Tackling Acute Lymphoblastic Leukemia—One Fish at a Time


Abstract
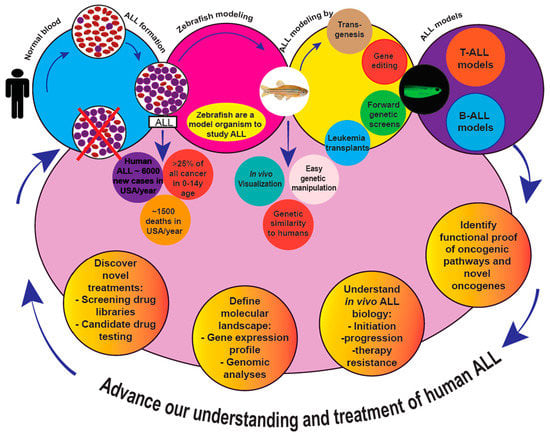
:
1. Introduction
Zebrafish are a Model Befitting Leukemia Modeling
2. Zebrafish Acute Lymphoblastic Leukemia Models
2.1. T-ALL: Introduction
2.2. Zebrafish T-ALL Models
2.2.1. Model 1: T-ALL Induced by Murine Myc – Tg(zrag2:EGFP-mMyc) Zebrafish
Key Discoveries
2.2.2. Model 2: T-ALL in Conditional mMyc Models
Model 2a: rag2:loxP-dsRED2-loxP-EGFP-mMyc [aka, Tg(rag2:LDL-EMyc)] Zebrafish
Model 2b: Double-Transgenic [Tg(hsp70:Cre;rag2:LDL-EMyc)] Zebrafish
Model 2c: Triple-Transgenic [Tg(hsp70:Cre;rag2:LDL-EMyc;rag2:EGFP-bcl2)] Zebrafish
2.2.3. Model 3: Co-Injection Strategies Pairing zrag2:EGFP-mMyc with Other Transgenes
Key Discoveries
2.2.4. Model 4: Inducible T-ALL in rag2:hMYC-ER Transgenic Zebrafish
Key Discoveries
2.2.5. Model 5: Zebrafish NOTCH T-ALL Models
Model 5a: Transgenic Human NOTCH1–Tg(rag2:hICN1-EGFP) Zebrafish
Model 5b: Transgenic Zebrafish Notch1–Tg (rag2:znotch1a ICD)
2.2.6. Models 6–8: Non-Transgenic T-ALL Models
Key Discoveries
2.2.7. Model 9: T-ALL Induced by ARID5B–Tg(rag2:hARID5B) Zebrafish
Key Discoveries
2.2.8. Model 10: T-ALL Induced by jdp2–Tg(rag2:zjdp2) Zebrafish
Key Discoveries
2.3. B-ALL: Introduction
2.4. B-ALL Zebrafish Models
2.4.1. Model 1: Transgenic hTEL-AML1 Zebrafish
Key Discoveries
2.4.2. Model 2: B-ALL in rag2:mMyc Transgenic Zebrafish
Key Discoveries
2.4.3. Model 3: B-ALL in rag2:hMYC-ER Transgenic Zebrafish
Key discoveries
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 4HT | 4-hydroxytamoxifen |
| aCGH | Array comparative genomic hybridization |
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloid leukemia |
| B-ALL | Pre-B acute lymphoblastic leukemia |
| CAN | Copy number aberration |
| CLL | Chronic lymphocytic leukemia |
| CML | Chronic myelogenous leukemia |
| CNS | Central Nervous System |
| CSR | Class switch recombination |
| CY | Cyclophosphamide |
| D. rerio | Danio rerio |
| dpf | Days post-fertilization |
| ENU | N-ethyl-N-nitrosourea |
| ER | Estrogen receptor |
| ETP | Early Thymocyte Progenitor |
| GFP | Green fluorescent protein |
| igm | Zebrafish immunoglobulin mu |
| igz | Zebrafish immunoglobulin zeta |
| ISH | In situ hybridization |
| IVF | In-vitro fertilization |
| LIC | Leukemia-initiating cell |
| LOF | Loss of function |
| LSC | Leukemic stem cell |
| PGE2 | Prostaglandin E2 |
| PI3K | Phophatidylinositol 3’-kinase |
| PRE | Prednisolone |
| PTEN | Phosphatase and tensin homolog |
| RT-PCR | Reverse transcription polymerase chain reaction |
| T-ALL | T cell acute lymphoblastic leukemia |
| T-LBL | T-cell lymphoblastic lymphoma |
| TCA | Tricarboxylic acid |
| TCR | T cell receptor |
| VCR | Vincristine |
References
- American Cancer Society. Cancer Facts & Figures 2019. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf (accessed on 10 August 2019).
