Meta-Analysis of Gene Expression Changes in the Blood of Patients with Mild Cognitive Impairment and Alzheimer’s Disease Dementia

Abstract
:
1. Introduction
2. Results
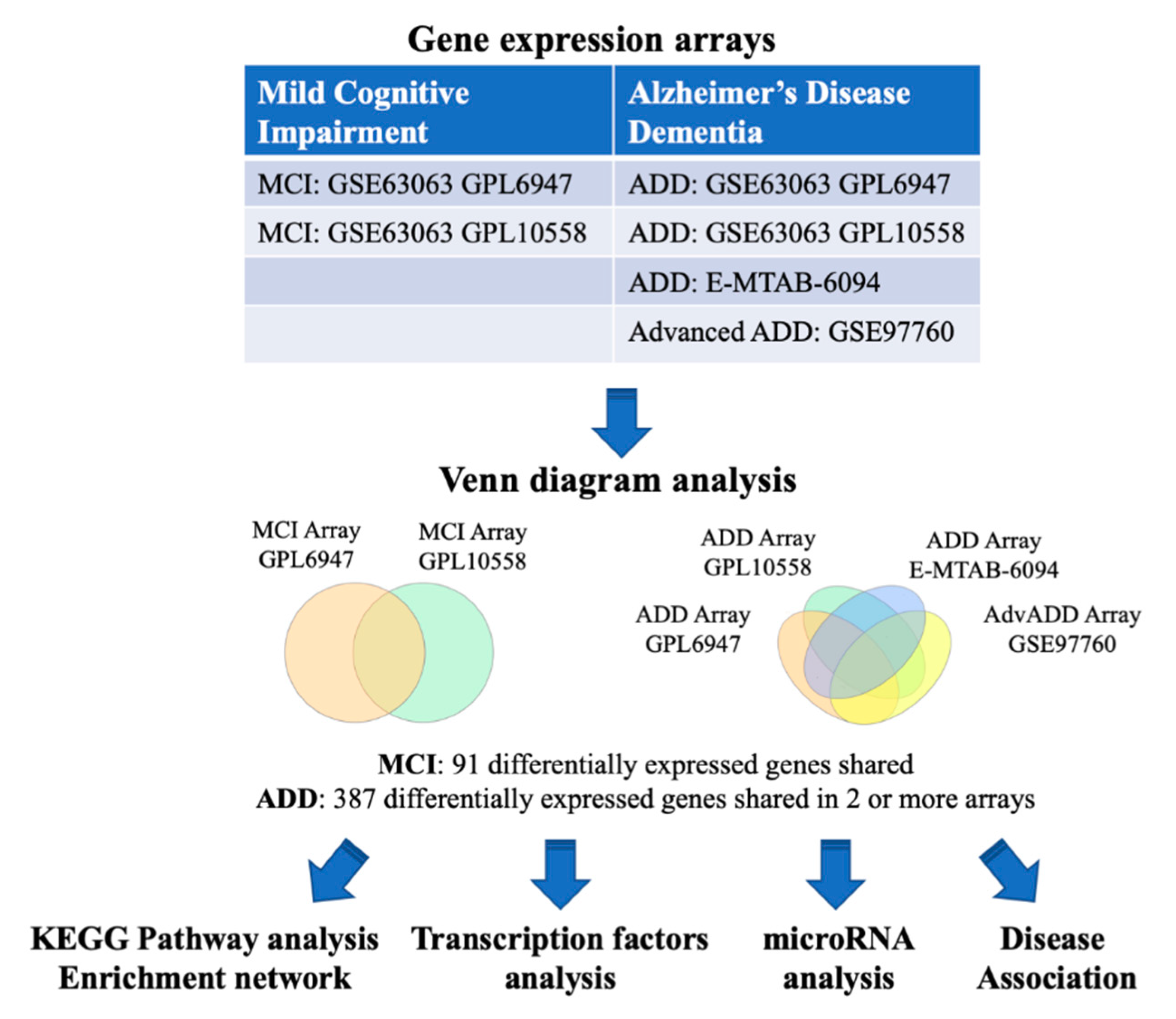
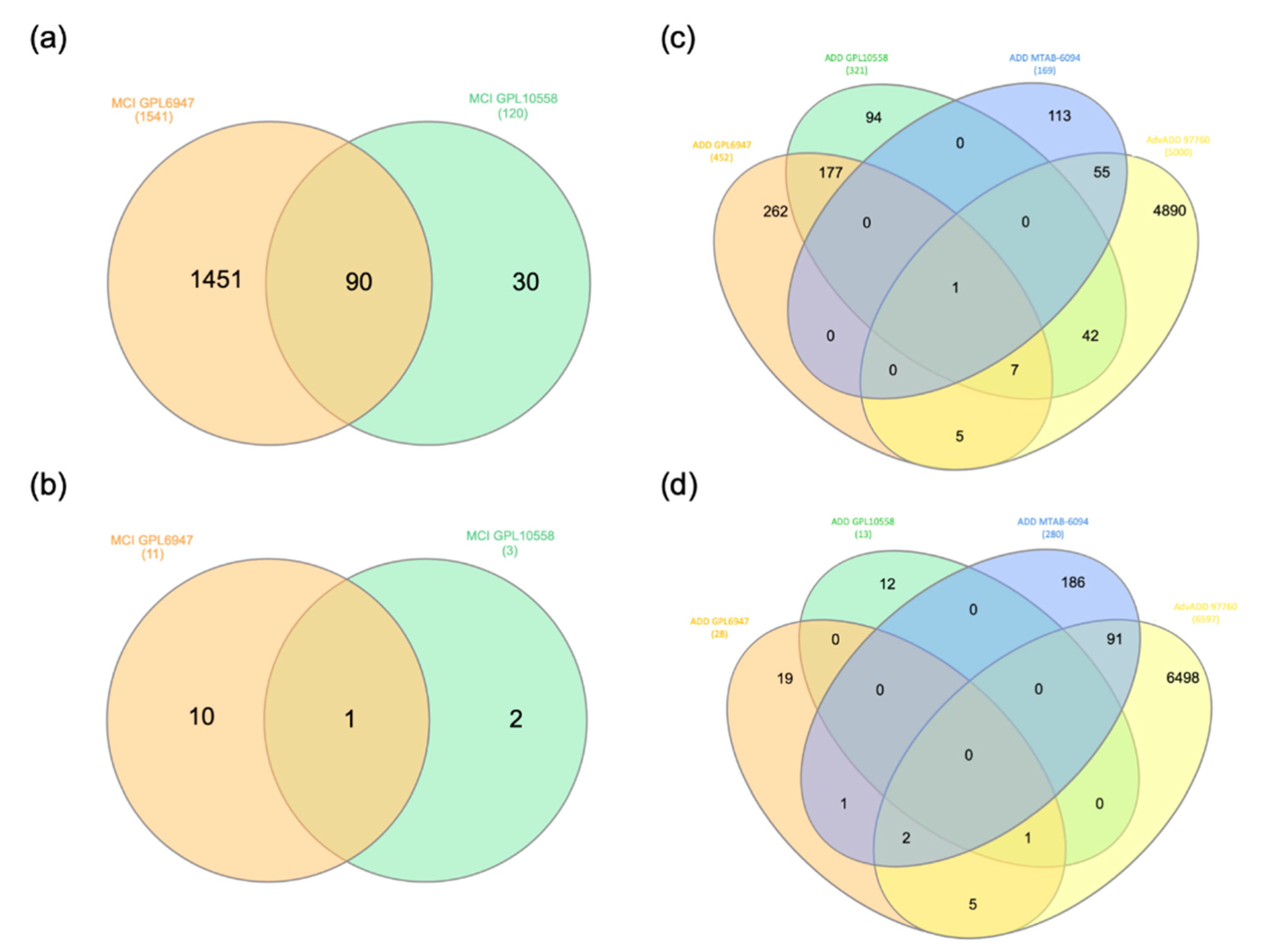
2.1. MCI and ADD Gene Expression Meta-Analysis
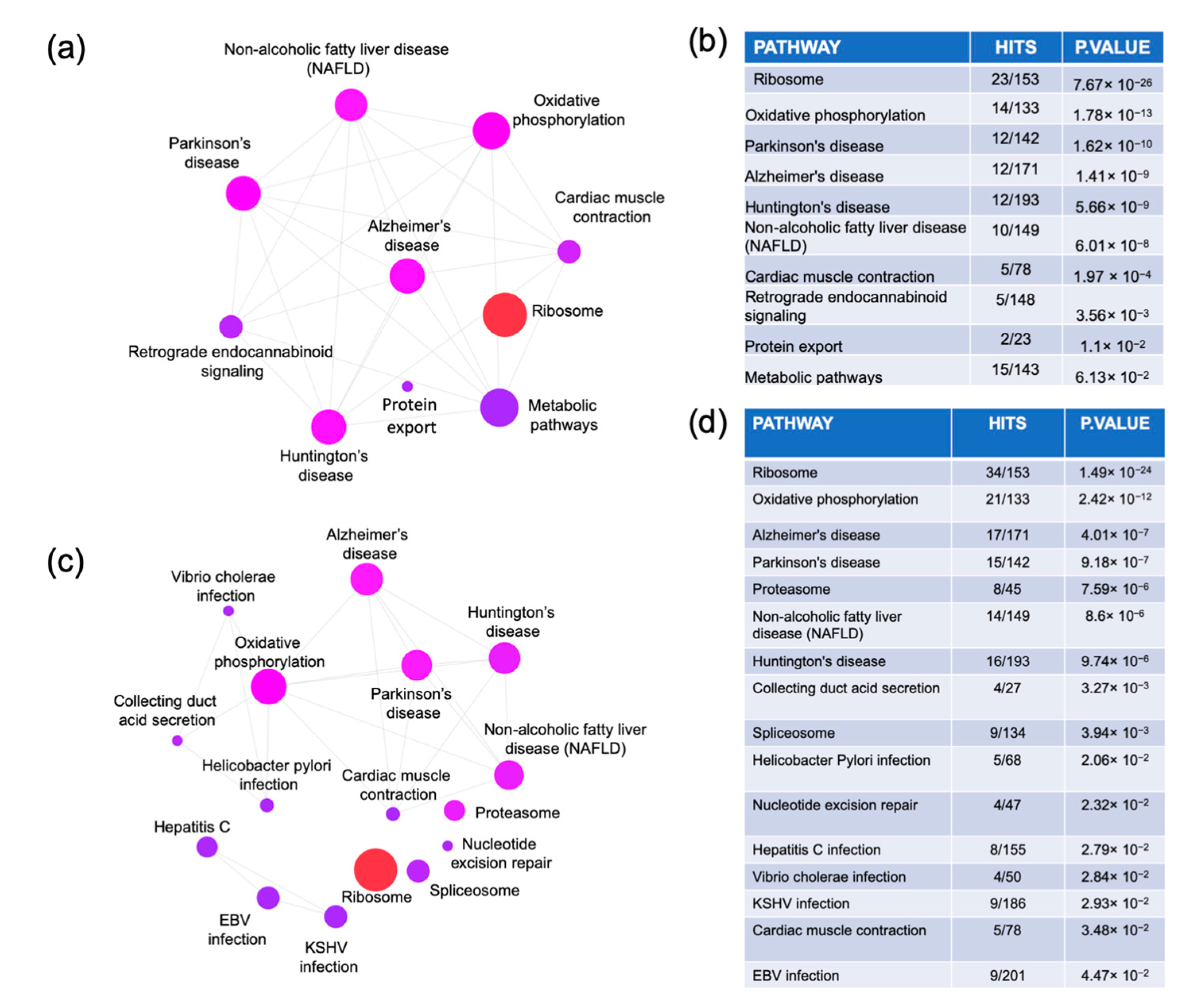
2.2. Pathway Enrichment Analysis
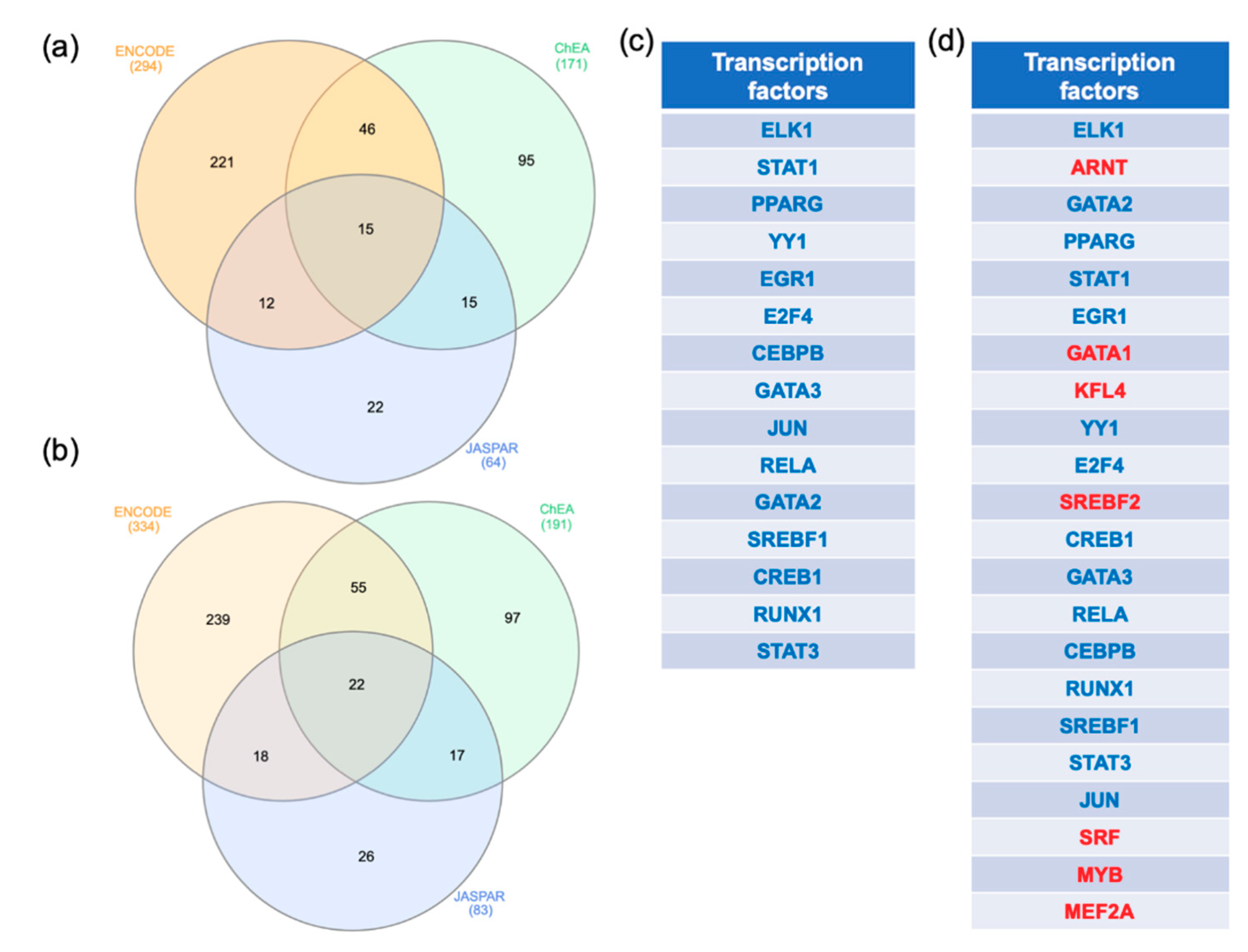
2.3. Gene-Transcription Factors Interaction Analysis
2.4. Gene-miRNA Interaction Analysis
2.5. Gene-Disease Association Analysis
3. Discussion
3.1. MCI and ADD Dysregulated Genes
3.2. MCI and ADD Enrichment Pathways
3.3. Transcription Factors that May Regulate MCI and ADD Genes Expression in Blood
3.4. miRNAs that May Regulate MCI and ADD Gene Expression in Blood
3.5. Limitations
4. Materials and Methods
4.1. Analysis of Blood Transcriptomic Studies
4.2. Pathway Enrichment Analysis
4.3. Gene-Transcription Factors Interaction Analysis
4.4. Gene-miRNA Interaction Analysis
4.5. Gene-Disease Association Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Robinson, L.; Tang, E.; Taylor, J.P. Dementia: Timely diagnosis and early intervention. BMJ 2015, 350, h3029. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Therneau, T.M.; Weigand, S.D.; Wiste, H.J.; Knopman, D.S.; Vemuri, P.; Lowe, V.J.; Mielke, M.M.; Roberts, R.O.; Machulda, M.M.; et al. Prevalence of Biologically vs Clinically Defined Alzheimer Spectrum Entities Using the National Institute on Aging-Alzheimer’s Association Research Framework. JAMA Neurol. 2019, 76, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Raber, J.; Huang, Y.; Ashford, J.W. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol. Aging 2004, 25, 641–650. [Google Scholar] [CrossRef]
- Bennett, D.A.; Wilson, R.S.; Schneider, J.A.; Evans, D.A.; Aggarwal, N.T.; Arnold, S.E.; Cochran, E.J.; Berry-Kravis, E.; Bienias, J.L. Apolipoprotein E epsilon4 allele, AD pathology, and the clinical expression of Alzheimer’s disease. Neurology 2003, 60, 246–252. [Google Scholar] [CrossRef]
- Bennett, D.A.; Schneider, J.A.; Wilson, R.S.; Bienias, J.L.; Berry-Kravis, E.; Arnold, S.E. Amyloid mediates the association of apolipoprotein E e4 allele to cognitive function in older people. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1194–1199. [Google Scholar] [CrossRef] [Green Version]
- Karch, C.M.; Cruchaga, C.; Goate, A.M. Alzheimer’s disease genetics: From the bench to the clinic. Neuron 2014, 83, 11–26. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hagg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Extance, A. Alzheimer’s failure raises questions about disease-modifying strategies. Nat. Rev. Drug Discov. 2010, 9, 749–751. [Google Scholar] [CrossRef]
- Alzheimer’s, A. Alzheimer’s disease facts and figures. Alzheimers Dement. 2011, 7, 208–244. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Tang, Y.; Gilbert, D.L.; Glauser, T.A.; Hershey, A.D.; Sharp, F.R. Blood gene expression profiling of neurologic diseases: A pilot microarray study. Arch. Neurol. 2005, 62, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Courtney, E.; Kornfeld, S.; Janitz, K.; Janitz, M. Transcriptome profiling in neurodegenerative disease. J. Neurosci. Methods 2010, 193, 189–202. [Google Scholar] [CrossRef]
- Wu, Y.; Le, W.; Jankovic, J. Preclinical biomarkers of Parkinson disease. Arch. Neurol. 2011, 68, 22–30. [Google Scholar] [CrossRef]
- Leuner, K.; Schulz, K.; Schutt, T.; Pantel, J.; Prvulovic, D.; Rhein, V.; Savaskan, E.; Czech, C.; Eckert, A.; Muller, W.E. Peripheral mitochondrial dysfunction in Alzheimer’s disease: Focus on lymphocytes. Mol. Neurobiol. 2012, 46, 194–204. [Google Scholar] [CrossRef]
- Santiago, J.A.; Potashkin, J.A. A network approach to clinical intervention in neurodegenerative diseases. Trends Mol. Med. 2014, 20, 694–703. [Google Scholar] [CrossRef]
