; Brand, M.D. UCPs—Unlikely calcium porters. Nat. Cell Biol. 2008, 10, 1235–1237. [Google Scholar] [CrossRef]
- Motloch, L.J.; Reda, S.; Wolny, M.; Hoppe, U.C. UCP2 Modulates Cardioprotective Effects of Ru360 in Isolated Cardiomyocytes during Ischemia. Pharm. Basel Switz. 2015, 8, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Motloch, L.J.; Gebing, T.; Reda, S.; Schwaiger, A.; Wolny, M.; Hoppe, U.C. UCP3 Regulates Single-Channel Activity of the Cardiac mCa1. J. Membr. Biol. 2016, 249, 577–584. [Google Scholar] [CrossRef]
- Waldeck-Weiermair, M.; Malli, R.; Naghdi, S.; Trenker, M.; Kahn, M.J.; Graier, W.F. The contribution of UCP2 and UCP3 to mitochondrial Ca2+ uptake is differentially determined by the source of supplied Ca2+. Cell Calcium 2010, 47, 433–440. [Google Scholar] [CrossRef]
- Waldeck-Weiermair, M.; Duan, X.; Naghdi, S.; Khan, M.J.; Trenker, M.; Malli, R.; Graier, W.F. Uncoupling protein 3 adjusts mitochondrial Ca2+ uptake to high and low Ca2+ signals. Cell Calcium 2010, 48, 288–301. [Google Scholar] [CrossRef]
- Larbig, R.; Reda, S.; Paar, V.; Trost, A.; Leitner, J.; Weichselbaumer, S.; Motloch, K.A.; Wernly, B.; Arrer, A.; Strauss, B.; et al. Through modulation of cardiac Ca2+ handling, UCP2 affects cardiac electrophysiology and influences the susceptibility for Ca2+-mediated arrhythmias. Exp. Physiol. 2017, 102, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E.; Tiozzo, R.; Lugli, G.; Crovetti, F.; Kratzing, C. The release of calcium from heart mitochondria by sodium. J. Mol. Cell. Cardiol. 1974, 6, 361–371. [Google Scholar] [CrossRef]
- Jung, D.W.; Baysal, K.; Brierley, G.P. The sodium-calcium antiport of heart mitochondria is not electroneutral. J. Biol. Chem. 1995, 270, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Kohlhaas, M.; Liu, T.; Knopp, A.; Zeller, T.; Ong, M.F.; Böhm, M.; O’Rourke, B.; Maack, C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 2010, 121, 1606–1613. [Google Scholar] [CrossRef]
- Smets, I.; Caplanusi, A.; Despa, S.; Molnar, Z.; Radu, M.; VandeVen, M.; Ameloot, M.; Steels, P. Ca2+ uptake in mitochondria occurs via the reverse action of the Na+/Ca2+ exchanger in metabolically inhibited MDCK cells. Am. J. Physiol. Renal Physiol. 2004, 286, F784–F794. [Google Scholar] [CrossRef]
- Kim, B.; Matsuoka, S. Cytoplasmic Na+-dependent modulation of mitochondrial Ca2+ via electrogenic mitochondrial Na+-Ca2+ exchange. J. Physiol. 2008, 586, 1683–1697. [Google Scholar] [CrossRef]
- Wit, A.L.; Rosen, M.R. Pathophysiologic mechanisms of cardiac arrhythmias. Am. Heart J. 1983, 106, 798–811. [Google Scholar] [CrossRef]
- Akar, F.G.; Hajjar, R.J. Gene therapies for arrhythmias in heart failure. Pflugers Arch. 2014, 466, 1211–1217. [Google Scholar] [CrossRef]
- Motloch, L.J.; Akar, F.G. Gene therapy to restore electrophysiological function in heart failure. Expert Opin. Biol. Ther. 2015, 15, 803–817. [Google Scholar] [CrossRef]
- Marban, E.; Robinson, S.W.; Wier, W.G. Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. J. Clin. Investig. 1986, 78, 1185–1192. [Google Scholar] [CrossRef]
- January, C.T.; Moscucci, A. Cellular mechanisms of early afterdepolarizations. Ann. N. Y. Acad. Sci. 1992, 644, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Tse, G. Mechanisms of cardiac arrhythmias. J. Arrhythm. 2016, 32, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Venetucci, L.A.; Trafford, A.W.; O’Neill, S.C.; Eisner, D.A. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc. Res. 2008, 77, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Venetucci, L.A.; Trafford, A.W.; Eisner, D.A. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: Threshold sarcoplasmic reticulum calcium content is required. Circ. Res. 2007, 100, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, P.J.; Brown, K.M.; Piippo, K.; Swan, H.; Devaney, J.M.; Brahmbhatt, B.; Donarum, E.A.; Marino, M.; Tiso, N.; Viitasalo, M.; et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001, 103, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Postma, A.V.; Denjoy, I.; Hoorntje, T.M.; Lupoglazoff, J.-M.; Da Costa, A.; Sebillon, P.; Mannens, M.M.A.M.; Wilde, A.A.M.; Guicheney, P. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2002, 91, e21–e26. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Xiao, B.; Yang, D.; Wang, R.; Choi, P.; Zhang, L.; Cheng, H.; Chen, S.R.W. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc. Natl. Acad. Sci. USA 2004, 101, 13062–13067. [Google Scholar] [CrossRef]
- Jiang, D.; Wang, R.; Xiao, B.; Kong, H.; Hunt, D.J.; Choi, P.; Zhang, L.; Chen, S.R.W. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ. Res. 2005, 97, 1173–1181. [Google Scholar] [CrossRef]
- Terentyev, D.; Nori, A.; Santoro, M.; Viatchenko-Karpinski, S.; Kubalova, Z.; Gyorke, I.; Terentyeva, R.; Vedamoorthyrao, S.; Blom, N.A.; Valle, G.; et al. Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ. Res. 2006, 98, 1151–1158. [Google Scholar] [CrossRef]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef]
- Ai, X.; Curran, J.W.; Shannon, T.R.; Bers, D.M.; Pogwizd, S.M. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 2005, 97, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Eisner, D.A.; Trafford, A.W.; Díaz, M.E.; Overend, C.L.; O’Neill, S.C. The control of Ca release from the cardiac sarcoplasmic reticulum: Regulation versus autoregulation. Cardiovasc. Res. 1998, 38, 589–604. [Google Scholar] [CrossRef]
- Liu, N.; Ruan, Y.; Denegri, M.; Bachetti, T.; Li, Y.; Colombi, B.; Napolitano, C.; Coetzee, W.A.; Priori, S.G. Calmodulin kinase II inhibition prevents arrhythmias in RyR2(R4496C+/-) mice with catecholaminergic polymorphic ventricular tachycardia. J. Mol. Cell. Cardiol. 2011, 50, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.S.; Bers, D.M. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc. Res. 2007, 73, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Brooksby, P.; Levi, A.J.; Jones, J.V. The electrophysiological characteristics of hypertrophied ventricular myocytes from the spontaneously hypertensive rat. J. Hypertens. 1993, 11, 611–622. [Google Scholar] [CrossRef]
- Cerbai, E.; Barbieri, M.; Li, Q.; Mugelli, A. Ionic basis of action potential prolongation of hypertrophied cardiac myocytes isolated from hypertensive rats of different ages. Cardiovasc. Res. 1994, 28, 1180–1187. [Google Scholar] [CrossRef]
- Richard, S.; Leclercq, F.; Lemaire, S.; Piot, C.; Nargeot, J. Ca2+ currents in compensated hypertrophy and heart failure. Cardiovasc. Res. 1998, 37, 300–311. [Google Scholar] [CrossRef]
