First Report of a Patient with MPS Type VII, Due to Novel Mutations in GUSB, Who Underwent Enzyme Replacement and Then Hematopoietic Stem Cell Transplantation

 ,
,
Abstract
:1. Introduction
Case Description
2. Results
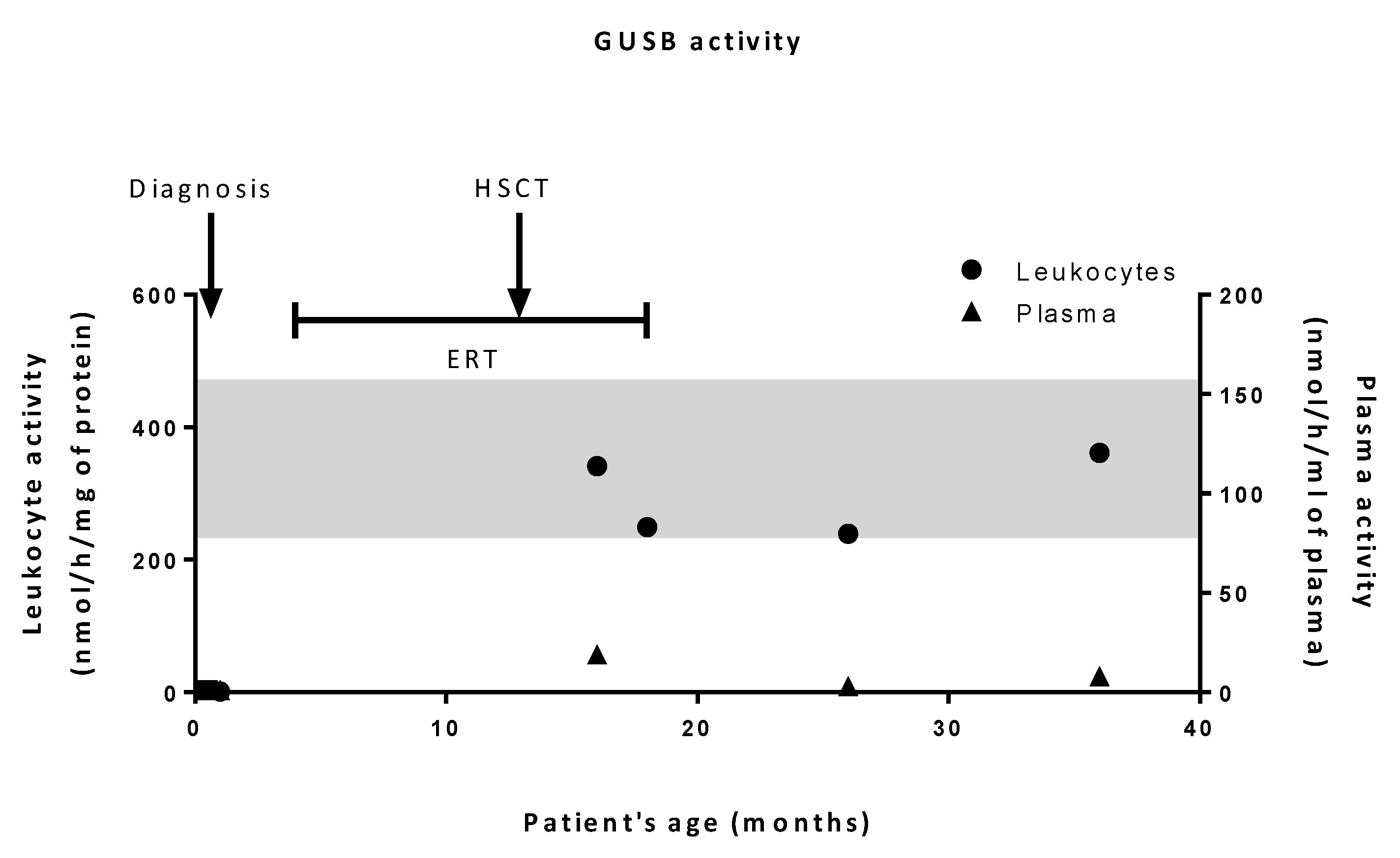
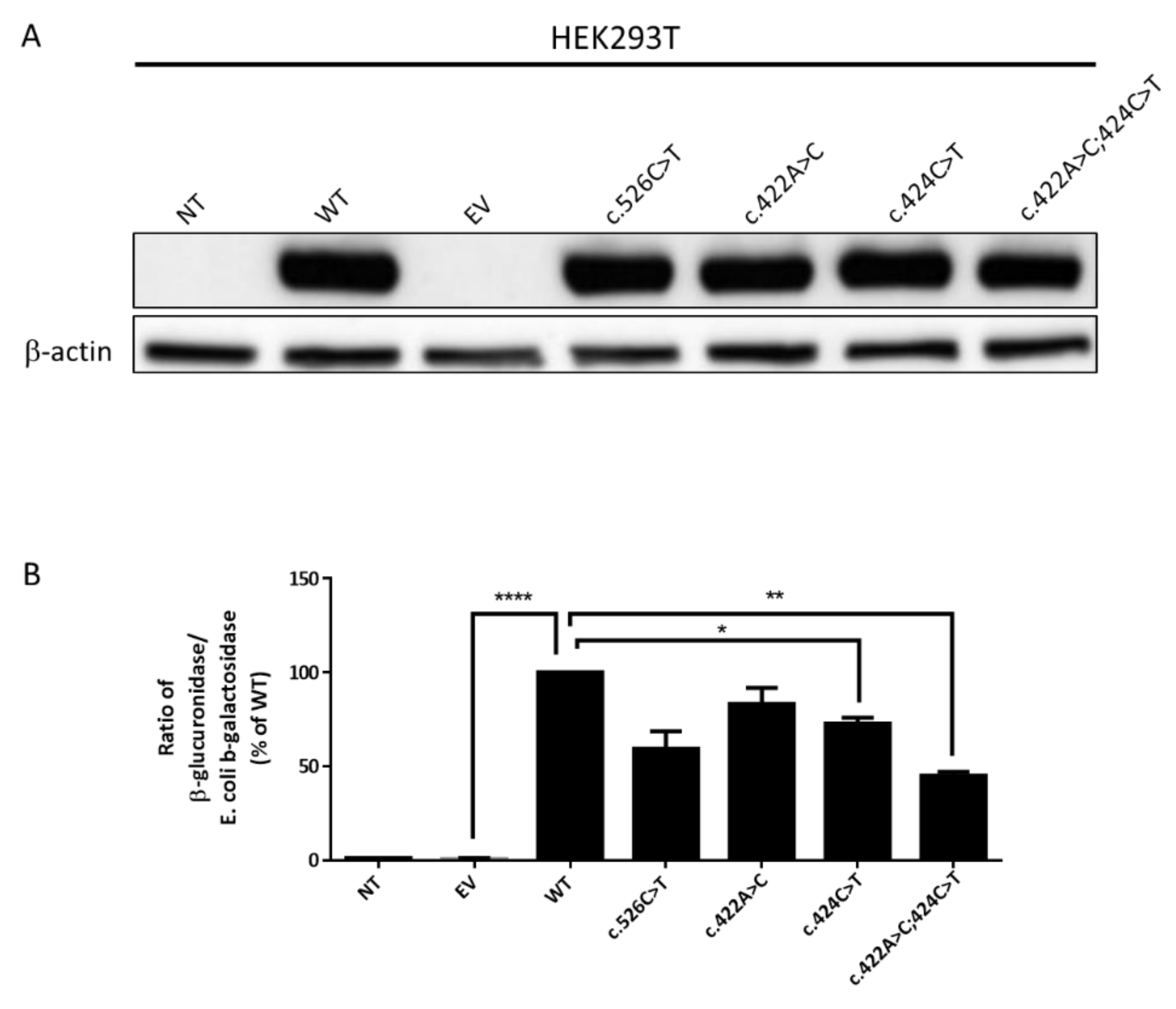
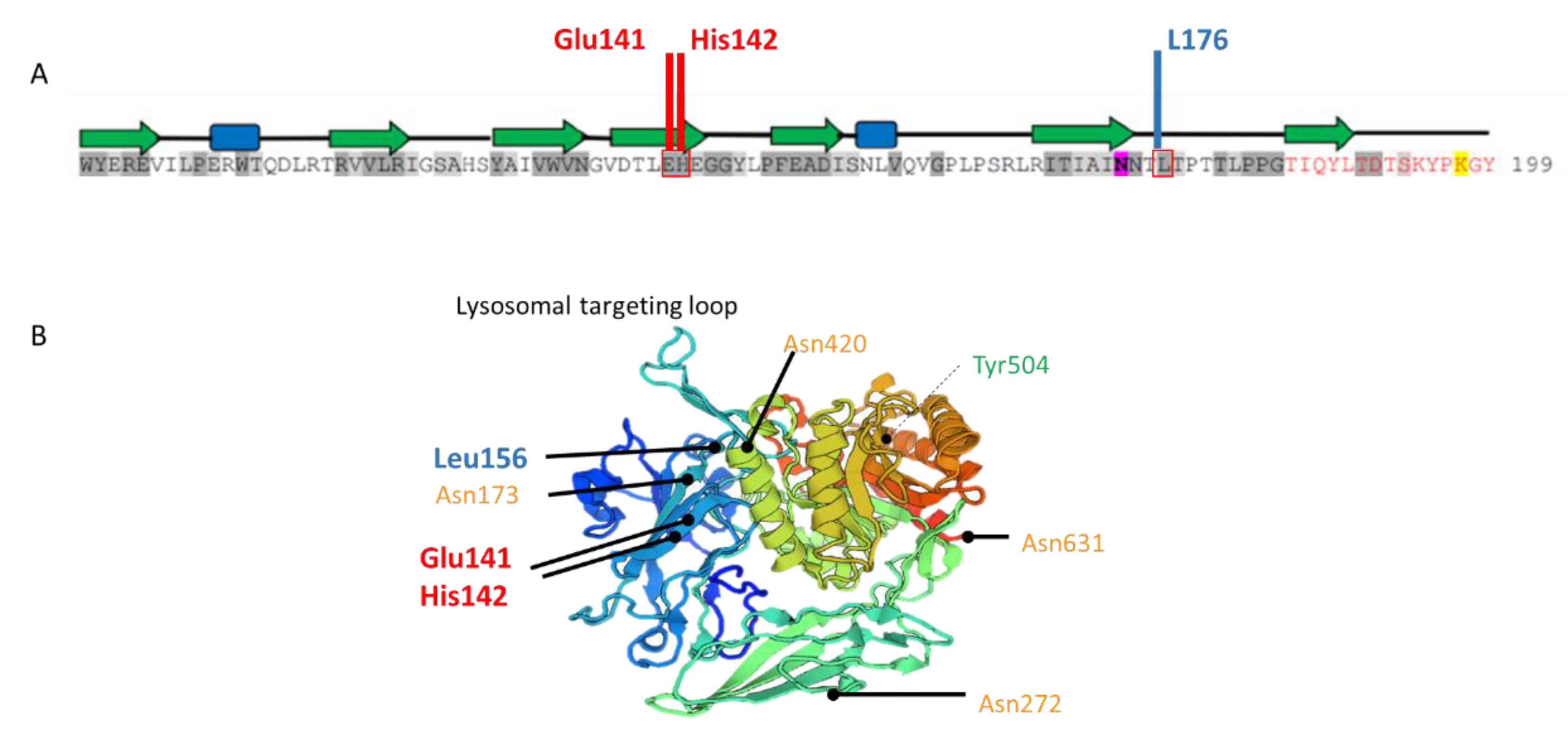
2.1. Biochemical Diagnosis and Characterization of Mutant GUSB Alleles
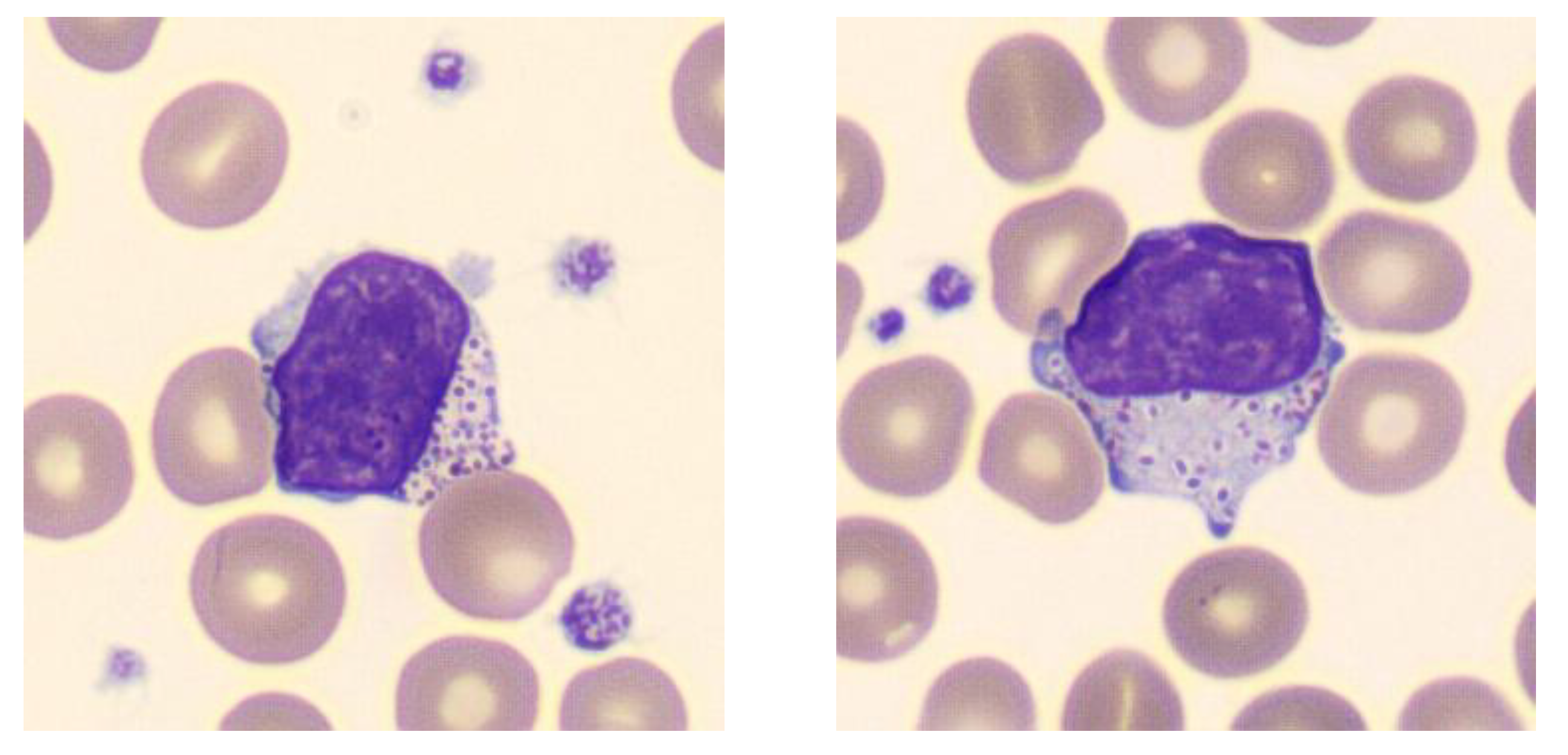
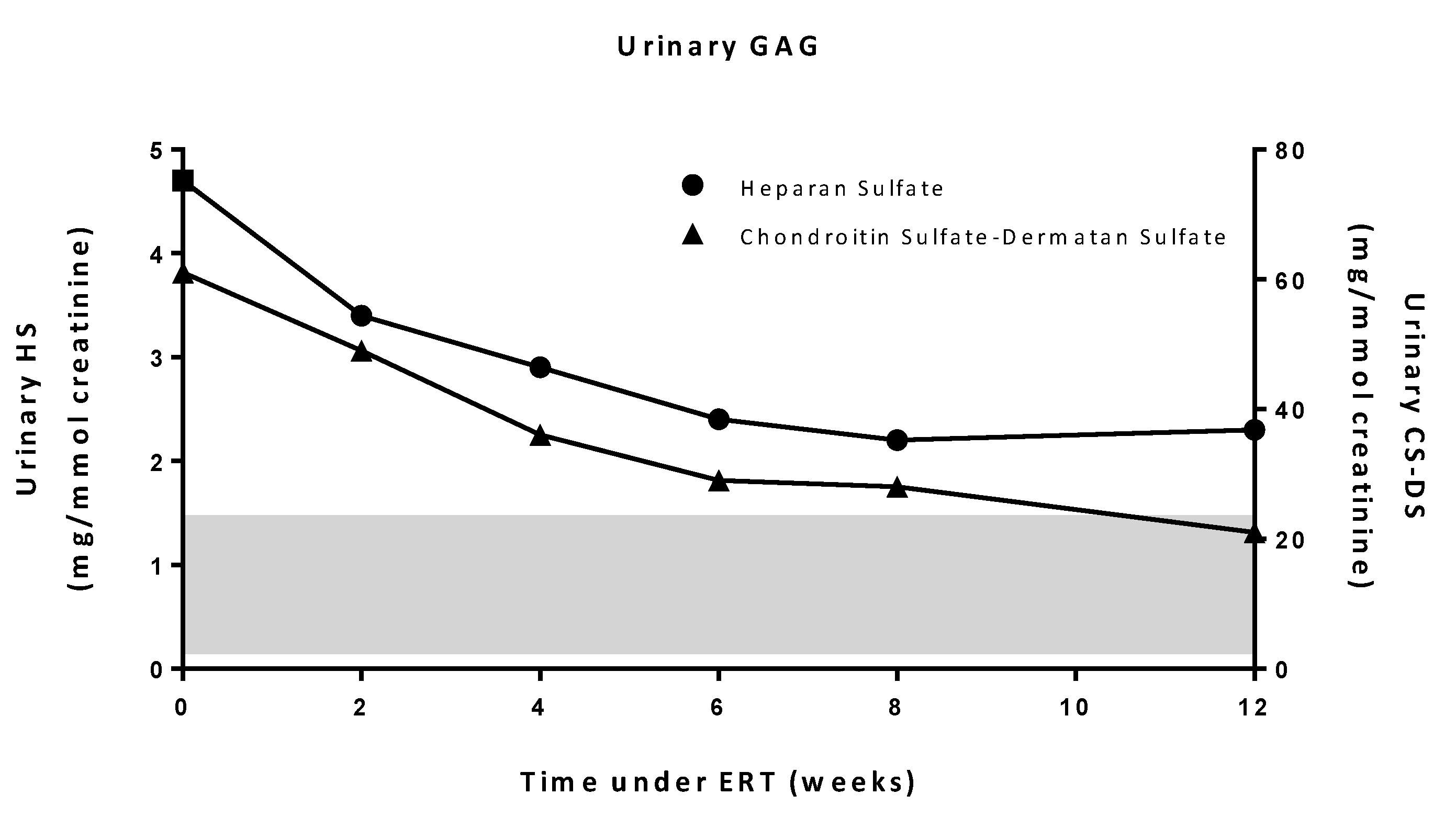
2.2. Patient’s History
3. Discussion