- Soler-Lopez, M.; Zanzoni, A.; Lluis, R.; Stelzl, U.; Aloy, P. Interactome mapping suggests new mechanistic details underlying Alzheimer’s disease. Genome Res. 2011, 21, 364–376. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.A.; Woltjer, R.L.; Goodenbour, J.M.; Horvath, S.; Geschwind, D.H. Genes and pathways underlying regional and cell type changes in Alzheimer’s disease. Genome Med. 2013, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Roussos, P.; McKenzie, A.; Zhou, X.; Kajiwara, Y.; Brennand, K.J.; De Luca, G.C.; Crary, J.F.; Casaccia, P.; Buxbaum, J.D.; et al. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Med. 2016, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- Grothe, M.J.; Sepulcre, J.; Gonzalez-Escamilla, G.; Jelistratova, I.; Scholl, M.; Hansson, O.; Teipel, S.J. Molecular properties underlying regional vulnerability to Alzheimer’s disease pathology. Brain 2018, 141, 2755–2771. [Google Scholar] [CrossRef]
- Liang, D.; Han, G.; Feng, X.; Sun, J.; Duan, Y.; Lei, H. Concerted perturbation observed in a hub network in Alzheimer’s disease. PLoS ONE 2012, 7, e40498. [Google Scholar] [CrossRef]
- Kikuchi, M.; Ogishima, S.; Miyamoto, T.; Miyashita, A.; Kuwano, R.; Nakaya, J.; Tanaka, H. Identification of unstable network modules reveals disease modules associated with the progression of Alzheimer’s disease. PLoS ONE 2013, 8, e76162. [Google Scholar] [CrossRef]
- Lau, P.; Bossers, K.; Janky, R.; Salta, E.; Frigerio, C.S.; Barbash, S.; Rothman, R.; Sierksma, A.S.; Thathiah, A.; Greenberg, D.; et al. Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 1613–1634. [Google Scholar] [CrossRef]
- Hallock, P.; Thomas, M.A. Integrating the Alzheimer’s disease proteome and transcriptome: A comprehensive network model of a complex disease. Omics J. Integr. Biol. 2012, 16, 37–49. [Google Scholar] [CrossRef]
- Raj, T.; Shulman, J.M.; Keenan, B.T.; Chibnik, L.B.; Evans, D.A.; Bennett, D.A.; Stranger, B.E.; De Jager, P.L. Alzheimer disease susceptibility loci: Evidence for a protein network under natural selection. Am. J. Hum. Genet. 2012, 90, 720–726. [Google Scholar] [CrossRef]
- Talwar, P.; Silla, Y.; Grover, S.; Gupta, M.; Agarwal, R.; Kushwaha, S.; Kukreti, R. Genomic convergence and network analysis approach to identify candidate genes in Alzheimer’s disease. BMC Genom. 2014, 15, 199. [Google Scholar] [CrossRef]
- Ciryam, P.; Kundra, R.; Freer, R.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M. A transcriptional signature of Alzheimer’s disease is associated with a metastable subproteome at risk for aggregation. Proc. Natl. Acad. Sci. USA 2014, 113, 4753–4758. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Chen, S.; Zheng, C.; Wei, H.; Song, X. Meta-Analysis of Gene Expression and Identification of Biological Regulatory Mechanisms in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 633. [Google Scholar] [CrossRef] [PubMed]
- Moradifard, S.; Hoseinbeyki, M.; Ganji, S.M.; Minuchehr, Z. Analysis of microRNA and Gene Expression Profiles in Alzheimer’s Disease: A Meta-Analysis Approach. Sci. Rep. 2018, 8, 4767. [Google Scholar] [CrossRef] [PubMed]
- Puthiyedth, N.; Riveros, C.; Berretta, R.; Moscato, P. Identification of Differentially Expressed Genes through Integrated Study of Alzheimer’s Disease Affected Brain Regions. PLoS ONE 2016, 11, e0152342. [Google Scholar] [CrossRef] [PubMed]
- Wruck, W.; Schroter, F.; Adjaye, J. Meta-Analysis of Transcriptome Data Related to Hippocampus Biopsies and iPSC-Derived Neuronal Cells from Alzheimer’s Disease Patients Reveals an Association with FOXA1 and FOXA2 Gene Regulatory Networks. J. Alzheimers Dis. 2016, 50, 1065–1082. [Google Scholar] [CrossRef]
- Barbash, S.; Soreq, H. Threshold-independent meta-analysis of Alzheimer’s disease transcriptomes shows progressive changes in hippocampal functions, epigenetics and microRNA regulation. Curr. Alzheimer Res. 2012, 9, 425–435. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, J.; Hao, M.; Bu, L. Long noncoding RNA Linc01296 promotes hepatocellular carcinoma development through regulation of the miR-26a/PTEN axis. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Patel, H.; Dobson, R.J.B.; Newhouse, S.J. A Meta-Analysis of Alzheimer’s Disease Brain Transcriptomic Data. J. Alzheimers Dis. 2019, 68, 1635–1656. [Google Scholar] [CrossRef]
- Kelly, J.; Moyeed, R.; Carroll, C.; Albani, D.; Li, X. Gene expression meta-analysis of Parkinson’s disease and its relationship with Alzheimer’s disease. Mol. Brain 2019, 12, 16. [Google Scholar] [CrossRef]