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhojwani, D.; Yang, J.J.; Pui, C.H. Biology of childhood acute lymphoblastic leukemia. Pediatr Clin. N. Am. 2015, 62, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Curtin, S.C.; Minino, A.M.; Anderson, R.N. Declines in Cancer Death Rates Among Children and Adolescents in the United States, 1999-2014. NCHS Data Brief. 2016, 257, 1–8. [Google Scholar]
- Locatelli, F.; Schrappe, M.; Bernardo, M.E.; Rutella, S. How I treat relapsed childhood acute lymphoblastic leukemia. Blood 2012, 120, 2807–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, J.M.; Silverman, L.B.; Levy, D.E.; Dalton, V.K.; Gelber, R.D.; Lehmann, L.; Cohen, H.J.; Sallan, S.E.; Asselin, B.L. Childhood T-cell acute lymphoblastic leukemia: The Dana-Farber Cancer Institute acute lymphoblastic leukemia consortium experience. J. Clin. Oncol. 2003, 21, 3616–3622. [Google Scholar] [CrossRef]
- Winter, S.S.; Dunsmore, K.P.; Devidas, M.; Wood, B.L.; Esiashvili, N.; Chen, Z.; Eisenberg, N.; Briegel, N.; Hayashi, R.J.; Gastier-Foster, J.M.; et al. Improved Survival for Children and Young Adults With T-Lineage Acute Lymphoblastic Leukemia: Results From the Children’s Oncology Group AALL0434 Methotrexate Randomization. J. Clin. Oncol. 2018, 36, 2926–2934. [Google Scholar] [CrossRef]
- Pui, C.H.; Carroll, W.L.; Meshinchi, S.; Arceci, R.J. Biology, risk stratification, and therapy of pediatric acute leukemias: An update. J. Clin. Oncol 2011, 29, 551–565. [Google Scholar] [CrossRef]
- Pui, C.H.; Mullighan, C.G.; Evans, W.E.; Relling, M.V. Pediatric acute lymphoblastic leukemia: Where are we going and how do we get there? Blood 2012, 120, 1165–1174. [Google Scholar] [CrossRef]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Baeten, J.T.; de Jong, J.L.O. Genetic Models of Leukemia in Zebrafish. Front. Cell Dev. Biol. 2018, 6, 115. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Jing, C.B.; Look, A.T. Zebrafish models of leukemia. Methods Cell Biol. 2017, 138, 563–592. [Google Scholar] [CrossRef] [PubMed]
- Payne, E.; Look, T. Zebrafish modelling of leukaemias. Br. J. Haematol. 2009, 146, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Squiban, B.; Frazer, J.K. Danio rerio: Small Fish Making a Big Splash in Leukemia. Curr. Pathobiol. Rep. 2014, 2, 61–73. [Google Scholar] [CrossRef]
- Avagyan, S.; Zon, L.I. Fish to Learn: Insights into Blood Development and Blood Disorders from Zebrafish Hematopoiesis. Hum. Gene. Ther. 2016, 27, 287–294. [Google Scholar] [CrossRef] [Green Version]
- Gore, A.V.; Pillay, L.M.; Venero Galanternik, M.; Weinstein, B.M. The zebrafish: A fintastic model for hematopoietic development and disease. Wiley Interdiscip. Rev. Dev. Biol. 2018, 7, e312. [Google Scholar] [CrossRef]
- Potts, K.S.; Bowman, T.V. Modeling Myeloid Malignancies Using Zebrafish. Front. Oncol. 2017, 7, 297. [Google Scholar] [CrossRef]
- Rissone, A.; Burgess, S.M. Rare Genetic Blood Disease Modeling in Zebrafish. Front. Genet. 2018, 9, 348. [Google Scholar] [CrossRef]
- Meeker, N.D.; Trede, N.S. Immunology and zebrafish: Spawning new models of human disease. Dev. Comp. Immunol. 2008, 32, 745–757. [Google Scholar] [CrossRef]
- Lam, S.H.; Chua, H.L.; Gong, Z.; Wen, Z.; Lam, T.J.; Sin, Y.M. Morphologic transformation of the thymus in developing zebrafish. Dev. Dyn. 2002, 225, 87–94. [Google Scholar] [CrossRef]