4. Materials and Methods
4.1. Lysosomal Enzyme Assays
4.2. Urinary GAG Analysis
4.3. DNA Studies
4.4. Cell Lines and Transfections
4.5. Western Blot Analysis
4.6. Treatments and Assessments
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Sly, W.S.; Quinton, B.A.; McAlister, W.H.; Rimoin, D.L. Beta glucuronidase deficiency: Report of clinical, radiologic, and biochemical features of a new mucopolysaccharidosis. J. Pediatr. 1973, 82, 249–257. [Google Scholar] [CrossRef]
- Zielonka, M.; Garbade, S.F.; Kölker, S.; Hoffmann, G.F.; Ries, M. Quantitative clinical characteristics of 53 patients with MPS VII: A cross-sectional analysis. Genet. Med. 2017, 19, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Montaño, A.M.; Lock-Hock, N.; Steiner, R.D.; Graham, B.H.; Szlago, M.; Greenstein, R.; Pineda, M.; Gonzalez-Meneses, A.; Çoker, M.; Bartholomew, D.; et al. Clinical course of sly syndrome (mucopolysaccharidosis type VII). J. Med. Genet. 2016, 53, 403–418. [Google Scholar] [CrossRef]
- McCafferty, E.H.; Scott, L.J. Vestronidase Alfa: A Review in Mucopolysaccharidosis VII. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2019, 33, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.E.; Volpe, L.; Bullaro, J.; Kakkis, E.D.; Sly, W.S. First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient. Mol. Genet. Metab. 2015, 114, 203–208. [Google Scholar] [CrossRef]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic stem cell transplantation for mucopolysaccharidoses: Past, present, and future. Biol. Blood Marrow Transplant. 2019, 25, e226–e246. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Dung, V.C.; Grubb, J.H.; Sly, W.S.; Grubb, J.H.; Dung, V.C.; Montaño, A.M.; Tomatsu, S. Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly Syndrome). Hum. Mutat. 2009, 30, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Khan, F.I.; Shahbaaz, M.; Bisetty, K.; Waheed, A.; Sly, W.S.; Ahmad, F.; Hassan, M.I. Large scale analysis of the mutational landscape in β-glucuronidase: A major player of mucopolysaccharidosis type VII. Gene 2016, 576, 36–44. [Google Scholar] [CrossRef]
- HGMD. Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 16 October 2019).