- Wei, C.J.; Cui, P.; Li, H.; Lang, W.J.; Liu, G.Y.; Ma, X.F. Shared genes between Alzheimer’s disease and ischemic stroke. CNS Neurosci. Ther. 2019, 25, 855–864. [Google Scholar] [CrossRef]
- Mirza, Z.; Kamal, M.A.; Buzenadah, A.M.; Al-Qahtani, M.H.; Karim, S. Establishing genomic/transcriptomic links between Alzheimer’s disease and type 2 diabetes mellitus by meta-analysis approach. CNS Neurol. Disord. Drug Targets 2014, 13, 501–516. [Google Scholar] [CrossRef] [PubMed]
- Caberlotto, L.; Nguyen, T.P.; Lauria, M.; Priami, C.; Rimondini, R.; Maioli, S.; Cedazo-Minguez, A.; Sita, G.; Morroni, F.; Corsi, M.; et al. Cross-disease analysis of Alzheimer’s disease and type-2 Diabetes highlights the role of autophagy in the pathophysiology of two highly comorbid diseases. Sci. Rep. 2019, 9, 3965. [Google Scholar] [CrossRef]
- Sood, S.; Gallagher, I.J.; Lunnon, K.; Rullman, E.; Keohane, A.; Crossland, H.; Phillips, B.E.; Cederholm, T.; Jensen, T.; van Loon, L.J.; et al. A novel multi-tissue RNA diagnostic of healthy ageing relates to cognitive health status. Genome Biol. 2015, 16, 185. [Google Scholar] [CrossRef]
- Lunnon, K.; Sattlecker, M.; Furney, S.J.; Coppola, G.; Simmons, A.; Proitsi, P.; Lupton, M.K.; Lourdusamy, A.; Johnston, C.; Soininen, H.; et al. A blood gene expression marker of early Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Lunnon, K.; Ibrahim, Z.; Proitsi, P.; Lourdusamy, A.; Newhouse, S.; Sattlecker, M.; Furney, S.; Saleem, M.; Soininen, H.; Kloszewska, I.; et al. Mitochondrial dysfunction and immune activation are detectable in early Alzheimer’s disease blood. J. Alzheimers Dis. 2012, 30, 685–710. [Google Scholar] [CrossRef] [PubMed]
- Naughton, B.J.; Duncan, F.J.; Murrey, D.A.; Meadows, A.S.; Newsom, D.E.; Stoicea, N.; White, P.; Scharre, D.W.; McCarty, D.M.; Fu, H. Blood genome-wide transcriptional profiles reflect broad molecular impairments and strong blood-brain links in Alzheimer’s disease. J. Alzheimers Dis. 2015, 43, 93–108. [Google Scholar] [CrossRef]
- Ferrarese, C.; Appollonio, I.; Frigo, M.; Meregalli, S.; Piolti, R.; Tamma, F.; Frattola, L. Cerebrospinal fluid levels of diazepam-binding inhibitor in neurodegenerative disorders with dementia. Neurology 1990, 40, 632–635. [Google Scholar] [CrossRef]
- Edgar, P.F.; Schonberger, S.J.; Dean, B.; Faull, R.L.; Kydd, R.; Cooper, G.J. A comparative proteome analysis of hippocampal tissue from schizophrenic and Alzheimer’s disease individuals. Mol. Psychiatry 1999, 4, 173–178. [Google Scholar] [CrossRef]
- Mills, J.D.; Nalpathamkalam, T.; Jacobs, H.I.; Janitz, C.; Merico, D.; Hu, P.; Janitz, M. RNA-Seq analysis of the parietal cortex in Alzheimer’s disease reveals alternatively spliced isoforms related to lipid metabolism. Neurosci. Lett. 2013, 536, 90–95. [Google Scholar] [CrossRef]
- Manners, H.N.; Roy, S.; Kalita, J.K. Intrinsic-overlapping co-expression module detection with application to Alzheimer’s Disease. Comput. Biol. Chem. 2018, 77, 373–389. [Google Scholar] [CrossRef]
- Adav, S.S.; Park, J.E.; Sze, S.K. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol. Brain 2019, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Armand-Ugon, M.; Ansoleaga, B.; Berjaoui, S.; Ferrer, I. Reduced Mitochondrial Activity is Early and Steady in the Entorhinal Cortex but it is Mainly Unmodified in the Frontal Cortex in Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic changes in Alzheimer’s disease: Decrements in DNA methylation. Neurobiol. Aging 2010, 31, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.; Pushpitha, K.; Deng, L.; Chitranshi, N.; Gupta, V.; Rajput, R.; Mangani, A.B.; Dheer, Y.; Godinez, A.; McKay, M.J.; et al. Upregulation of Proteolytic Pathways and Altered Protein Biosynthesis Underlie Retinal Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 6017–6034. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Long, J.; Pan, G.; He, T.; Anichtchik, O.; Belshaw, R.; Albani, D.; Edison, P.; Green, E.K.; et al. Systematic Analysis and Biomarker Study for Alzheimer’s Disease. Sci. Rep. 2018, 8, 17394. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Liao, L.; Chen, C.; Guo, Y.; Song, D.; Wang, Y.; Chen, Y.; Zhang, K.; Ying, M.; Li, S.; et al. Proteomics Analysis of Blood Serums from Alzheimer’s Disease Patients Using iTRAQ Labeling Technology. J. Alzheimers Dis. 2017, 56, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Hagmeyer, S.; Romao, M.A.; Cristovao, J.S.; Vilella, A.; Zoli, M.; Gomes, C.M.; Grabrucker, A.M. Distribution and Relative Abundance of S100 Proteins in the Brain of the APP23 Alzheimer’s Disease Model Mice. Front. Neurosci. 2019, 13, 640. [Google Scholar] [CrossRef]