- Paik, E.J.; Zon, L.I. Hematopoietic development in the zebrafish. Int. J. Dev. Biol. 2010, 54, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Jing, L.; Zon, L.I. Zebrafish as a model for normal and malignant hematopoiesis. Dis. Model. Mech. 2011, 4, 433–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentley, V.L.; Veinotte, C.J.; Corkery, D.P.; Pinder, J.B.; LeBlanc, M.A.; Bedard, K.; Weng, A.P.; Berman, J.N.; Dellaire, G. Focused chemical genomics using zebrafish xenotransplantation as a pre-clinical therapeutic platform for T-cell acute lymphoblastic leukemia. Haematologica 2015, 100, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Corkery, D.P.; Dellaire, G.; Berman, J.N. Leukaemia xenotransplantation in zebrafish--chemotherapy response assay in vivo. Br. J. Haematol. 2011, 153, 786–789. [Google Scholar] [CrossRef]
- Veinotte, C.J.; Dellaire, G.; Berman, J.N. Hooking the big one: The potential of zebrafish xenotransplantation to reform cancer drug screening in the genomic era. Dis. Model. Mech. 2014, 7, 745–754. [Google Scholar] [CrossRef]
- Yan, C.; Brunson, D.C.; Tang, Q.; Do, D.; Iftimia, N.A.; Moore, J.C.; Hayes, M.N.; Welker, A.M.; Garcia, E.G.; Dubash, T.D.; et al. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell 2019, 177, 1903–1914.e14. [Google Scholar] [CrossRef]
- Pruvot, B.; Jacquel, A.; Droin, N.; Auberger, P.; Bouscary, D.; Tamburini, J.; Muller, M.; Fontenay, M.; Chluba, J.; Solary, E. Leukemic cell xenograft in zebrafish embryo for investigating drug efficacy. Haematologica 2011, 96, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Durinck, K.; Goossens, S.; Peirs, S.; Wallaert, A.; Van Loocke, W.; Matthijssens, F.; Pieters, T.; Milani, G.; Lammens, T.; Rondou, P.; et al. Novel biological insights in T-cell acute lymphoblastic leukemia. Exp. Hematol. 2015, 43, 625–639. [Google Scholar] [CrossRef]
- Litzow, M.R.; Ferrando, A.A. How I treat T-cell acute lymphoblastic leukemia in adults. Blood 2015, 126, 833–841. [Google Scholar] [CrossRef] [Green Version]
- Hoelzer, D.; Gokbuget, N. T-cell lymphoblastic lymphoma and T-cell acute lymphoblastic leukemia: A separate entity? Clin. Lymphoma Myeloma 2009, 9, S214–S221. [Google Scholar] [CrossRef]
- Langenau, D.M.; Traver, D.; Ferrando, A.A.; Kutok, J.L.; Aster, J.C.; Kanki, J.P.; Lin, S.; Prochownik, E.; Trede, N.S.; Zon, L.I.; et al. Myc-induced T cell leukemia in transgenic zebrafish. Science 2003, 299, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.A.; Cleveland, J.L. Myc pathways provoking cell suicide and cancer. Oncogene 2003, 22, 9007–9021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.M.; Calvo, J.A.; Draheim, K.M.; Cunningham, L.A.; Hermance, N.; Beverly, L.; Krishnamoorthy, V.; Bhasin, M.; Capobianco, A.J.; Kelliher, M.A. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol. Cell Biol. 2006, 26, 8022–8031. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.L.; Lau, A.; Wai, C.; Del Bianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef]
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.H.; Downing, J.R.; Gilliland, D.G.; et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002, 1, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- La Starza, R.; Borga, C.; Barba, G.; Pierini, V.; Schwab, C.; Matteucci, C.; Lema Fernandez, A.G.; Leszl, A.; Cazzaniga, G.; Chiaretti, S.; et al. Genetic profile of T-cell acute lymphoblastic leukemias with MYC translocations. Blood 2014, 124, 3577–3582. [Google Scholar] [CrossRef]
- Langenau, D.M.; Feng, H.; Berghmans, S.; Kanki, J.P.; Kutok, J.L.; Look, A.T. Cre/lox-regulated transgenic zebrafish model with conditional myc-induced T cell acute lymphoblastic leukemia. Proc. Natl. Acad Sci. USA 2005, 102, 6068–6073. [Google Scholar] [CrossRef] [Green Version]