- Hill, J.A. Electrical remodeling in cardiac hypertrophy. Trends Cardiovasc. Med. 2003, 13, 316–322. [Google Scholar] [CrossRef]
- Ryder, K.O.; Bryant, S.M.; Hart, G. Membrane current changes in left ventricular myocytes isolated from guinea pigs after abdominal aortic coarctation. Cardiovasc. Res. 1993, 27, 1278–1287. [Google Scholar] [CrossRef]
- Ouadid, H.; Albat, B.; Nargeot, J. Calcium currents in diseased human cardiac cells. J. Cardiovasc. Pharmacol. 1995, 25, 282–291. [Google Scholar] [CrossRef]
- de Lucia, C.; Eguchi, A.; Koch, W.J. New Insights in Cardiac β-Adrenergic Signaling During Heart Failure and Aging. Front. Pharmacol. 2018, 9, 904. [Google Scholar] [CrossRef] [PubMed]
- Letsas, K.P.; Sacher, F.; Probst, V.; Weber, R.; Knecht, S.; Kalusche, D.; Haïssaguerre, M.; Arentz, T. Prevalence of early repolarization pattern in inferolateral leads in patients with Brugada syndrome. Heart Rhythm 2008, 5, 1685–1689. [Google Scholar] [CrossRef] [PubMed]
- Burashnikov, E.; Pfeiffer, R.; Barajas-Martinez, H.; Delpón, E.; Hu, D.; Desai, M.; Borggrefe, M.; Häissaguerre, M.; Kanter, R.; Pollevick, G.D.; et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 2010, 7, 1872–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Templin, C.; Ghadri, J.-R.; Rougier, J.-S.; Baumer, A.; Kaplan, V.; Albesa, M.; Sticht, H.; Rauch, A.; Puleo, C.; Hu, D.; et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur. Heart J. 2011, 32, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Bjerregaard, P.; Nallapaneni, H.; Gussak, I. Short QT interval in clinical practice. J. Electrocardiol. 2010, 43, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Makiyama, T.; Koyama, T.; Kannankeril, P.J.; Seto, S.; Okamura, K.; Oda, H.; Itoh, H.; Okada, M.; Tanabe, N.; et al. High prevalence of early repolarization in short QT syndrome. Heart Rhythm 2010, 7, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, C.; Antzelevitch, C. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac voltage-dependent L-type calcium channel. Circ. Res. 2011, 108, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Pogwizd, S.M.; Schlotthauer, K. Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res. Cardiol. 2002, 97 (Suppl. 1), I36–I42. [Google Scholar] [CrossRef] [PubMed]
- Lederer, W.J.; Tsien, R.W. Transient inward current underlying arrhythmogenic effects of cardiotonic steroids in Purkinje fibres. J. Physiol. 1976, 263, 73–100. [Google Scholar] [CrossRef]
- Rahm, A.-K.; Lugenbiel, P.; Schweizer, P.A.; Katus, H.A.; Thomas, D. Role of ion channels in heart failure and channelopathies. Biophys. Rev. 2018, 10, 1097–1106. [Google Scholar] [CrossRef]
- Shaw, R.M.; Rudy, Y. The vulnerable window for unidirectional block in cardiac tissue: Characterization and dependence on membrane excitability and intercellular coupling. J. Cardiovasc. Electrophysiol. 1995, 6, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabò, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Sommakia, S.; Houlihan, P.R.; Deane, S.S.; Simcox, J.A.; Torres, N.S.; Jeong, M.-Y.; Winge, D.R.; Villanueva, C.J.; Chaudhuri, D. Mitochondrial cardiomyopathies feature increased uptake and diminished efflux of mitochondrial calcium. J. Mol. Cell. Cardiol. 2017, 113, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [Green Version]
- Odagiri, K.; Katoh, H.; Kawashima, H.; Tanaka, T.; Ohtani, H.; Saotome, M.; Urushida, T.; Satoh, H.; Hayashi, H. Local control of mitochondrial membrane potential, permeability transition pore and reactive oxygen species by calcium and calmodulin in rat ventricular myocytes. J. Mol. Cell. Cardiol. 2009, 46, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: An update and review. Biochim. Biophys. Acta 2006, 1757, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Aon, M.A.; Cortassa, S.; Marbán, E.; O’Rourke, B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 2003, 278, 44735–44744. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; O’Rourke, B. The Fundamental Organization of Cardiac Mitochondria as a Network of Coupled Oscillators. Biophys. J. 2006, 91, 4317–4327. [Google Scholar] [CrossRef] [Green Version]
- Aon, M.A.; Cortassa, S.; Maack, C.; O’Rourke, B. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J. Biol. Chem. 2007, 282, 21889–21900. [Google Scholar] [CrossRef] [PubMed]
- Hopper, R.K.; Carroll, S.; Aponte, A.M.; Johnson, D.T.; French, S.; Shen, R.-F.; Witzmann, F.A.; Harris, R.A.; Balaban, R.S. Mitochondrial matrix phosphoproteome: Effect of extra mitochondrial calcium. Biochemistry 2006, 45, 2524–2536. [Google Scholar] [CrossRef] [PubMed]
- Motloch, L.J.; Hu, J.; Akar, F.G. The Mitochondrial Translocator Protein and Arrhythmogenesis in Ischemic Heart Disease. Oxid. Med. Cell. Longev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Sorriento, D.; Ciccarelli, M.; Del Giudice, C.; Fiordelisi, A.; Napolitano, L.; Trimarco, B.; Iaccarino, G.; Santulli, G. Functional Role of Mitochondria in Arrhythmogenesis. Adv. Exp. Med. Biol. 2017, 982, 191–202. [Google Scholar]
- Nakayama, H.; Chen, X.; Baines, C.P.; Klevitsky, R.; Zhang, X.; Zhang, H.; Jaleel, N.; Chua, B.H.L.; Hewett, T.E.; Robbins, J.; et al. Ca2+-and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J. Clin. Investig. 2007, 117, 2431–2444. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Fozzard, H.A. Cardiac sodium and calcium channels: A history of excitatory currents. Cardiovasc. Res. 2002, 55, 1–8. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef]
- Briot, J.; Tétreault, M.-P.; Bourdin, B.; Parent, L. Inherited Ventricular Arrhythmias: The Role of the Multi-Subunit Structure of the L-Type Calcium Channel Complex. Adv. Exp. Med. Biol. 2017, 966, 55–64. [Google Scholar]
- Hamilton, S.; Terentyev, D. Altered Intracellular Calcium Homeostasis and Arrhythmogenesis in the Aged Heart. Int. J. Mol. Sci. 2019, 20, 2386. [Google Scholar] [CrossRef]
- Tomaselli, G.F.; Beuckelmann, D.J.; Calkins, H.G.; Berger, R.D.; Kessler, P.D.; Lawrence, J.H.; Kass, D.; Feldman, A.M.; Marban, E. Sudden cardiac death in heart failure. The role of abnormal repolarization. Circulation 1994, 90, 2534–2539. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, A. History of calcium antagonists. Circ. Res. 1983, 52, I3–I16. [Google Scholar]