- Di Domenico, F.; Sultana, R.; Tiu, G.F.; Scheff, N.N.; Perluigi, M.; Cini, C.; Butterfield, D.A. Protein levels of heat shock proteins 27, 32, 60, 70, 90 and thioredoxin-1 in amnestic mild cognitive impairment: An investigation on the role of cellular stress response in the progression of Alzheimer disease. Brain Res. 2010, 1333, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Lovell, M.A.; Xie, C.; Gabbita, S.P.; Markesbery, W.R. Decreased thioredoxin and increased thioredoxin reductase levels in Alzheimer’s disease brain. Free Radic. Biol. Med. 2000, 28, 418–427. [Google Scholar] [CrossRef]
- Arodin, L.; Lamparter, H.; Karlsson, H.; Nennesmo, I.; Bjornstedt, M.; Schroder, J.; Fernandes, A.P. Alteration of thioredoxin and glutaredoxin in the progression of Alzheimer’s disease. J. Alzheimers Dis. 2014, 39, 787–797. [Google Scholar] [CrossRef]
- Akterin, S.; Cowburn, R.F.; Miranda-Vizuete, A.; Jimenez, A.; Bogdanovic, N.; Winblad, B.; Cedazo-Minguez, A. Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer’s disease. Cell Death Differ. 2006, 13, 1454–1465. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Cao, M.; Zheng, H.; Tan, X.; Li, L.; Cui, G.; Xu, J.; Cao, J.; Ke, K.; Wu, Q. Upregulation of SYF2 is associated with neuronal apoptosis caused by reactive astrogliosis to neuroinflammation. J. Neurosci. Res. 2014, 92, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; McCoy, M.K.; Harms, A.S.; Ruhn, K.A.; Gold, S.J.; Tansey, M.G. Regulator of G-protein signaling 10 promotes dopaminergic neuron survival via regulation of the microglial inflammatory response. J. Neurosci. 2008, 28, 8517–8528. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.R.; Islam, T.; Zaman, T.; Shahjaman, M.; Karim, M.R.; Huq, F.; Quinn, J.M.W.; Holsinger, R.M.D.; Gov, E.; Moni, M.A. Identification of molecular signatures and pathways to identify novel therapeutic targets in Alzheimer’s disease: Insights from a systems biomedicine perspective. Genomics 2019. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.R.; Islam, T.; Turanli, B.; Zaman, T.; Faruquee, H.M.; Rahman, M.M.; Mollah, M.N.H.; Nanda, R.K.; Arga, K.Y.; Gov, E.; et al. Network-based approach to identify molecular signatures and therapeutic agents in Alzheimer’s disease. Comput. Biol. Chem. 2019, 78, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Ortega, K.; Garcia-Esparcia, P.; Gil, L.; Lucas, J.J.; Ferrer, I. Altered Machinery of Protein Synthesis in Alzheimer’s: From the Nucleolus to the Ribosome. Brain Pathol. 2016, 26, 593–605. [Google Scholar] [CrossRef]
- Ding, Q.; Markesbery, W.R.; Chen, Q.; Li, F.; Keller, J.N. Ribosome dysfunction is an early event in Alzheimer’s disease. J. Neurosci. 2005, 25, 9171–9175. [Google Scholar] [CrossRef]
- Meier, S.; Bell, M.; Lyons, D.N.; Rodriguez-Rivera, J.; Ingram, A.; Fontaine, S.N.; Mechas, E.; Chen, J.; Wolozin, B.; LeVine, H., 3rd; et al. Pathological Tau Promotes Neuronal Damage by Impairing Ribosomal Function and Decreasing Protein Synthesis. J. Neurosci. 2016, 36, 1001–1007. [Google Scholar] [CrossRef] [Green Version]
- He, K.; Nie, L.; Zhou, Q.; Rahman, S.U.; Liu, J.; Yang, X.; Li, S. Proteomic Profiles of the Early Mitochondrial Changes in APP/PS1 and ApoE4 Transgenic Mice Models of Alzheimer’s Disease. J. Proteome Res. 2019, 18, 2632–2642. [Google Scholar] [CrossRef]
- Lanzillotta, C.; Di Domenico, F.; Perluigi, M.; Butterfield, D.A. Targeting Mitochondria in Alzheimer Disease: Rationale and Perspectives. CNS Drugs 2019. [Google Scholar] [CrossRef]
- Gibson, G.E.; Shi, Q. A mitocentric view of Alzheimer’s disease suggests multi-faceted treatments. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S591–S607. [Google Scholar] [CrossRef]