- Ferrando, A.A.; Look, A.T. Gene expression profiling in T-cell acute lymphoblastic leukemia. Semin. Hematol. 2003, 40, 274–280. [Google Scholar] [CrossRef]
- Anderson, N.M.; Li, D.; Peng, H.L.; Laroche, F.J.; Mansour, M.R.; Gjini, E.; Aioub, M.; Helman, D.J.; Roderick, J.E.; Cheng, T.; et al. The TCA cycle transferase DLST is important for MYC-mediated leukemogenesis. Leukemia 2016, 30, 1365–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizgirev, I.V.; Revskoy, S. A new zebrafish model for experimental leukemia therapy. Cancer Biol. Ther. 2010, 9, 895–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.C.; Raimondi, A.R.; Salthouse, C.D.; Ignatius, M.S.; Blackburn, J.S.; Mizgirev, I.V.; Storer, N.Y.; de Jong, J.L.; Chen, A.T.; Zhou, Y.; et al. High-throughput cell transplantation establishes that tumor-initiating cells are abundant in zebrafish T-cell acute lymphoblastic leukemia. Blood 2010, 115, 3296–3303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizgireuv, I.V.; Revskoy, S.Y. Transplantable tumor lines generated in clonal zebrafish. Cancer Res. 2006, 66, 3120–3125. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Langenau, D.M.; Madge, J.A.; Quinkertz, A.; Gutierrez, A.; Neuberg, D.S.; Kanki, J.P.; Look, A.T. Heat-shock induction of T-cell lymphoma/leukaemia in conditional Cre/lox-regulated transgenic zebrafish. Br. J. Haematol. 2007, 138, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Huiting, L.N.; Samaha, Y.; Zhang, G.L.; Roderick, J.E.; Li, B.; Anderson, N.M.; Wang, Y.W.; Wang, L.; Laroche, F.; Choi, J.W.; et al. UFD1 contributes to MYC-mediated leukemia aggressiveness through suppression of the proapoptotic unfolded protein response. Leukemia 2018, 32, 2339–2351. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Stachura, D.L.; White, R.M.; Gutierrez, A.; Zhang, L.; Sanda, T.; Jette, C.A.; Testa, J.R.; Neuberg, D.S.; Langenau, D.M.; et al. T-lymphoblastic lymphoma cells express high levels of BCL2, S1P1, and ICAM1, leading to a blockade of tumor cell intravasation. Cancer Cell 2010, 18, 353–366. [Google Scholar] [CrossRef]
- Langenau, D.M.; Keefe, M.D.; Storer, N.Y.; Jette, C.A.; Smith, A.C.; Ceol, C.J.; Bourque, C.; Look, A.T.; Zon, L.I. Co-injection strategies to modify radiation sensitivity and tumor initiation in transgenic Zebrafish. Oncogene 2008, 27, 4242–4248. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, J.S.; Liu, S.; Wilder, J.L.; Dobrinski, K.P.; Lobbardi, R.; Moore, F.E.; Martinez, S.A.; Chen, E.Y.; Lee, C.; Langenau, D.M. Clonal evolution enhances leukemia-propagating cell frequency in T cell acute lymphoblastic leukemia through Akt/mTORC1 pathway activation. Cancer Cell 2014, 25, 366–378. [Google Scholar] [CrossRef]
- Gutierrez, A.; Sanda, T.; Grebliunaite, R.; Carracedo, A.; Salmena, L.; Ahn, Y.; Dahlberg, S.; Neuberg, D.; Moreau, L.A.; Winter, S.S.; et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 2009, 114, 647–650. [Google Scholar] [CrossRef]