- Rosso, R.; Kalman, J.M.; Rogowski, O.; Diamant, S.; Birger, A.; Biner, S.; Belhassen, B.; Viskin, S. Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2007, 4, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N. A fourth class of anti-dysrhythmic action? Effect of verapamil on ouabain toxicity, on atrial and ventricular intracellular potentials, and on other features of cardiac function. Cardiovasc. Res. 1972, 6, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Padial, L.; Barón-Esquivias, G.; Hernández Madrid, A.; Marzal Martín, D.; Pallarés-Carratalá, V.; de la Sierra, A. Clinical Experience with Diltiazem in the Treatment of Cardiovascular Diseases. Cardiol. Ther. 2016, 5, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Page, R.L.; Joglar, J.A.; Caldwell, M.A.; Calkins, H.; Conti, J.B.; Deal, B.J.; Estes, N.A.M.; Field, M.E.; Goldberger, Z.D.; Hammill, S.C.; et al. 2015 ACC/AHA/HRS Guideline for the Management of Adult Patients With Supraventricular Tachycardia: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J. Am. Coll. Cardiol. 2016, 67, e27–e115. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Chopra, N.; Laver, D.; Hwang, H.S.; Davies, S.S.; Roach, D.E.; Duff, H.J.; Roden, D.M.; Wilde, A.A.M.; Knollmann, B.C. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat. Med. 2009, 15, 380–383. [Google Scholar] [CrossRef]
- van der Werf, C.; Kannankeril, P.J.; Sacher, F.; Krahn, A.D.; Viskin, S.; Leenhardt, A.; Shimizu, W.; Sumitomo, N.; Fish, F.A.; Bhuiyan, Z.A.; et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J. Am. Coll. Cardiol. 2011, 57, 2244–2254. [Google Scholar] [CrossRef]
- Singh, S.N.; Fletcher, R.D.; Fisher, S.G.; Singh, B.N.; Lewis, H.D.; Deedwania, P.C.; Massie, B.M.; Colling, C.; Lazzeri, D. Amiodarone in patients with congestive heart failure and asymptomatic ventricular arrhythmia. Survival Trial of Antiarrhythmic Therapy in Congestive Heart Failure. N. Engl. J. Med. 1995, 333, 77–82. [Google Scholar] [CrossRef]
- Waldo, A.L.; Camm, A.J.; deRuyter, H.; Friedman, P.L.; MacNeil, D.J.; Pauls, J.F.; Pitt, B.; Pratt, C.M.; Schwartz, P.J.; Veltri, E.P. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. The SWORD Investigators. Survival With Oral d-Sotalol. Lancet Lond. Engl. 1996, 348, 7–12. [Google Scholar] [CrossRef]
- Køber, L.; Thomsen, P.E.B.; Møller, M.; Torp-Pedersen, C.; Carlsen, J.; Sandøe, E.; Egstrup, K.; Agner, E.; Videbaek, J.; Marchant, B.; et al. Effect of dofetilide in patients with recent myocardial infarction and left-ventricular dysfunction: A randomised trial. Lancet Lond. Engl. 2000, 356, 2052–2058. [Google Scholar] [CrossRef]
- Kuck, K.H.; Cappato, R.; Siebels, J.; Rüppel, R. Randomized comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from cardiac arrest: The Cardiac Arrest Study Hamburg (CASH). Circulation 2000, 102, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Donahue, J.K. Gene therapy for ventricular tachyarrhythmias. Gene Ther. 2012, 19, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Gould, P.A.; Krahn, A.D. Canadian Heart Rhythm Society Working Group on Device Advisories Complications associated with implantable cardioverter-defibrillator replacement in response to device advisories. JAMA 2006, 295, 1907–1911. [Google Scholar] [CrossRef] [PubMed]
- Bonci, D.; Cittadini, A.; Latronico, M.V.G.; Borello, U.; Aycock, J.K.; Drusco, A.; Innocenzi, A.; Follenzi, A.; Lavitrano, M.; Monti, M.G.; et al. “Advanced” generation lentiviruses as efficient vectors for cardiomyocyte gene transduction in vitro and in vivo. Gene Ther. 2003, 10, 630–636. [Google Scholar] [CrossRef]
- Cockrell, A.S.; Kafri, T. Gene delivery by lentivirus vectors. Mol. Biotechnol. 2007, 36, 184–204. [Google Scholar] [CrossRef]
- Bongianino, R.; Priori, S.G. Gene therapy to treat cardiac arrhythmias. Nat. Rev. Cardiol. 2015, 12, 531–546. [Google Scholar] [CrossRef]
- Hulot, J.-S.; Ishikawa, K.; Hajjar, R.J. Gene therapy for the treatment of heart failure: Promise postponed. Eur. Heart J. 2016, 37, 1651–1658. [Google Scholar] [CrossRef]
- Greenberg, B. Gene therapy for heart failure. Trends Cardiovasc. Med. 2017, 27, 216–222. [Google Scholar] [CrossRef]
- Alba, R.; Bosch, A.; Chillon, M. Gutless adenovirus: Last-generation adenovirus for gene therapy. Gene Ther. 2005, 12 (Suppl. 1), S18–S27. [Google Scholar] [CrossRef]
- Liu, Z.; Donahue, J.K. The Use of Gene Therapy for Ablation of Atrial Fibrillation. Arrhythmia Electrophysiol. Rev. 2014, 3, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Rabinowitz, J.E. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gao, G. State-of-the-art human gene therapy: Part I. Gene delivery technologies. Discov. Med. 2014, 18, 67–77. [Google Scholar] [PubMed]
- Wang, D.; Gao, G. State-of-the-art human gene therapy: Part II. Gene therapy strategies and clinical applications. Discov. Med. 2014, 18, 151–161. [Google Scholar] [PubMed]
- Fish, K.M.; Ishikawa, K. Advances in gene therapy for heart failure. Discov. Med. 2015, 19, 285–291. [Google Scholar] [PubMed]
- Hammoudi, N.; Ishikawa, K.; Hajjar, R.J. Adeno-associated virus-mediated gene therapy in cardiovascular disease. Curr. Opin. Cardiol. 2015, 30, 228–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Gori, J.L.; Hsu, P.D.; Maeder, M.L.; Shen, S.; Welstead, G.G.; Bumcrot, D. Delivery and Specificity of CRISPR-Cas9 Genome Editing Technologies for Human Gene Therapy. Hum. Gene Ther. 2015, 26, 443–451. [Google Scholar] [CrossRef]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Castellanos Rivera, R.M.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Keeler, A.M.; ElMallah, M.K.; Flotte, T.R. Gene Therapy 2017: Progress and Future Directions. Clin. Transl. Sci. 2017, 10, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.H.; Marsh, P.; Schmiess-Heine, L.; Burke, P.J.; Lee, A.; Lee, J.; Cao, H. Cardiac tissue engineering: State-of-the-art methods and outlook. J. Biol. Eng. 2019, 13, 57. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, R.E.; Boccuzzi, S.J.; Cruess, D.; Nattel, S. Diltiazem increases late-onset congestive heart failure in postinfarction patients with early reduction in ejection fraction. The Adverse Experience Committee; and the Multicenter Diltiazem Postinfarction Research Group. Circulation 1991, 83, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Cingolani, E.; McDonald, A.D.; Donahue, J.K.; Marbán, E. Creation of a genetic calcium channel blocker by targeted gem gene transfer in the heart. Circ. Res. 2004, 95, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, E.; Ramirez Correa, G.A.; Kizana, E.; Murata, M.; Cho, H.C.; Marbán, E. Gene therapy to inhibit the calcium channel beta subunit: Physiological consequences and pathophysiological effects in models of cardiac hypertrophy. Circ. Res. 2007, 101, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Subramanyam, P.; Chang, D.D.; Fang, K.; Xie, W.; Marks, A.R.; Colecraft, H.M. Manipulating L-type calcium channels in cardiomyocytes using split-intein protein transsplicing. Proc. Natl. Acad. Sci. USA 2013, 110, 15461–15466. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, B.; Kass, D.A.; Tomaselli, G.F.; Kääb, S.; Tunin, R.; Marbán, E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: Experimental studies. Circ. Res. 1999, 84, 562–570. [Google Scholar] [CrossRef]