- Delbarba, A.; Abate, G.; Prandelli, C.; Marziano, M.; Buizza, L.; Arce Varas, N.; Novelli, A.; Cuetos, F.; Martinez, C.; Lanni, C.; et al. Mitochondrial Alterations in Peripheral Mononuclear Blood Cells from Alzheimer’s Disease and Mild Cognitive Impairment Patients. Oxid. Med. Cell. Longev. 2016, 2016, 5923938. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.R.; Wang, Q.H.; Zhang, T.; Liu, Y.H.; Yao, X.Q.; Zeng, F.; Li, J.; Zhou, F.Y.; Wang, L.; Yan, J.C.; et al. Associations Between Hepatic Functions and Plasma Amyloid-Beta Levels-Implications for the Capacity of Liver in Peripheral Amyloid-Beta Clearance. Mol. Neurobiol. 2017, 54, 2338–2344. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.W.; Gottesman, R.F.; Clark, J.M.; Hernaez, R.; Chang, Y.; Kim, C.; Ha, K.H.; Guallar, E.; Lazo, M. Nonalcoholic fatty liver disease is associated with cognitive function in adults. Neurology 2016, 86, 1136–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astarita, G.; Jung, K.M.; Berchtold, N.C.; Nguyen, V.Q.; Gillen, D.L.; Head, E.; Cotman, C.W.; Piomelli, D. Deficient liver biosynthesis of docosahexaenoic acid correlates with cognitive impairment in Alzheimer’s disease. PLoS ONE 2010, 5, e12538. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Parks, S.B.; Kushner, J.D.; Nauman, D.; Burgess, D.; Ludwigsen, S.; Partain, J.; Nixon, R.R.; Allen, C.N.; Irwin, R.P.; et al. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am. J. Hum. Genet. 2006, 79, 1030–1039. [Google Scholar] [CrossRef]
- Turdi, S.; Guo, R.; Huff, A.F.; Wolf, E.M.; Culver, B.; Ren, J. Cardiomyocyte contractile dysfunction in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. PLoS ONE 2009, 4, e6033. [Google Scholar] [CrossRef]
- Kim, T.W.; Song, I.U.; Jeong, D.S.; Lee, K.S. Clinical effect of cerebrovascular atherosclerosis on cognition in Alzheimer’s disease. Arch. Gerontol. Geriatr. 2016, 63, 55–58. [Google Scholar] [CrossRef]
- Hong, L.; Huang, H.C.; Jiang, Z.F. Relationship between amyloid-beta and the ubiquitin-proteasome system in Alzheimer’s disease. Neurol. Res. 2014, 36, 276–282. [Google Scholar] [CrossRef]
- Riederer, B.M.; Leuba, G.; Vernay, A.; Riederer, I.M. The role of the ubiquitin proteasome system in Alzheimer’s disease. Exp. Biol. Med. 2011, 236, 268–276. [Google Scholar] [CrossRef]
- Almeida, C.G.; Takahashi, R.H.; Gouras, G.K. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J. Neurosci. 2006, 26, 4277–4288. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, J. Amyloid-beta peptide is a substrate of the human 20S proteasome. ACS Chem. Neurosci. 2010, 1, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Gregori, L.; Hainfeld, J.F.; Simon, M.N.; Goldgaber, D. Binding of amyloid beta protein to the 20 S proteasome. J. Biol. Chem. 1997, 272, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Lopez Salon, M.; Pasquini, L.; Besio Moreno, M.; Pasquini, J.M.; Soto, E. Relationship between beta-amyloid degradation and the 26S proteasome in neural cells. Exp. Neurol. 2003, 180, 131–143. [Google Scholar] [CrossRef]
- Diner, I.; Hales, C.M.; Bishof, I.; Rabenold, L.; Duong, D.M.; Yi, H.; Laur, O.; Gearing, M.; Troncoso, J.; Thambisetty, M.; et al. Aggregation properties of the small nuclear ribonucleoprotein U1-70K in Alzheimer disease. J. Biol. Chem. 2014, 289, 35296–35313. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Hales, C.M.; Chen, P.C.; Gozal, Y.; Dammer, E.B.; Fritz, J.J.; Wang, X.; Xia, Q.; Duong, D.M.; Street, C.; et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16562–16567. [Google Scholar] [CrossRef]
- Hales, C.M.; Dammer, E.B.; Diner, I.; Yi, H.; Seyfried, N.T.; Gearing, M.; Glass, J.D.; Montine, T.J.; Levey, A.I.; Lah, J.J. Aggregates of small nuclear ribonucleic acids (snRNAs) in Alzheimer’s disease. Brain Pathol. 2014, 24, 344–351. [Google Scholar] [CrossRef]
- Hales, C.M.; Dammer, E.B.; Deng, Q.; Duong, D.M.; Gearing, M.; Troncoso, J.C.; Thambisetty, M.; Lah, J.J.; Shulman, J.M.; Levey, A.I.; et al. Changes in the detergent-insoluble brain proteome linked to amyloid and tau in Alzheimer’s Disease progression. Proteomics 2016, 16, 3042–3053. [Google Scholar] [CrossRef]