- Palomero, T.; Dominguez, M.; Ferrando, A.A. The role of the PTEN/AKT Pathway in NOTCH1-induced leukemia. Cell Cycle 2008, 7, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Borga, C.; Foster, C.A.; Iyer, S.; Garcia, S.P.; Langenau, D.M.; Frazer, J.K. Molecularly distinct models of zebrafish Myc-induced B cell leukemia. Leukemia 2019, 33, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Borga, C.; Park, G.; Foster, C.; Burroughs-Garcia, J.; Marchesin, M.; Shah, R.; Hasan, A.; Ahmed, S.T.; Bresolin, S.; Batchelor, L.; et al. Simultaneous B and T cell acute lymphoblastic leukemias in zebrafish driven by transgenic MYC: Implications for oncogenesis and lymphopoiesis. Leukemia 2019, 33, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Grebliunaite, R.; Feng, H.; Kozakewich, E.; Zhu, S.; Guo, F.; Payne, E.; Mansour, M.; Dahlberg, S.E.; Neuberg, D.S.; et al. Pten mediates Myc oncogene dependence in a conditional zebrafish model of T cell acute lymphoblastic leukemia. J. Exp. Med. 2011, 208, 1595–1603. [Google Scholar] [CrossRef]
- Reynolds, C.; Roderick, J.E.; LaBelle, J.L.; Bird, G.; Mathieu, R.; Bodaar, K.; Colon, D.; Pyati, U.; Stevenson, K.E.; Qi, J.; et al. Repression of BIM mediates survival signaling by MYC and AKT in high-risk T-cell acute lymphoblastic leukemia. Leukemia 2014, 28, 1819–1827. [Google Scholar] [CrossRef]
- Gutierrez, A.; Pan, L.; Groen, R.W.; Baleydier, F.; Kentsis, A.; Marineau, J.; Grebliunaite, R.; Kozakewich, E.; Reed, C.; Pflumio, F.; et al. Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J. Clin. Investig. 2014, 124, 644–655. [Google Scholar] [CrossRef] [Green Version]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.t.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef]
- Chen, J.; Jette, C.; Kanki, J.P.; Aster, J.C.; Look, A.T.; Griffin, J.D. NOTCH1-induced T-cell leukemia in transgenic zebrafish. Leukemia 2007, 21, 462–471. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, J.S.; Liu, S.; Raiser, D.M.; Martinez, S.A.; Feng, H.; Meeker, N.D.; Gentry, J.; Neuberg, D.; Look, A.T.; Ramaswamy, S.; et al. Notch signaling expands a pre-malignant pool of T-cell acute lymphoblastic leukemia clones without affecting leukemia-propagating cell frequency. Leukemia 2012, 26, 2069–2078. [Google Scholar] [CrossRef]
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133. [Google Scholar] [CrossRef] [Green Version]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navas, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazer, J.K.; Meeker, N.D.; Rudner, L.; Bradley, D.F.; Smith, A.C.; Demarest, B.; Joshi, D.; Locke, E.E.; Hutchinson, S.A.; Tripp, S.; et al. Heritable T-cell malignancy models established in a zebrafish phenotypic screen. Leukemia 2009, 23, 1825–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langenau, D.M.; Ferrando, A.A.; Traver, D.; Kutok, J.L.; Hezel, J.P.; Kanki, J.P.; Zon, L.I.; Look, A.T.; Trede, N.S. In vivo tracking of T cell development, ablation, and engraftment in transgenic zebrafish. Proc. Natl. Acad. Sci. USA 2004, 101, 7369–7374. [Google Scholar] [CrossRef] [PubMed]
- Rudner, L.A.; Brown, K.H.; Dobrinski, K.P.; Bradley, D.F.; Garcia, M.I.; Smith, A.C.; Downie, J.M.; Meeker, N.D.; Look, A.T.; Downing, J.R.; et al. Shared acquired genomic changes in zebrafish and human T-ALL. Oncogene 2011, 30, 4289–4296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, W.Z.; Tan, S.H.; Ngoc, P.C.T.; Amanda, S.; Yam, A.W.Y.; Liau, W.S.; Gong, Z.; Lawton, L.N.; Tenen, D.G.; Sanda, T. ARID5B as a critical downstream target of the TAL1 complex that activates the oncogenic transcriptional program and promotes T-cell leukemogenesis. Genes Dev. 2017, 31, 2343–2360. [Google Scholar] [CrossRef]
- Mansour, M.R.; He, S.; Li, Z.; Lobbardi, R.; Abraham, B.J.; Hug, C.; Rahman, S.; Leon, T.E.; Kuang, Y.Y.; Zimmerman, M.W.; et al. JDP2: An oncogenic bZIP transcription factor in T cell acute lymphoblastic leukemia. J. Exp. Med. 2018, 215, 1929–1945. [Google Scholar] [CrossRef]