- Wu, B.M.; Tomatsu, S.; Fukuda, S.; Sukegawa, K.; Orii, T.; Sly, W.S. Overexpression rescues the mutant phenotype of L176F mutation causing beta-glucuronidase deficiency mucopolysaccharidosis in two mennonite siblings. J. Biol. Chem. 1994, 269, 23681–23688. [Google Scholar]
- Hassan, M.I.; Waheed, A.; Grubb, J.H.; Klei, H.E.; Korolev, S.; Sly, W.S. High Resolution Crystal Structure of Human β-Glucuronidase Reveals Structural Basis of Lysosome Targeting. PLoS ONE 2013, 8, e79687. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.I.; Waheed, A.; Grubb, J.H.; Klei, H.E.; Korolev, S.; Sly, W.S. Correction: High Resolution Crystal Structure of Human β-Glucuronidase Reveals Structural Basis of Lysosome Targeting. PLoS ONE 2015, 10, e0138401. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.C.; Abe, T.; Hill, W.D.; Studdard, A.M.; Carothers, J.; Masuya, M.; Fleming, P.A.; Drake, C.J.; Ogawa, M. Hematopoietic origin of microglial and perivascular cells in brain. Exp. Neurol. 2004, 186, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Kato, K.; Sukegawa, K.; Tomatsu, S.; Fukuda, S.; Emura, S.; Kojima, S.; Matsuyama, T.; Sly, W.S.; Kondo, N. Treatment of MPS VII (Sly disease) by allogeneic BMT in a female with homozygous A619V mutation. Bone Marrow Transplant. 1998, 21, 629. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Vervoort, R.; Lissens, W.; Hoo, J.J.; Valentino, L.A.; Sly, W.S. Beta-glucuronidase P408S, P415L mutations: Evidence that both mutations combine to produce an MPS VII allele in certain Mexican patients. Hum. Genet. 1996, 98, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Furlan, F.; Rovelli, A.; Rigoldi, M.; Filocamo, M.; Tappino, B.; Friday, D.; Gasperini, S.; Mariani, S.; Izzi, C.; Bondioni, M.P.; et al. A new case report of severe mucopolysaccharidosis type VII: Diagnosis, treatment with haematopoietic cell transplantation and prenatal diagnosis in a second pregnancy. Ital. J. Pediatr. 2018, 44, 128. [Google Scholar] [CrossRef]
- Sisinni, L.; Pineda, M.; Coll, M.J.; Gort, L.; Turon, E.; Torrent, M.; Ey, A.; Tobajas, E.; Badell, I. Haematopoietic stem cell transplantation for mucopolysaccharidosis type VII: A case report. Pediatr. Transplant. 2018, 22, e13278. [Google Scholar] [CrossRef]
- Peck, S.H.; O’Donnell, P.J.M.; Kang, J.L.; Malhotra, N.R.; Dodge, G.R.; Pacifici, M.; Shore, E.M.; Haskins, M.E.; Smith, L.J. Delayed hypertrophic differentiation of epiphyseal chondrocytes contributes to failed secondary ossification in mucopolysaccharidosis VII dogs. Mol. Genet. Metab. 2015, 116, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.; Suzuki, Y.; Tanaka, A.; Yabe, H.; Kato, S.; Shimada, T.; Mason, R.W.; Orii, K.E.; Fukao, T.; Orii, T.; et al. Impact of enzyme replacement therapy and hematopoietic stem cell therapy on growth in patients with Hunter syndrome. Mol. Genet. Metab. Rep. 2014, 1, 184–196. [Google Scholar] [CrossRef]
- Tanjuakio, J.; Suzuki, Y.; Patel, P.; Yasuda, E.; Kubaski, F.; Tanaka, A.; Yabe, H.; Mason, R.W.; Montaño, A.M.; Orii, K.E.; et al. Activities of daily living in patients with Hunter syndrome: Impact of enzyme replacement therapy and hematopoietic stem cell transplantation. Mol. Genet. Metab. 2015, 114, 161–169. [Google Scholar] [CrossRef]