- Cheng, Z.; Shang, Y.; Gao, S.; Zhang, T. Overexpression of U1 snRNA induces decrease of U1 spliceosome function associated with Alzheimer’s disease. J. Neurogenet. 2017, 31, 337–343. [Google Scholar] [CrossRef]
- Cheng, Z.; Du, Z.; Zhai, B.; Yang, Z.; Zhang, T. U1 small nuclear RNA overexpression implicates autophagic-lysosomal system associated with AD. Neurosci. Res. 2018, 136, 48–55. [Google Scholar] [CrossRef]
- Ohe, K.; Mayeda, A. HMGA1a trapping of U1 snRNP at an authentic 5’ splice site induces aberrant exon skipping in sporadic Alzheimer’s disease. Mol. Cell. Biol. 2010, 30, 2220–2228. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Li, H.L.; Su, J.B.; Ding, F.H.; Zhao, J.J.; Chai, F.; Li, Y.X.; Cui, S.C.; Sun, F.Y.; Wu, Z.Y.; et al. Regulation of RAGE splicing by hnRNP A1 and Tra2beta-1 and its potential role in AD pathogenesis. J. Neurochem. 2015, 133, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, D.; Inguglia, L.; Walters, J.; Picone, P.; Di Carlo, M. A Shotgun Proteomics Approach Reveals a New Toxic Role for Alzheimer’s Disease Abeta Peptide: Spliceosome Impairment. J. Proteome Res. 2017, 16, 1526–1541. [Google Scholar] [CrossRef] [PubMed]
- Bu, X.L.; Yao, X.Q.; Jiao, S.S.; Zeng, F.; Liu, Y.H.; Xiang, Y.; Liang, C.R.; Wang, Q.H.; Wang, X.; Cao, H.Y.; et al. A study on the association between infectious burden and Alzheimer’s disease. Eur. J. Neurol. 2015, 22, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Li, Y. Herpesviral infections and antimicrobial protection for Alzheimer’s disease: Implications for prevention and treatment. J. Med. Virol. 2019, 91, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Zwolinska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef]
- Licastro, F.; Carbone, I.; Raschi, E.; Porcellini, E. The 21st century epidemic: Infections as inductors of neuro-degeneration associated with Alzheimer’s Disease. Immun. Ageing 2014, 11, 22. [Google Scholar] [CrossRef]
- Hemling, N.; Roytta, M.; Rinne, J.; Pollanen, P.; Broberg, E.; Tapio, V.; Vahlberg, T.; Hukkanen, V. Herpesviruses in brains in Alzheimer’s and Parkinson’s diseases. Ann. Neurol. 2003, 54, 267–271. [Google Scholar] [CrossRef]
- Malaguarnera, M.; Bella, R.; Alagona, G.; Ferri, R.; Carnemolla, A.; Pennisi, G. Helicobacter pylori and Alzheimer’s disease: A possible link. Eur. J. Intern. Med. 2004, 15, 381–386. [Google Scholar] [CrossRef]
- Bibi, F.; Yasir, M.; Sohrab, S.S.; Azhar, E.I.; Al-Qahtani, M.H.; Abuzenadah, A.M.; Kamal, M.A.; Naseer, M.I. Link between chronic bacterial inflammation and Alzheimer disease. CNS Neurol. Disord. Drug Targets 2014, 13, 1140–1147. [Google Scholar] [CrossRef]
- Miklossy, J. Historic evidence to support a causal relationship between spirochetal infections and Alzheimer’s disease. Front. Aging Neurosci. 2015, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Boziki, M.; Gavalas, E.; Zavos, C.; Deretzi, G.; Grigoriadis, N.; Tsolaki, M.; Chatzopoulos, D.; Katsinelos, P.; Tzilves, D.; et al. Increased cerebrospinal fluid Helicobacter pylori antibody in Alzheimer’s disease. Int. J. Neurosci. 2009, 119, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Zeng, J.; Yang, Y.; Xiong, Y.; Zhang, Z.H.; Qiu, M.; Yan, X.; Sun, X.Y.; Tuo, Q.Z.; Liu, R.; et al. Helicobacter pylori filtrate induces Alzheimer-like tau hyperphosphorylation by activating glycogen synthase kinase-3beta. J. Alzheimers Dis. 2015, 43, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adhes. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lillenes, M.S.; Rabano, A.; Stoen, M.; Riaz, T.; Misaghian, D.; Mollersen, L.; Esbensen, Y.; Gunther, C.C.; Selnes, P.; Stenset, V.T.; et al. Altered DNA base excision repair profile in brain tissue and blood in Alzheimer’s disease. Mol. Brain 2016, 9, 61. [Google Scholar] [CrossRef]
- Chen, Z.S.; Chan, H.Y.E. Transcriptional dysregulation in neurodegenerative diseases: Who tipped the balance of Yin Yang 1 in the brain? Neural Regen. Res. 2019, 14, 1148–1151. [Google Scholar] [CrossRef]
- Li, M.D.; Burns, T.C.; Morgan, A.A.; Khatri, P. Integrated multi-cohort transcriptional meta-analysis of neurodegenerative diseases. Acta Neuropathol. Commun. 2014, 2, 93. [Google Scholar] [CrossRef]