- Mullighan, C.G. Molecular genetics of B-precursor acute lymphoblastic leukemia. J. Clin. Investig. 2012, 122, 3407–3415. [Google Scholar] [CrossRef] [Green Version]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef]
- Chiang, M.Y.; Wang, Q.; Gormley, A.C.; Stein, S.J.; Xu, L.; Shestova, O.; Aster, J.C.; Pear, W.S. High selective pressure for Notch1 mutations that induce Myc in T-cell acute lymphoblastic leukemia. Blood 2016, 128, 2229–2240. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef]
- Lilljebjorn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G.; et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat. Commun 2016, 7, 11790. [Google Scholar] [CrossRef] [PubMed]
- Lilljebjorn, H.; Fioretos, T. New oncogenic subtypes in pediatric B-cell precursor acute lymphoblastic leukemia. Blood 2017, 130, 1395–1401. [Google Scholar] [CrossRef]
- Tamai, H.; Miyake, K.; Takatori, M.; Miyake, N.; Yamaguchi, H.; Dan, K.; Shimada, T.; Inokuchi, K. Activated K-Ras protein accelerates human MLL/AF4-induced leukemo-lymphomogenicity in a transgenic mouse model. Leukemia 2011, 25, 888–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacoby, E.; Chien, C.D.; Fry, T.J. Murine models of acute leukemia: Important tools in current pediatric leukemia research. Front. Oncol. 2014, 4, 95. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Feng, Z.; Lemieux, M.E.; Faber, J.; Vempati, S.; Sinha, A.U.; Xia, X.; Jesneck, J.; Bracken, A.P.; Silverman, L.B.; et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 2008, 14, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Trede, N.S.; Langenau, D.M.; Traver, D.; Look, A.T.; Zon, L.I. The use of zebrafish to understand immunity. Immunity 2004, 20, 367–379. [Google Scholar] [CrossRef]
- Sabaawy, H.E.; Azuma, M.; Embree, L.J.; Tsai, H.J.; Starost, M.F.; Hickstein, D.D. TEL-AML1 transgenic zebrafish model of precursor B cell acute lymphoblastic leukemia. Proc. Natl. Acad Sci. USA 2006, 103, 15166–15171. [Google Scholar] [CrossRef]
- Wiemels, J.L.; Ford, A.M.; Van Wering, E.R.; Postma, A.; Greaves, M. Protracted and variable latency of acute lymphoblastic leukemia after TEL-AML1 gene fusion in utero. Blood 1999, 94, 1057–1062. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Seto, M. TEL (ETV6)-AML1 (RUNX1) initiates self-renewing fetal pro-B cells in association with a transcriptional program shared with embryonic stem cells in mice. Stem Cells 2013, 31, 236–247. [Google Scholar] [CrossRef]
- Greaves, M.F.; Maia, A.T.; Wiemels, J.L.; Ford, A.M. Leukemia in twins: Lessons in natural history. Blood 2003, 102, 2321–2333. [Google Scholar] [CrossRef]
- Raess, P.W.; Moore, S.R.; Cascio, M.J.; Dunlap, J.; Fan, G.; Gatter, K.; Olson, S.B.; Braziel, R.M. MYC immunohistochemical and cytogenetic analysis are required for identification of clinically relevant aggressive B cell lymphoma subtypes. Leuk Lymphoma 2018, 59, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Navid, F.; Mosijczuk, A.D.; Head, D.R.; Borowitz, M.J.; Carroll, A.J.; Brandt, J.M.; Link, M.P.; Rozans, M.K.; Thomas, G.A.; Schwenn, M.R.; et al. Acute lymphoblastic leukemia with the (8;14)(q24;q32) translocation and FAB L3 morphology associated with a B-precursor immunophenotype: The Pediatric Oncology Group experience. Leukemia 1999, 13, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.V.; Chilton, L.; Wilkinson, J.; Ensor, H.M.; Bown, N.; Proctor, S.J. A population-based cytogenetic study