- Sands, M.S.; Vogler, C.; Torrey, A.; Levy, B.; Gwynn, B.; Grubb, J.; Sly, W.S.; Birkenmeier, E.H. Murine mucopolysaccharidosis type VII: Long term therapeutic effects of enzyme replacement and enzyme replacement followed by bone marrow transplantation. J. Clin. Invest. 1997, 99, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Barth, A.L.; de Magalhães, T.S.P.C.; Reis, A.B.R.; de Oliveira, M.L.; Scalco, F.B.; Cavalcanti, N.C.; Silva, D.S.e; Torres, D.A.; Costa, A.A.P.; Bonfim, C.; et al. Early hematopoietic stem cell transplantation in a patient with severe mucopolysaccharidosis II: A 7 years follow-up. Mol. Genet. Metab. Rep. 2017, 12, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Moreau, J.; Brassier, A.; Amaddeo, A.; Neven, B.; Caillaud, C.; Chabli, A.; Fernandez-Bolanos, M.; Olmo, J.; Valayannopoulos, V.; Fauroux, B. Obstructive sleep apnea syndrome after hematopoietic stem cell transplantation in children with mucopolysaccharidosis type I. Mol. Genet. Metab. 2015, 116, 275–280. [Google Scholar] [CrossRef]
- Hall, C.W.; Cantz, M.; Neufeld, E.F. A β-Glucuronidase Deficiency Mucopolysaccharidosis: Studies in Cultured Fibroblasts. Arch. Biochem. Biophys. 1973, 155, 32–38. [Google Scholar] [CrossRef]
- Glaser, J.H.; Sly, W.S. Beta-glucuronidase deficiency mucopolysaccharidosis: Methods for enzymatic diagnosis. J. Lab. Clin. Med. 1973, 82, 969–977. [Google Scholar] [PubMed]
- De Jong, J.G.; Wevers, R.A.; Laarakkers, C.; Poorthuis, B.J. Dimethylmethylene blue-based spectrophotometry of glycosaminoglycans in untreated urine: A rapid screening procedure for mucopolysaccharidoses. Clin. Chem. 1989, 35, 1472–1477. [Google Scholar] [PubMed]
- Hopwood, J.J.; Harrison, J.R. High-resolution electrophoresis of urinary glycosaminoglycans: An improved screening test for the mucopolysaccharidoses. Anal. Biochem. 1982, 119, 120–127. [Google Scholar] [CrossRef]
- Shipley, J.M.; Klinkenberg, M.; Wu, B.M.; Bachinsky, D.R.; Grubb, J.H.; Sly, W.S. Mutational analysis of a patient with mucopolysaccharidosis type VII, and identification of pseudogenes. Am. J. Hum. Genet. 1993, 52, 517–526. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| References | Yamada et al., 1998 [15] | Montano et al., 2016 [4] | Sisinni et al., 2018 [18] | Furlan et al., 2018 [17] | Present Case | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Age at diagnosis (months) | 1 (girl) | 22 (girl) | 24 (boy) | 4 (boy) | 26 (boy) | 0.5 (girl) | 11 (girl) | 1 (boy) | 0.5 (boy) | |
| Clinical signs | Hydrops fetalis | NM | NM | NM | NM | Yes | Yes | No | Yes | No |
| Head, eyes, ear-nose-throat | Coarse face Mild deafness | NM | Coarse face | Coarse face | NM | Coarse face | Coarse face | Coarse face Bilateral severe hypoacusia | Coarse face Acute otitis media Mild deafness | |
| Cardio-respiratory | Recurrent respiratory infections Peripheral pulmonary stenosis Ventricular septal defect | NM | Recurrent respiratory infections Cardiac valve disease | Recurrent respiratory infections Cardiac valve disease | Breathing difficulty | Cardiac distress | Recurrent respiratory infections No cardiac disease | Respiratory distress recurrent infections | None | |