- Huang, N.Q.; Jin, H.; Zhou, S.Y.; Shi, J.S.; Jin, F. TLR4 is a link between diabetes and Alzheimer’s disease. Behav. Brain Res. 2017, 316, 234–244. [Google Scholar] [CrossRef]
- Aubry, S.; Shin, W.; Crary, J.F.; Lefort, R.; Qureshi, Y.H.; Lefebvre, C.; Califano, A.; Shelanski, M.L. Assembly and interrogation of Alzheimer’s disease genetic networks reveal novel regulators of progression. PLoS ONE 2015, 10, e0120352. [Google Scholar] [CrossRef]
- Nowak, K.; Lange-Dohna, C.; Zeitschel, U.; Gunther, A.; Luscher, B.; Robitzki, A.; Perez-Polo, R.; Rossner, S. The transcription factor Yin Yang 1 is an activator of BACE1 expression. J. Neurochem. 2006, 96, 1696–1707. [Google Scholar] [CrossRef]
- Rossner, S.; Sastre, M.; Bourne, K.; Lichtenthaler, S.F. Transcriptional and translational regulation of BACE1 expression—implications for Alzheimer’s disease. Prog. Neurobiol. 2006, 79, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, D.K.; Ge, Y.W.; Rogers, J.T.; Sambamurti, K.; Greig, N.H.; Maloney, B. Taking down the unindicted co-conspirators of amyloid beta-peptide-mediated neuronal death: Shared gene regulation of BACE1 and APP genes interacting with CREB, Fe65 and YY1 transcription factors. Curr. Alzheimer Res. 2006, 3, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Lanke, V.; Moolamalla, S.T.R.; Roy, D.; Vinod, P.K. Integrative Analysis of Hippocampus Gene Expression Profiles Identifies Network Alterations in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 153. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.W.; Ghosh, C.; Song, W.; Maloney, B.; Lahiri, D.K. Mechanism of promoter activity of the beta-amyloid precursor protein gene in different cell lines: Identification of a specific 30 bp fragment in the proximal promoter region. J. Neurochem. 2004, 90, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Guglielmotto, M.; Medana, C.; Catalano, M.G.; Cutrupi, S.; Borghi, R.; Tamagno, E.; Boccuzzi, G.; Aragno, M. Dysregulation of SREBP2 induces BACE1 expression. Neurobiol. Dis. 2011, 44, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Barbero-Camps, E.; Fernandez, A.; Martinez, L.; Fernandez-Checa, J.C.; Colell, A. APP/PS1 mice overexpressing SREBP-2 exhibit combined Abeta accumulation and tau pathology underlying Alzheimer’s disease. Hum. Mol. Genet. 2013, 22, 3460–3476. [Google Scholar] [CrossRef]
- Picard, C.; Julien, C.; Frappier, J.; Miron, J.; Theroux, L.; Dea, D.; Breitner, J.C.S.; Poirier, J. Alterations in cholesterol metabolism-related genes in sporadic Alzheimer’s disease. Neurobiol. Aging 2018, 66, 180.e181–180.e189. [Google Scholar] [CrossRef]
- Zhang, J.; Shu, Y.; Qu, Y.; Zhang, L.; Chu, T.; Zheng, Y.; Zhao, H. C-myb Plays an Essential Role in the Protective Function of IGF-1 on Cytotoxicity Induced by Abeta25-35 via the PI3K/Akt Pathway. J. Mol. Neurosci. 2017, 63, 412–418. [Google Scholar] [CrossRef]
- Gonzalez, P.; Alvarez, V.; Menendez, M.; Lahoz, C.H.; Martinez, C.; Corao, A.I.; Calatayud, M.T.; Pena, J.; Garcia-Castro, M.; Coto, E. Myocyte enhancing factor-2A in Alzheimer’s disease: Genetic analysis and association with MEF2A-polymorphisms. Neurosci. Lett. 2007, 411, 47–51. [Google Scholar] [CrossRef]
- McKeever, P.M.; Schneider, R.; Taghdiri, F.; Weichert, A.; Multani, N.; Brown, R.A.; Boxer, A.L.; Karydas, A.; Miller, B.; Robertson, J.; et al. MicroRNA Expression Levels Are Altered in the Cerebrospinal Fluid of Patients with Young-Onset Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 8826–8841. [Google Scholar] [CrossRef]
- Herrera-Espejo, S.; Santos-Zorrozua, B.; Alvarez-Gonzalez, P.; Lopez-Lopez, E.; Garcia-Orad, A. A Systematic Review of MicroRNA Expression as Biomarker of Late-Onset Alzheimer’s Disease. Mol. Neurobiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Siedlecki-Wullich, D.; Catala-Solsona, J.; Fabregas, C.; Hernandez, I.; Clarimon, J.; Lleo, A.; Boada, M.; Saura, C.A.; Rodriguez-Alvarez, J.; Minano-Molina, A.J. Altered microRNAs related to synaptic function as potential plasma biomarkers for Alzheimer’s disease. Alzheimers Res. Ther. 2019, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Dursun, E.; Candas, E.; Yilmazer, S.; Gezen-Ak, D. Amyloid Beta 1-42 Alters the Expression of miRNAs in Cortical Neurons. J. Mol. Neurosci. 