- Kawase, Y.; Ly, H.Q.; Prunier, F.; Lebeche, D.; Shi, Y.; Jin, H.; Hadri, L.; Yoneyama, R.; Hoshino, K.; Takewa, Y.; et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J. Am. Coll. Cardiol. 2008, 51, 1112–1119. [Google Scholar] [CrossRef]
- Mariani, J.A.; Smolic, A.; Preovolos, A.; Byrne, M.J.; Power, J.M.; Kaye, D.M. Augmentation of left ventricular mechanics by recirculation-mediated AAV2/1-SERCA2a gene delivery in experimental heart failure. Eur. J. Heart Fail. 2011, 13, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Byrne, M.J.; Power, J.M.; Preovolos, A.; Mariani, J.A.; Hajjar, R.J.; Kaye, D.M. Recirculating cardiac delivery of AAV2/1SERCA2a improves myocardial function in an experimental model of heart failure in large animals. Gene Ther. 2008, 15, 1550–1557. [Google Scholar] [CrossRef] [Green Version]
- del Monte, F.; Lebeche, D.; Guerrero, J.L.; Tsuji, T.; Doye, A.A.; Gwathmey, J.K.; Hajjar, R.J. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc. Natl. Acad. Sci. USA 2004, 101, 5622–5627. [Google Scholar] [CrossRef] [Green Version]
- Prunier, F.; Kawase, Y.; Gianni, D.; Scapin, C.; Danik, S.B.; Ellinor, P.T.; Hajjar, R.J.; Del Monte, F. Prevention of ventricular arrhythmias with sarcoplasmic reticulum Ca2+ ATPase pump overexpression in a porcine model of ischemia reperfusion. Circulation 2008, 118, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Cutler, M.J.; Wan, X.; Laurita, K.R.; Hajjar, R.J.; Rosenbaum, D.S. Targeted SERCA2a gene expression identifies molecular mechanism and therapeutic target for arrhythmogenic cardiac alternans. Circ. Arrhythm. Electrophysiol. 2009, 2, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.R.; Bannister, M.L.; Collins, T.; Pearce, E.; Sepehripour, A.H.; Dubb, S.S.; Garcia, E.; O’Gara, P.; Liang, L.; Kohlbrenner, E.; et al. SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ. Arrhythm. Electrophysiol. 2011, 4, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Cutler, M.J.; Wan, X.; Plummer, B.N.; Liu, H.; Deschenes, I.; Laurita, K.R.; Hajjar, R.J.; Rosenbaum, D.S. Targeted sarcoplasmic reticulum Ca2+ ATPase 2a gene delivery to restore electrical stability in the failing heart. Circulation 2012, 126, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Davia, K.; Bernobich, E.; Ranu, H.K.; del Monte, F.; Terracciano, C.M.; MacLeod, K.T.; Adamson, D.L.; Chaudhri, B.; Hajjar, R.J.; Harding, S.E. SERCA2A overexpression decreases the incidence of aftercontractions in adult rabbit ventricular myocytes. J. Mol. Cell. Cardiol. 2001, 33, 1005–1015. [Google Scholar] [CrossRef]
- Xie, L.-H.; Sato, D.; Garfinkel, A.; Qu, Z.; Weiss, J.N. Intracellular Ca alternans: Coordinated regulation by sarcoplasmic reticulum release, uptake, and leak. Biophys. J. 2008, 95, 3100–3110. [Google Scholar] [CrossRef]
- Giordano, F.J.; He, H.; McDonough, P.; Meyer, M.; Sayen, M.R.; Dillmann, W.H. Adenovirus-mediated gene transfer reconstitutes depressed sarcoplasmic reticulum Ca2+-ATPase levels and shortens prolonged cardiac myocyte Ca2+ transients. Circulation 1997, 96, 400–403. [Google Scholar] [CrossRef]
- Hajjar, R.J.; Kang, J.X.; Gwathmey, J.K.; Rosenzweig, A. Physiological effects of adenoviral gene transfer of sarcoplasmic reticulum calcium ATPase in isolated rat myocytes. Circulation 1997, 95, 423–429. [Google Scholar] [CrossRef]
- Terracciano, C.M.N.; Hajjar, R.J.; Harding, S.E. Overexpression of SERCA2a accelerates repolarisation in rabbit ventricular myocytes. Cell Calcium 2002, 31, 299–305. [Google Scholar] [CrossRef]
- Motloch, L.J.; Cacheux, M.; Ishikawa, K.; Xie, C.; Hu, J.; Aguero, J.; Fish, K.M.; Hajjar, R.J.; Akar, F.G. Primary Effect of SERCA 2a Gene Transfer on Conduction Reserve in Chronic Myocardial Infarction. J. Am. Heart Assoc. 2018, 7, e009598. [Google Scholar] [CrossRef]
- Strauss, B.; Sassi, Y.; Bueno-Beti, C.; Ilkan, Z.; Raad, N.; Cacheux, M.; Bisserier, M.; Turnbull, I.C.; Kohlbrenner, E.; Hajjar, R.J.; et al. Intra-tracheal gene delivery of aerosolized SERCA2a to the lung suppresses ventricular arrhythmias in a model of pulmonary arterial hypertension. J. Mol. Cell. Cardiol. 2019, 127, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Motloch, L.J.; Ishikawa, K.; Xie, C.; Hu, J.; Aguero, J.; Fish, K.M.; Hajjar, R.J.; Akar, F.G. Increased afterload following myocardial infarction promotes conduction-dependent arrhythmias that are unmasked by hypokalemia. JACC Basic Transl. Sci. 2017, 2, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yano, M.; Uchinoumi, H.; Hino, A.; Suetomi, T.; Ono, M.; Tateishi, H.; Oda, T.; Okuda, S.; Doi, M.; et al. Defective calmodulin binding to the cardiac ryanodine receptor plays a key role in CPVT-associated channel dysfunction. Biochem. Biophys. Res. Commun. 2010, 394, 660–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, M.; Yano, M.; Hino, A.; Suetomi, T.; Xu, X.; Susa, T.; Uchinoumi, H.; Tateishi, H.; Oda, T.; Okuda, S.; et al. Dissociation of calmodulin from cardiac ryanodine receptor causes aberrant Ca2+ release in heart failure. Cardiovasc. Res. 2010, 87, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Kushnir, A.; Santulli, G.; Reiken, S.R.; Coromilas, E.; Godfrey, S.J.; Brunjes, D.L.; Colombo, P.C.; Yuzefpolskaya, M.; Sokol, S.I.; Kitsis, R.N.; et al. Ryanodine Receptor Calcium Leak in Circulating B-Lymphocytes as a Biomarker in Heart Failure. Circulation 2018, 138, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Hasenfuss, G.; Teerlink, J.R. Cardiac inotropes: Current agents and future directions. Eur. Heart J. 2011, 32, 1838–1845. [Google Scholar] [CrossRef]
- Bongianino, R.; Denegri, M.; Mazzanti, A.; Lodola, F.; Vollero, A.; Boncompagni, S.; Fasciano, S.; Rizzo, G.; Mangione, D.; Barbaro, S.; et al. Allele-Specific Silencing of Mutant mRNA Rescues Ultrastructural and Arrhythmic Phenotype in Mice Carriers of the R4496C Mutation in the Ryanodine Receptor Gene (RYR2). Circ. Res. 2017, 121, 525–536. [Google Scholar] [CrossRef]
- Pan, X.; Philippen, L.; Lahiri, S.K.; Lee, C.; Park, S.H.; Word, T.A.; Li, N.; Jarrett, K.E.; Gupta, R.; Reynolds, J.O.; et al. In Vivo Ryr2 Editing Corrects Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2018, 123, 953–963. [Google Scholar] [CrossRef]