| Musculo-skeletal | Lumbar gibbus, bilateral femoral head hypoplasia Wheel-chair bound | Kyphosis and talipes equinovarus | NM | NM | NM | NM | Short trunk with pectus carinatum Scoliosis and kyphosis Talipes equinovarus Acetabular hip dysplasia Joint contractures and stiffness Genu valgum Clawed hands | Bilateral club-foot Pectus carinatum Gibbus Joint stiffness Dysostosis multiplex | Talipes equinovarus Kyphosis | |
| Thoraco-lumbar and abdominal | Hepatosplenomegaly Umbilical hernia | NM | NM | Hepato-splenomegaly | Hepatosplenomegaly | Hepatomegaly Inguinal hernia | Hepatomegaly Umbilical hernia | Hepatomegaly Bilateral inguinal hernias | Hepato-splenomegaly | |
| Neurological | Mental retardation (IQ: 50) | Normal intelligence | Mental retardation | Mental retardation | Mental retardation Development delay | NM | No alterations (IQ:100) | Axial hypotonia Hypertonia in limbs | Slight axial hypotonia | |
| Age at transplantation (years) | 12 | 2 and 4 | 7 | NM | 3 | 0.5 | 2 and 3.5 | 1.2 | 1.3 | |
| Evolution | Stabilization of symptoms Stop of recurrent infections Improved motor function (ability to walk short distances without assistance) | Moderate clinical manifestations Slow progression Persistent kyphosis and talipes equinovarus Normal intelligence | Deceased (from complications of the procedure) | Deceased (from complications of the procedure) | Moderate clinical phenotype Swallowing difficulties Recurrent respiratory infections Skeletal abnormalities Restrictive and obstructive airway disease | No clinical manifestations | Normal motor function Stabilization of musculoskeletal symptoms Improvement of coarse face and hepatomegaly Good respiratory function IQ: 109 | Worsening respiratory disease Deceased at 25 months of age | Stabilized growth curve No hepatosplenomegaly Normal psychomotor development | |
| Age at last follow-up (years) | 14.5 | NM | 15 | 1.25 | 9 | 4 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubot, P.; Sabourdy, F.; Plat, G.; Jubert, C.; Cancès, C.; Broué, P.; Touati, G.; Levade, T. First Report of a Patient with MPS Type VII, Due to Novel Mutations in GUSB, Who Underwent Enzyme Replacement and Then Hematopoietic Stem Cell Transplantation. Int. J. Mol. Sci. 2019, 20, 5345. https://doi.org/10.3390/ijms20215345
Dubot P, Sabourdy F, Plat G, Jubert C, Cancès C, Broué P, Touati G, Levade T. First Report of a Patient with MPS Type VII, Due to Novel Mutations in GUSB, Who Underwent Enzyme Replacement and Then Hematopoietic Stem Cell Transplantation. International Journal of Molecular Sciences. 2019; 20(21):5345. https://doi.org/10.3390/ijms20215345
Chicago/Turabian StyleDubot, Patricia, Frédérique Sabourdy, Geneviève Plat, Charlotte Jubert, Claude Cancès, Pierre Broué, Guy Touati, and Thierry Levade. 2019. "First Report of a Patient with MPS Type VII, Due to Novel Mutations in GUSB, Who Underwent Enzyme Replacement and Then Hematopoietic Stem Cell Transplantation" International Journal of Molecular Sciences 20, no. 21: 5345. https://doi.org/10.3390/ijms20215345
APA StyleDubot, P., Sabourdy, F., Plat, G., Jubert, C., Cancès, C., Broué, P., Touati, G., & Levade, T. (2019). First Report of a Patient with MPS Type VII, Due to Novel Mutations in GUSB, Who Underwent Enzyme Replacement and Then Hematopoietic Stem Cell Transplantation. International Journal of Molecular Sciences, 20(21), 5345. https://doi.org/10.3390/ijms20215345