of adults with acute lymphoblastic leukemia. Blood 2010, 115, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.; Gill, K.; Hoehn, D.; Sulis, M.; Bhagat, G.; Alobeid, B. C-myc protein expression in B-cell acute lymphoblastic leukemia, prognostic significance? Leuk. Res. 2014, 38, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.G.; Iyer, S.; Garcia, S.P.; Loontiens, S.; Sadreyev, R.I.; Speleman, F.; Langenau, D.M. Cell of origin dictates aggression and stem cell number in acute lymphoblastic leukemia. Leukemia 2018, 32, 1860–1865. [Google Scholar] [CrossRef]
- Ono, M.; Nose, M. Persistent expression of an unproductive immunoglobulin heavy chain allele with DH-JH-gamma configuration in peripheral tissues. APMIS 2007, 115, 1350–1356. [Google Scholar] [CrossRef]
- Nelson, K.J.; Haimovich, J.; Perry, R.P. Characterization of productive and sterile transcripts from the immunoglobulin heavy-chain locus: Processing of micron and muS mRNA. Mol. Cell Biol. 1983, 3, 1317–1332. [Google Scholar] [CrossRef]
- Neale, G.A.; Kitchingman, G.R. mRNA transcripts initiating within the human immunoglobulin mu heavy chain enhancer region contain a non-translatable exon and are extremely heterogeneous at the 5′ end. Nucleic Acids Res. 1991, 19, 2427–2433. [Google Scholar] [CrossRef]
- Burroughs-Garcia, J.; Hasan, A.; Park, G.; Borga, C.; Frazer, J.K. Isolating Malignant and Non-Malignant B Cells from lck:eGFP Zebrafish. J. Vis. Exp. 2019. [Google Scholar] [CrossRef]
- Kohrer, S.; Havranek, O.; Seyfried, F.; Hurtz, C.; Coffey, G.P.; Kim, E.; Ten Hacken, E.; Jager, U.; Vanura, K.; O’Brien, S.; et al. Pre-BCR signaling in precursor B-cell acute lymphoblastic leukemia regulates PI3K/AKT, FOXO1 and MYC, and can be targeted by SYK inhibition. Leukemia 2016, 30, 1246–1254. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.S.; Shinton, S.A.; Rhodes, J.; Tang, L.; Feng, H.; Jette, C.A.; Look, A.T.; Hayakawa, K.; Hardy, R.R. Zebrafish B Cell Development without a Pre-B Cell Stage, Revealed by CD79 Fluorescence Reporter Transgenes. J. Immunol. 2017, 199, 1706–1715. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| T-ALL Models | Transgenes (Promoter:Oncogene) | Incidence (%) | Mean Latency (dpf) | Key Features | Other Remarks | |||
|---|---|---|---|---|---|---|---|---|
| Promoter | Oncogene(s) | Full Construct | ||||||
| Model 1 | Murine Myc (mMyc) | rag2 | mMyc | rag2:EGFP-mMyc | 100 | 52 | T-ALL mirrored most common TAL1 human T-ALL subtype; used as a foundation for several ensuing T-ALL studies | First transgenic zebrafish ALL and cancer model; complete penetrance and short latency created challenges [32] |
| Model 2a | Cre-Lox conditional mMyc | rag2 | mMyc | rag2:loxP-dsRED2-loxP-EGFP-mMyc | 7 | 151 | Used Cre recombinase injection to induce EGFP-mMyc expression | Low penetrance [40] |
| Model 2b | Heat shock inducible Cre with mMyc | rag2 | mMyc | hsp70:Cre;rag2:LDL-EGFP-mMyc | 80 | 120 | Used heat shock-inducible Cre transgene to markedly improve T-ALL penetrance | Easier to maintain and amenable to forward-genetic screens [46] |
| Model 2c | Above line with bcl2 | rag2 | mMYC/bcl2 | hsp70:Cre;rag2:LDL-EGFP-mMyc;rag2:EGFP-bcl2 | >80 | <120 | T-LBL favored over T-ALL due to constitutive bcl2 expression | Used to study differences between T-LBL and T-ALL [48] |
| Model 3 | Co-injection of multiple transgenes | rag2 | mMyc | rag2:mMyc and rag2:dsRED2 co-injected into stably transgenic rag2:GFP fish | N/A | N/A | Proved use of co-injection, allowing hundreds of embryos to be injected with multiple transgene combinations | Used to color-code T-ALL in the syngeneic CG1 background for LIC and cancer heterogeneity studies [49] |