- Denegri, M.; Bongianino, R.; Lodola, F.; Boncompagni, S.; De Giusti, V.C.; Avelino-Cruz, J.E.; Liu, N.; Persampieri, S.; Curcio, A.; Esposito, F.; et al. Single delivery of an adeno-associated viral construct to transfer the CASQ2 gene to knock-in mice affected by catecholaminergic polymorphic ventricular tachycardia is able to cure the disease from birth to advanced age. Circulation 2014, 129, 2673–2681. [Google Scholar] [CrossRef]
- Lodola, F.; Morone, D.; Denegri, M.; Bongianino, R.; Nakahama, H.; Rutigliano, L.; Gosetti, R.; Rizzo, G.; Vollero, A.; Buonocore, M.; et al. Adeno-associated virus-mediated CASQ2 delivery rescues phenotypic alterations in a patient-specific model of recessive catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 2016, 7, e2393. [Google Scholar] [CrossRef]
- Kurtzwald-Josefson, E.; Yadin, D.; Harun-Khun, S.; Waldman, M.; Aravot, D.; Shainberg, A.; Eldar, M.; Hochhauser, E.; Arad, M. Viral delivered gene therapy to treat catecholaminergic polymorphic ventricular tachycardia (CPVT2) in mouse models. Heart Rhythm 2017, 14, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Walton, S.D.; Ho, H.-T.; Belevych, A.E.; Tikunova, S.B.; Bonilla, I.; Shettigar, V.; Knollmann, B.C.; Priori, S.G.; Volpe, P.; et al. Gene Transfer of Engineered Calmodulin Alleviates Ventricular Arrhythmias in a Calsequestrin-Associated Mouse Model of Catecholaminergic Polymorphic Ventricular Tachycardia. J. Am. Heart Assoc. 2018, 7, e008155. [Google Scholar] [CrossRef] [PubMed]
- Bezzerides, V.J.; Caballero, A.; Wang, S.; Ai, Y.; Hylind, R.J.; Lu, F.; Heims-Waldron, D.A.; Chambers, K.D.; Zhang, D.; Abrams, D.J.; et al. Gene Therapy for Catecholaminergic Polymorphic Ventricular Tachycardia by Inhibition of Ca2+/Calmodulin-Dependent Kinase II. Circulation 2019, 140, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Zhang, D.; Qi, Y.; Li, Y.; Lee, K.Y.; Bezzerides, V.J.; Yang, P.; Xia, S.; Kim, S.L.; Liu, X.; et al. Insights Into the Pathogenesis of Catecholaminergic Polymorphic Ventricular Tachycardia From Engineered Human Heart Tissue. Circulation 2019, 140, 390–404. [Google Scholar] [CrossRef]
- Di Pasquale, E.; Lodola, F.; Miragoli, M.; Denegri, M.; Avelino-Cruz, J.E.; Buonocore, M.; Nakahama, H.; Portararo, P.; Bloise, R.; Napolitano, C.; et al. CaMKII inhibition rectifies arrhythmic phenotype in a patient-specific model of catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 2013, 4, e843. [Google Scholar] [CrossRef] [PubMed]
- Knollmann, B.C.; Chopra, N.; Hlaing, T.; Akin, B.; Yang, T.; Ettensohn, K.; Knollmann, B.E.C.; Horton, K.D.; Weissman, N.J.; Holinstat, I.; et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2006, 116, 2510–2520. [Google Scholar] [CrossRef] [PubMed]
- Viatchenko-Karpinski, S.; Terentyev, D.; Györke, I.; Terentyeva, R.; Volpe, P.; Priori, S.G.; Napolitano, C.; Nori, A.; Williams, S.C.; Györke, S. Abnormal calcium signaling and sudden cardiac death associated with mutation of calsequestrin. Circ. Res. 2004, 94, 471–477. [Google Scholar] [CrossRef]
- Faggioni, M.; Kryshtal, D.O.; Knollmann, B.C. Calsequestrin mutations and catecholaminergic polymorphic ventricular tachycardia. Pediatr. Cardiol. 2012, 33, 959–967. [Google Scholar] [CrossRef]
- Adachi-Akahane, S.; Lu, L.; Li, Z.; Frank, J.S.; Philipson, K.D.; Morad, M. Calcium Signaling in Transgenic Mice Overexpressing Cardiac Na+-Ca2+ Exchanger. J. Gen. Physiol. 1997, 109, 717–729. [Google Scholar] [CrossRef]
- Yao, A.; Su, Z.; Nonaka, A.; Zubair, I.; Lu, L.; Philipson, K.D.; Bridge, J.H.; Barry, W.H. Effects of overexpression of the Na+-Ca2+ exchanger on [Ca2+] i transients in murine ventricular myocytes. Circ. Res. 1998, 82, 657–665. [Google Scholar] [CrossRef]
- Terracciano, C.M.; Souza, A.I.; Philipson, K.D.; MacLeod, K.T. Na+-Ca2+ exchange and sarcoplasmic reticular Ca2+ regulation in ventricular myocytes from transgenic mice overexpressing the Na+-Ca2+ exchanger. J. Physiol. 1998, 512 Pt 3, 651–667. [Google Scholar] [CrossRef] [PubMed]
- Reuter, H.; Han, T.; Motter, C.; Philipson, K.D.; Goldhaber, J.I. Mice overexpressing the cardiac sodium-calcium exchanger: Defects in excitation–contraction coupling. J. Physiol. 2004, 554, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, K.; Kobayashi, K.; Kuro, O.M.; Yao, A.; Iwamoto, T.; Yanaka, N.; Kita, S.; Nishida, A.; Azuma, S.; Toyoda, Y.; et al. Targeted disruption of Na+/Ca2+ exchanger gene leads to cardiomyocyte apoptosis and defects in heartbeat. J. Biol. Chem. 2000, 275, 36991–36998. [Google Scholar] [CrossRef] [PubMed]
- Schillinger, W.; Janssen, P.M.; Emami, S.; Henderson, S.A.; Ross, R.S.; Teucher, N.; Zeitz, O.; Philipson, K.D.; Prestle, J.; Hasenfuss, G. Impaired contractile performance of cultured rabbit ventricular myocytes after adenoviral gene transfer of Na+-Ca2+ exchanger. Circ. Res. 2000, 87, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Ranu, H.K.; Terracciano, C.M.N.; Davia, K.; Bernobich, E.; Chaudhri, B.; Robinson, S.E.; Bin Kang, Z.; Hajjar, R.J.; MacLeod, K.T.; Harding, S.E. Effects of Na+/Ca2+-exchanger overexpression on excitation-contraction coupling in adult rabbit ventricular myocytes. J. Mol. Cell. Cardiol. 2002, 34, 389–400. [Google Scholar] [CrossRef]
- Tadros, G.M.; Zhang, X.-Q.; Song, J.; Carl, L.L.; Rothblum, L.I.; Tian, Q.; Dunn, J.; Lytton, J.; Cheung, J.Y. Effects of Na+/Ca2+ exchanger downregulation on contractility and [Ca2+]i transients in adult rat myocytes. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1616–H1626. [Google Scholar] [CrossRef]
- Schillinger, W.; Ohler, A.; Emami, S.; Müller, F.; Christians, C.; Janssen, P.M.L.; Kögler, H.; Teucher, N.; Pieske, B.; Seidler, T.; et al. The functional effect of adenoviral Na+/Ca2+ exchanger overexpression in rabbit myocytes depends on the activity of the Na+/K+-ATPase. Cardiovasc. Res. 2003, 57, 996–1003. [Google Scholar] [CrossRef]
- Bölck, B.; Münch, G.; Mackenstein, P.; Hellmich, M.; Hirsch, I.; Reuter, H.; Hattebuhr, N.; Weig, H.-J.; Ungerer, M.; Brixius, K.; et al. Na+/Ca2+ exchanger overexpression impairs frequency—And ouabain—Dependent cell shortening in adult rat cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1435–H1445. [Google Scholar] [CrossRef]
- Shamraj, O.I.; Grupp, I.L.; Grupp, G.; Melvin, D.; Gradoux, N.; Kremers, W.; Lingrel, J.B.; De Pover, A. Characterisation of Na/K-ATPase, its isoforms, and the inotropic response to ouabain in isolated failing human hearts. Cardiovasc. Res. 1993, 27, 2229–2237. [Google Scholar] [CrossRef] [Green Version]
- Nørgaard, A.; Bagger, J.P.; Bjerregaard, P.; Baandrup, U.; Kjeldsen, K.; Thomsen, P.E. Relation of left ventricular function and Na,K-pump concentration in suspected idiopathic dilated cardiomyopathy. Am. J. Cardiol. 1988, 61, 1312–1315. [Google Scholar] [CrossRef]
- Schwinger, R.H.; Wang, J.; Frank, K.; Müller-Ehmsen, J.; Brixius, K.; McDonough, A.A.; Erdmann, E. Reduced sodium pump alpha1, alpha3, and beta1-isoform protein levels and Na+, K+-ATPase activity but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation 1999, 99, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Viscomi, C.; Simard, M.-L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef] [PubMed]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Rasmussen, T.P.; Koval, O.M.; Joiner, M.-L.A.; Hall, D.D.; Chen, B.; Luczak, E.D.; Wang, Q.; Rokita, A.G.; Wehrens, X.H.T.; et al. The mitochondrial uniporter controls fight or flight heart rate increases. Nat. Commun. 2015, 6, 6081. [Google Scholar] [CrossRef]
- Oropeza-Almazán, Y.; Vázquez-Garza, E.; Chapoy-Villanueva, H.; Torre-Amione, G.; García-Rivas, G. Small Interfering RNA Targeting Mitochondrial Calcium Uniporter Improves Cardiomyocyte Cell Viability in Hypoxia/Reoxygenation Injury by Reducing Calcium Overload. Oxid. Med. Cell. Longev. 2017, 2017, 5750897. [Google Scholar] [CrossRef]
- Suarez, J.; Cividini, F.; Scott, B.T.; Lehmann, K.; Diaz-Juarez, J.; Diemer, T.; Dai, A.; Suarez, J.A.; Jain, M.; Dillmann, W.H. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J. Biol. Chem. 2018, 293, 8182–8195. [Google Scholar] [CrossRef] [Green Version]
- Flarsheim, C.E.; Grupp, I.L.; Matlib, M.A. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am. J. Physiol. 1996, 271, H192–H202. [Google Scholar] [CrossRef]
- Tanaka, Y.; Konno, N.; Kako, K.J. Mitochondrial dysfunction observed in situ in cardiomyocytes of rats in experimental diabetes. Cardiovasc. Res. 1992, 26, 409–414. [Google Scholar] [CrossRef]
- Liu, T.; Takimoto, E.; Dimaano, V.L.; DeMazumder, D.; Kettlewell, S.; Smith, G.; Sidor, A.; Abraham, T.P.; O’Rourke, B. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ. Res. 2014, 115, 44–54. [Google Scholar] [CrossRef]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Chaanine, A.H.; Kizana, E.; Park, W.J.; Hajjar, R.J. SUMO1-dependent modulation of SERCA2a in heart failure. Nature 2011, 477, 601–605. [Google Scholar] [CrossRef]
- Tilemann, L.; Lee, A.; Ishikawa, K.; Aguero, J.; Rapti, K.; Santos-Gallego, C.; Kohlbrenner, E.; Fish, K.M.; Kho, C.; Hajjar, R.J. SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Jeong, D.; Mitsuyama, S.; Oh, J.G.; Liang, L.; Ikeda, Y.; Sadoshima, J.; Hajjar, R.J.; Kho, C. The role of SUMO-1 in cardiac oxidative stress and hypertrophy. Antioxid. Redox Signal. 2014, 21, 1986–2001. [Google Scholar] [CrossRef] [PubMed]
- Rohde, D.; Ritterhoff, J.; Voelkers, M.; Katus, H.A.; Parker, T.G.; Most, P. S100A1: A multifaceted therapeutic target in cardiovascular disease. J. Cardiovasc. Transl. Res. 2010, 3, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Most, P.; Pleger, S.T.; Völkers, M.; Heidt, B.; Boerries, M.; Weichenhan, D.; Löffler, E.; Janssen, P.M.L.; Eckhart, A.D.; Martini, J.; et al. Cardiac adenoviral S100A1 gene delivery rescues failing myocardium. J. Clin. Investig. 2004, 114, 1550–1563. [Google Scholar] [CrossRef] [PubMed]
- Pleger, S.T.; Remppis, A.; Heidt, B.; Völkers, M.; Chuprun, J.K.; Kuhn, M.; Zhou, R.-H.; Gao, E.; Szabo, G.; Weichenhan, D.; et al. S100A1 gene therapy preserves in vivo cardiac function after myocardial infarction. Mol. Ther. 2005, 12, 1120–1129. [Google Scholar] [CrossRef]
- Pleger, S.T.; Most, P.; Boucher, M.; Soltys, S.; Chuprun, J.K.; Pleger, W.; Gao, E.; Dasgupta, A.; Rengo, G.; Remppis, A.; et al. Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation 2007, 115, 2506–2515. [Google Scholar] [CrossRef]
- Pleger, S.T.; Shan, C.; Ksienzyk, J.; Bekeredjian, R.; Boekstegers, P.; Hinkel, R.; Schinkel, S.; Leuchs, B.; Ludwig, J.; Qiu, G.; et al. Cardiac AAV9-S100A1 gene therapy rescues post-ischemic heart failure in a preclinical large animal model. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. CB 2005, 15, 1235–1241. [Google Scholar] [CrossRef]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef]
- Bergmeier, W.; Weidinger, C.; Zee, I.; Feske, S. Emerging roles of store-operated Ca2+ entry through STIM and ORAI proteins in immunity, hemostasis and cancer. Channels 2013, 7, 379–391. [Google Scholar] [CrossRef]
- Ong, H.L.; Liu, X.; Sharma, A.; Hegde, R.S.; Ambudkar, I.S. Intracellular Ca2+ release via the ER translocon activates store-operated calcium entry. Pflugers Arch. 2007, 453, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Soda, T.; Tanzi, F.; Guerra, G.; Mapelli, L.; Lodola, F.; D’Angelo, E. Stim and Orai proteins in neuronal Ca2+ signaling and excitability. Front. Cell. Neurosci. 2015, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.; Cuk, M.; Patel, B.; Lian, J.; Ouimet, M.; Kaufmann, U.; Yang, J.; Horvath, R.; Hornig-Do, H.-T.; Chrzanowska-Lightowlers, Z.M.; et al. Store-Operated Ca2+ Entry Controls Induction of Lipolysis and the Transcriptional Reprogramming to Lipid Metabolism. Cell Metab. 2017, 25, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Lodola, F.; Laforenza, U.; Bonetti, E.; Lim, D.; Dragoni, S.; Bottino, C.; Ong, H.L.; Guerra, G.; Ganini, C.; Massa, M.; et al. Store-operated Ca2+ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS ONE 2012, 7, e42541. [Google Scholar] [CrossRef]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Reforgiato, M.; Poletto, V.; Lodola, F.; Bottino, C.; Guido, D.; Rappa, A.; Pareek, S.; et al. Enhanced expression of Stim, Orai, and TRPC transcripts and proteins in endothelial progenitor cells isolated from patients with primary myelofibrosis. PLoS ONE 2014, 9, e91099. [Google Scholar] [CrossRef]
- Zuccolo, E.; Laforenza, U.; Ferulli, F.; Pellavio, G.; Scarpellino, G.; Tanzi, M.; Turin, I.; Faris, P.; Lucariello, A.; Maestri, M.; et al. Stim and Orai mediate constitutive Ca2+ entry and control endoplasmic reticulum Ca2+ refilling in primary cultures of colorectal carcinoma cells. Oncotarget 2018, 9, 31098–31119. [Google Scholar] [CrossRef]
- Xia, J.; Wang, H.; Huang, H.; Sun, L.; Dong, S.; Huang, N.; Shi, M.; Bin, J.; Liao, Y.; Liao, W. Elevated Orai1 and STIM1 expressions upregulate MACC1 expression to promote tumor cell proliferation, metabolism, migration, and invasion in human gastric cancer. Cancer Lett. 2016, 381, 31–40. [Google Scholar] [CrossRef]
- Moccia, F.; Berra-Romani, R.; Rosti, V. Manipulating Intracellular Ca2+ Signals to Stimulate Therapeutic Angiogenesis in Cardiovascular Disorders. Curr. Pharm. Biotechnol. 2018, 19, 686–699. [Google Scholar] [CrossRef]