| Model 4 | Human MYC (hMYC) | rag2 | hMYC | rag2:hMYC-ER | 100 (4HT treated fish) | 37 | 4HT-inducible hMYC expressing T-ALL used to study MYC and PI3K-AKT interaction. | Showed BIM repression is a key event downstream of MYC and PI3K-AKT in resistant T-ALL; also used to show therapeutic potential for PP2A in T-ALL [55] |
| Myristoylated murine Akt2 (myr-mAkt2) | rag2 | myr-mAkt2 | rag2:myr-mAkt2 | 17 | 140 | Showed myr-mAkt2 could induce T-ALL in isolation | Low incidence and has not been extensively studied [55] | |
| Model 5a | Human NOTCH1 (hICN1) | rag2 | hICN1 | rag2:hICN1-EGFP | 40 | 330 | NOTCH1 induced T-ALL without up-regulating endogenous myca and mycb | Allows NOTCH1 and MYC to be studied separately, not possible in mammals [59] |
| Model 5b | Zebrafish notch1a (znotch1a) | rag2 | znotch1aICD | rag2:znotch1aICD | 100 | <52 | hICN1/znotch1aICD accelerated T-ALL, but did not alter T-ALL proliferation or apoptosis | Showed NOTCH1 activation alone was insufficient to induce T-ALL [60] |
| Model 6–8 | Germline mutants: hlk, srk, and otg | N/A | N/A | N/A | 40–50 in homozygotes | 150–270 | Showed endogenous mutations could also drive zebrafish T-ALL | Mutant genes were never identified [63] |
| Model 9 | Human ARID5B | rag2 | hARID5B | rag2:hARID5B | 5 | 180 | Proved role of ARID5B in T-ALL | Fish also showed delayed thymic involution, and thymocytes showed radiation resistance. [66] |
| Model 10 | Zebrafish jdp2 | rag2 | zjdp2 | rag2:zjdp2 | 50 | 270–360 | Proved role of jdp2 in T-ALL and linked mcl1 to drug resistance | Fish also showed thymic hyperplasia and delayed thymic involution [67] |
| B-ALL Models | Transgenes (promoter:oncogene) | Incidence | Mean Latency (dpf) | Key Features | Other Remarks | |||
| Promoter | Oncogene(s) | Full Construct | ||||||
| Model 1 | Human TEL-AML1 (ETV6-RUNX1) | Xenopus ef1a (Xef1a)/zebrafish beta-actin (zba) | TEL-AML1 | Xef1a:TEL-AML1 and zba:TEL-AML1 | 3 | 360 | Long latency and low incidence recapitulate human TEL-AML1 B-ALL | Models the most prevalent type of pediatric ALL [78] |
| Model 2 | Murine Myc (mMyc) | rag2 | mMyc | rag2:EGFP-mMyc | Not reported | Not reported | Develops ighm+ B-ALL (in addition to T-ALL) | Unexpectedly unique expression signature from hMYC B-ALL [79] |
| Model 3 | Human MYC (hMYC) | rag2 | hMYC | rag2:hMYC-ER | Not reported | Not reported | Develops ighz+ B-ALL (in addition to T-ALL) | Only large zebrafish B-ALL study; unexpectedly unique expression signature from mMyc B-ALL [54] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, A.A.; Park, G.; Frazer, J.K. Tackling Acute Lymphoblastic Leukemia—One Fish at a Time. Int. J. Mol. Sci. 2019, 20, 5313. https://doi.org/10.3390/ijms20215313
Sinha AA, Park G, Frazer JK. Tackling Acute Lymphoblastic Leukemia—One Fish at a Time. International Journal of Molecular Sciences. 2019; 20(21):5313. https://doi.org/10.3390/ijms20215313
Chicago/Turabian StyleSinha, Arpan A., Gilseung Park, and J. Kimble Frazer. 2019. "Tackling Acute Lymphoblastic Leukemia—One Fish at a Time" International Journal of Molecular Sciences 20, no. 21: 5313. https://doi.org/10.3390/ijms20215313
APA StyleSinha, A. A., Park, G., & Frazer, J. K. (2019). Tackling Acute Lymphoblastic Leukemia—One Fish at a Time. International Journal of Molecular Sciences, 20(21), 5313. https://doi.org/10.3390/ijms20215313