- Collins, H.E.; Pat, B.M.; Zou, L.; Litovsky, S.H.; Wende, A.R.; Young, M.E.; Chatham, J.C. Novel role of the ER/SR Ca2+ sensor STIM1 in the regulation of cardiac metabolism. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1014–H1026. [Google Scholar] [CrossRef]
- Bartoli, F.; Sabourin, J. Cardiac Remodeling and Disease: Current Understanding of STIM1/Orai1-Mediated Store-Operated Ca2+ Entry in Cardiac Function and Pathology. Adv. Exp. Med. Biol. 2017, 993, 523–534. [Google Scholar]
- Ji, Y.; Guo, X.; Zhang, Z.; Huang, Z.; Zhu, J.; Chen, Q.-H.; Gui, L. CaMKIIδ meditates phenylephrine induced cardiomyocyte hypertrophy through store-operated Ca2+ entry. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2017, 27, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Jiang, J.; Yue, Z.; Liu, S.; Ma, Y.; Yu, N.; Gao, Y.; Sun, S.; Chen, S.; Liu, P. Store-Operated Ca2+ Entry (SOCE) contributes to angiotensin II-induced cardiac fibrosis in cardiac fibroblasts. J. Pharmacol. Sci. 2016, 132, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Li, T.; Brochet, D.X.P.; Rosenberg, P.B.; Lederer, W.J. STIM1 enhances SR Ca2+ content through binding phospholamban in rat ventricular myocytes. Proc. Natl. Acad. Sci. USA 2015, 112, E4792–E4801. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Zhang, Y.; Wang, Q.; Li, D.; Yang, Y.; Ma, S.; Yang, D. Overexpression of SARAF Ameliorates Pressure Overload-Induced Cardiac Hypertrophy Through Suppressing STIM1-Orai1 in Mice. Cell. Physiol. Biochem. 2018, 47, 817–826. [Google Scholar] [CrossRef]
- Sabourin, J.; Boet, A.; Rucker-Martin, C.; Lambert, M.; Gomez, A.-M.; Benitah, J.-P.; Perros, F.; Humbert, M.; Antigny, F. Ca2+ handling remodeling and STIM1L/Orai1/TRPC1/TRPC4 upregulation in monocrotaline-induced right ventricular hypertrophy. J. Mol. Cell. Cardiol. 2018, 118, 208–224. [Google Scholar] [CrossRef]
- Troupes, C.D.; Wallner, M.; Borghetti, G.; Zhang, C.; Mohsin, S.; von Lewinski, D.; Berretta, R.M.; Kubo, H.; Chen, X.; Soboloff, J.; et al. Role of STIM1 (Stromal Interaction Molecule 1) in Hypertrophy-Related Contractile Dysfunction. Circ. Res. 2017, 121, 125–136. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, A.Y.; Kim, J.J.; Graham, V.; Finch, E.A.; Nepliouev, I.; Zhao, G.; Li, T.; Lederer, W.J.; Stiber, J.A.; et al. STIM1-Ca2+ signaling modulates automaticity of the mouse sinoatrial node. Proc. Natl. Acad. Sci. USA 2015, 112, E5618–E5627. [Google Scholar] [CrossRef]
- Li, P.; Bian, X.-Y.; Chen, Q.; Yao, X.-F.; Wang, X.-D.; Zhang, W.-C.; Tao, Y.-J.; Jin, R.; Zhang, L. Blocking of stromal interaction molecule 1 expression influence cell proliferation and promote cell apoptosis in vitro and inhibit tumor growth in vivo in head and neck squamous cell carcinoma. PLoS ONE 2017, 12, e0177484. [Google Scholar] [CrossRef]
- Schmidt, S.; Liu, G.; Liu, G.; Yang, W.; Honisch, S.; Pantelakos, S.; Stournaras, C.; Hönig, A.; Lang, F. Enhanced Orai1 and STIM1 expression as well as store operated Ca2+ entry in therapy resistant ovary carcinoma cells. Oncotarget 2014, 5, 4799–4810. [Google Scholar] [CrossRef]
- Moccia, F.; Lodola, F.; Dragoni, S.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Ca2+ signalling in endothelial progenitor cells: A novel means to improve cell-based therapy and impair tumour vascularisation. Curr. Vasc. Pharmacol. 2014, 12, 87–105. [Google Scholar] [CrossRef]
- del Monte, F.; Harding, S.E.; Dec, G.W.; Gwathmey, J.K.; Hajjar, R.J. Targeting phospholamban by gene transfer in human heart failure. Circulation 2002, 105, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Kaye, D.M.; Preovolos, A.; Marshall, T.; Byrne, M.; Hoshijima, M.; Hajjar, R.; Mariani, J.A.; Pepe, S.; Chien, K.R.; Power, J.M. Percutaneous cardiac recirculation-mediated gene transfer of an inhibitory phospholamban peptide reverses advanced heart failure in large animals. J. Am. Coll. Cardiol. 2007, 50, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Suckau, L.; Fechner, H.; Chemaly, E.; Krohn, S.; Hadri, L.; Kockskämper, J.; Westermann, D.; Bisping, E.; Ly, H.; Wang, X.; et al. Long-term cardiac-targeted RNA interference for the treatment of heart failure restores cardiac function and reduces pathological hypertrophy. Circulation 2009, 119, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Ziolo, M.T.; Martin, J.L.; Bossuyt, J.; Bers, D.M.; Pogwizd, S.M. Adenoviral gene transfer of mutant phospholamban rescues contractile dysfunction in failing rabbit myocytes with relatively preserved SERCA function. Circ. Res. 2005, 96, 815–817. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.O.; Quick, A.P.; Wang, Q.; Beavers, D.L.; Philippen, L.E.; Showell, J.; Barreto-Torres, G.; Thuerauf, D.J.; Doroudgar, S.; Glembotski, C.C.; et al. Junctophilin-2 gene therapy rescues heart failure by normalizing RyR2-mediated Ca2+ release. Int. J. Cardiol. 2016, 225, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Beavers, D.L.; Landstrom, A.P.; Chiang, D.Y.; Wehrens, X.H.T. Emerging roles of junctophilin-2 in the heart and implications for cardiac diseases. Cardiovasc. Res. 2014, 103, 198–205. [Google Scholar] [CrossRef]
- Guo, A.; Zhang, X.; Iyer, V.R.; Chen, B.; Zhang, C.; Kutschke, W.J.; Weiss, R.M.; Franzini-Armstrong, C.; Song, L.-S. Overexpression of junctophilin-2 does not enhance baseline function but attenuates heart failure development after cardiac stress. Proc. Natl. Acad. Sci. USA. 2014, 111, 12240–12245. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Ishikawa, K.; Fish, K.; Oh, J.G.; Motloch, L.J.; Kohlbrenner, E.; Lee, P.; Xie, C.; Lee, A.; Liang, L.; et al. Protein Phosphatase Inhibitor-1 Gene Therapy in a Swine Model of Nonischemic Heart Failure. J. Am. Coll. Cardiol. 2017, 70, 1744–1756. [Google Scholar] [CrossRef]


{kind=link}
| Method | Advantages | Limitations | References |
|---|---|---|---|
| Lentivirus | Broad host range Infection of dividing and non-dividing cells Low cytotoxicity Long-term expression (integration into genome) Insert capacity: 8 kb | No specific tropism for CMs (requires direct injection into the heart) Risk of insertional mutagenesis (integration into genome) | [125,126,127,128,129] |
| AD | Broad host range High level of gene expression No host genome integration Insert capacity: <35 kb | Short-term expression Strong immunogenicity No specific tropism for CMs | [127,129,130,131] |
| AAV | Relatively broad host range Low pathogenicity and toxicity Infection of dividing and non-dividing cells Long-term expression Serotype modulation for organ specificity | Difficulties in high transgene expression Delayed expression Presence of NAbs Small insert capacity of <5 kb | [127,129,132,133,134,135,136] |
| CRISPR/Cas9 | Targeting specific DNA sequences Any organism Simple and precise (compared to gene targeting) Inactivation, integration and allele substitution possible Reactivation of non-dividing cells Low immunogenicity | Difficulties in off-target effects (nonspecific and mismatched genetic modifications) Difficulties in delivery of large Cas9 sequences | [137,138,139,140,141] |
| Author (year) | Vector | Delivery Technique | Genetic Information | Species/Model | Outcomes |
|---|---|---|---|---|---|
| Murata et al. (2004) [143] | AD | Injection into LV cavity | Mutant Ras-related G-protein Gem W296G | Guinea pig/wt | ↓ ICa-L in CMs ↓ QT in vivo |
| Cingolani et al. (2007) [144] | Lentivirus | Injection into LV cavity | Hairpin RNA for β2 | Rat/aortic-banded model of LV hypertrophy | ↓ ICa-L in CMs |
| Subramanyam et al. (2013) [145] | AD | In vitro | Split-intein-tagged α1C-fragments | Rat/wt | ↑ Ca2+ transients β-adrenergic regulation |
| Author (year) | Vector | Delivery Technique | Species/Model | Outcomes |
|---|---|---|---|---|
| Giordano et al. (1997) [157] | AD | In vitro | Rat— ↓ SERCA2a expression | ↑ SERCA2a expression ↓ Ca2+ transients ↑ SR Ca2+ uptake |
| Hajjar et al. (1997) [158] | AD | In vitro | Rat— wt | ↑ peak Ca2+ release ↓ resting Ca2+ levels |
| Terracciano et al. (2002) [159] | AD | In vitro | Rabbit— wt | ↓ APD ↑ SR Ca2+ content |
| del Monte et al. (2004) [150] | AD | Intramyocardial injection | Rat— wt | ↓ VT after I/R |
| Prunier et al. (2008) [151] | AD | Anterograde coronary injection | Swine— wt | ↓ VT after I/R |
| Cutler et al. (2009) [152] | AD | Injection into aortic root | Guinea pig— wt | ↓ APD alternans in vitro and ex vivo ↓ VT ex vivo |
| Lyon et al. (2011) [153] | AD AAV9 | Intramyocardial (AV), or tail vein (AAV9) | Rat— HF | ↓ VT ex vivo ↓ spontaneous and isoproterenol triggered VT in vivo |
| Cutler et al. (2012) [154] | AAV9 | Injection into aortic root | Guinea pig— HF | ↓ APD alternans ↓ VT ex vivo |
| Motloch et al. (2018) [160] | AAV1 | Intracoronary injection | Swine— MI | ↓ QRS duration in vivo ↓ VT in vivo and ex vivo |
| Strauss et al. (2019) [161] | AAV1 | Aerosolized | Rat— PAH | ↓ VT in vivo ↓ APD duration Reversed spatial APD heterogeneity ↑ Electrophysiological remodeling |
| Author (year) | Vector | Delivery Technique | Genetic Information | Species/Model | Outcomes |
|---|---|---|---|---|---|
| Bongianino et al. (2017) [167] | AAV9 | Intraperitoneal injection | miRyR2-U10 | Mouse/wt | ↓ DADs ↓ VT in vivo |
| Pan et al. (2018) [168] | AAV9 | Subcutaneous injection | RyR2 | Mouse/CPVT (R176Q/+) | ↓ arrhythmias in vivo |
| Denegri et al. (2014) [169] | AAV9 | Intraperitoneal injection | CASQ2 | Mouse/CPVT (R33Q) | ↓ VT in vivo |
| Lodola et al. (2016) [170] | AAV9 | In vitro | CASQ2 | Human/CPVT; iPSCs (CASQ2-G112+5X) | ↓ DADs ↑ Ca2+ transient amplitude and duration of Ca2+ sparks |
| Kurtzwald-Josefson et al. (2017) [171] | AAV9 | Intraperitoneal injection | CASQ2 | Mouse/CPVT (CASQ2D307H or CASQ2Δ/Δ) | ↓ VT in vivo |
| Liu et al. (2018) [172] | AAV9 | Intra-thoracic cavity injection | CaM | Mouse/CPVT (R33Q) | ↑ Ca2+ handling ↓ VT in vivo |
| Bezzerides et al. (2019) [173] | AAV9 | Subcutaneous injection In vitro | CaMKII | Mouse/ CPVT (RYR2R176Q/+) Human/CPVT; iPSCs (different mutations) | ↓ ventricular arrhythmia in vivo |
| Author (year) | Vector | Delivery Technique | Species/Model | Expression Properties | Outcomes |
|---|---|---|---|---|---|
| Schillinger et al. (2000) [184] | AD | In vitro | Rabbit/wt | OE | ↓ contractile function |
| Terracciano et al. (2001) [175] | Transfection reagent | In vitro Injected into nuclei | Mouse/wt | OE | ↑ Ca2+ handling and homeostasis |
| Ranu et al. (2002) [185] | AD | In vitro | Rabbit/wt | OE | ↓ contraction amplitude |
| Tadros et al. (2002) [186] | AD | In vitro | Rat/MI | DR | ↓ Ca2+ influx and efflux |
| Schillinger et al. (2003) [187] | AD | In vitro | Rabbit/wt | OE | Systolic and diastolic dysfunction |
| Bölck et al. (2004) [188] | AD | In vitro | Rat/wt | OE | ↓ cell shortening at higher stimulation frequencies ↑ intracellular systolic Ca2+ and contractile amplitude at low stimulation rates |
| Author (year) | Vector | Delivery Technique | Genetic Information | Species/Model | Outcomes |
|---|---|---|---|---|---|
| Wu et al. (2015) [194] | AD | In vitro, Mouse embryonic stem cells | DN-MCU | Mouse/wt | MCU is necessary for physiological heart rate acceleration |
| Oropeza-Almazán et al. (2017) [195] | Transfection reagent | In vitro | siRNA targeting MCU | Rat/H/R injury | ↓ mitochondrial permeability pore opening ↓ oxidative stress |
| Suarez et al. (2018) [196] | AAV9 | Direct jugular vein injection | MCU | Mouse/Diabetic | Restoration of cardiac myocyte and heart function |
| Larbig et al. (2017) [51] | Knock-out model | UCP2-/- | Mouse/Knock-out | ↓ ICa-L in CM ↑ slope factor of action potential upstrokes ↑ hyperpolarized resting membrane potential ↓ PR, WRS and QTc interval ↑ after-depolarizations ↑ arrhythmias | |
| Luongo et al. (2017) [193] | Knock-out and OE model | SLC8B1 (NCLX) | Mouse/Knock-out and OE | ↑ mCa2+ clearance Prevention of heart failure | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paar, V.; Jirak, P.; Larbig, R.; Zagidullin, N.S.; Brandt, M.C.; Lichtenauer, M.; Hoppe, U.C.; Motloch, L.J. Pathophysiology of Calcium Mediated Ventricular Arrhythmias and Novel Therapeutic Options with Focus on Gene Therapy. Int. J. Mol. Sci. 2019, 20, 5304. https://doi.org/10.3390/ijms20215304
Paar V, Jirak P, Larbig R, Zagidullin NS, Brandt MC, Lichtenauer M, Hoppe UC, Motloch LJ. Pathophysiology of Calcium Mediated Ventricular Arrhythmias and Novel Therapeutic Options with Focus on Gene Therapy. International Journal of Molecular Sciences. 2019; 20(21):5304. https://doi.org/10.3390/ijms20215304
Chicago/Turabian StylePaar, Vera, Peter Jirak, Robert Larbig, Naufal Shamilevich Zagidullin, Mathias C. Brandt, Michael Lichtenauer, Uta C. Hoppe, and Lukas J. Motloch. 2019. "Pathophysiology of Calcium Mediated Ventricular Arrhythmias and Novel Therapeutic Options with Focus on Gene Therapy" International Journal of Molecular Sciences 20, no. 21: 5304. https://doi.org/10.3390/ijms20215304
APA StylePaar, V., Jirak, P., Larbig, R., Zagidullin, N. S., Brandt, M. C., Lichtenauer, M., Hoppe, U. C., & Motloch, L. J. (2019). Pathophysiology of Calcium Mediated Ventricular Arrhythmias and Novel Therapeutic Options with Focus on Gene Therapy. International Journal of Molecular Sciences, 20(21), 5304. https://doi.org/10.3390/ijms20215304