2019, 67, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, S.G.; Erdal, M.E.; Ozge, A.A.; Sungur, M.A. Can Peripheral MicroRNA Expression Data Serve as Epigenomic (Upstream) Biomarkers of Alzheimer’s Disease? Omics J. Integr. Biol. 2016, 20, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Kino, Y.; Niida, S. MicroRNA-Seq Data Analysis Pipeline to Identify Blood Biomarkers for Alzheimer’s Disease from Public Data. Biomark Insights 2015, 10, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Hebert, S.S.; Horre, K.; Nicolai, L.; Bergmans, B.; Papadopoulou, A.S.; Delacourte, A.; De Strooper, B. MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol. Dis. 2009, 33, 422–428. [Google Scholar] [CrossRef]
- Kume, K.; Iwama, H.; Deguchi, K.; Ikeda, K.; Takata, T.; Kokudo, Y.; Kamada, M.; Fujikawa, K.; Hirose, K.; Masugata, H.; et al. Serum microRNA expression profiling in patients with multiple system atrophy. Mol. Med. Rep. 2018, 17, 852–860. [Google Scholar] [CrossRef]
- Bofill-De Ros, X.; Santos, M.; Vila-Casadesus, M.; Villanueva, E.; Andreu, N.; Dierssen, M.; Fillat, C. Genome-wide miR-155 and miR-802 target gene identification in the hippocampus of Ts65Dn Down syndrome mouse model by miRNA sponges. BMC Genom. 2015, 16, 907. [Google Scholar] [CrossRef]
- Keck-Wherley, J.; Grover, D.; Bhattacharyya, S.; Xu, X.; Holman, D.; Lombardini, E.D.; Verma, R.; Biswas, R.; Galdzicki, Z. Abnormal microRNA expression in Ts65Dn hippocampus and whole blood: Contributions to Down syndrome phenotypes. Dev. Neurosci. 2011, 33, 451–467. [Google Scholar] [CrossRef]
- Nuovo, G.; Tili, E.; Awad, H.; Michaille, J.J. Roles of miR-155 microRNA in dementia associated with Down’s syndrome. Med. Sci. 2018, 34, 922–924. [Google Scholar] [CrossRef]
- Tili, E.; Mezache, L.; Michaille, J.J.; Amann, V.; Williams, J.; Vandiver, P.; Quinonez, M.; Fadda, P.; Mikhail, A.; Nuovo, G. microRNA 155 up regulation in the CNS is strongly correlated to Down’s syndrome dementia. Ann. Diagn. Pathol. 2018, 34, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhao, D.; Zhao, Y.; Wang, Y.; Wen, C. Inhibition of microRNA-155 Alleviates Cognitive Impairment in Alzheimer’s Disease and Involvement of Neuroinflammation. Curr. Alzheimer Res. 2019, 16, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lee, J.E. miR-155 is involved in Alzheimer’s disease by regulating T lymphocyte function. Front. Aging Neurosci. 2015, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Guedes, J.R.; Custodia, C.M.; Silva, R.J.; de Almeida, L.P.; Pedroso de Lima, M.C.; Cardoso, A.L. Early miR-155 upregulation contributes to neuroinflammation in Alzheimer’s disease triple transgenic mouse model. Hum. Mol. Genet. 2014, 23, 6286–6301. [Google Scholar] [CrossRef] [PubMed]
- Sierksma, A.; Lu, A.; Salta, E.; Vanden Eynden, E.; Callaerts-Vegh, Z.; D’Hooge, R.; Blum, D.; Buee, L.; Fiers, M.; De Strooper, B. Deregulation of neuronal miRNAs induced by amyloid-beta or TAU pathology. Mol. Neurodegener. 2018, 13, 54. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Qi, Y.; Liu, C.F.; Lu, J.M.; Shi, J.; Shi, Y. MicroRNA expression data analysis to identify key miRNAs associated with Alzheimer’s disease. J. Gene Med. 2018, 20, e3014. [Google Scholar] [CrossRef]
- Caggiu, E.; Paulus, K.; Mameli, G.; Arru, G.; Sechi, G.P.; Sechi, L.A. Differential expression of miRNA 155 and miRNA 146a in Parkinson’s disease patients. eNeurologicalSci 2018, 13, 1–4. [Google Scholar] [CrossRef]
- Cheng, X.R.; Cui, X.L.; Zheng, Y.; Zhang, G.R.; Li, P.; Huang, H.; Zhao, Y.Y.; Bo, X.C.; Wang, S.Q.; Zhou, W.X.; et al. Nodes and biological processes identified on the basis of network analysis in the brain of the senescence accelerated mice as an Alzheimer’s disease animal model. Front. Aging Neurosci. 2013, 5, 65. [Google Scholar] [CrossRef]
- Bennett, D.A.; Schneider, J.A.; Arvanitakis, Z.; Kelly, J.F.; Aggarwal, N.T.; Shah, R.C.; Wilson, R.S. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006, 66, 1837–1844. [Google Scholar] [CrossRef]
- Jendroska, K.; Poewe, W.; Daniel, S.E.; Pluess, J.; Iwerssen-Schmidt, H.; Paulsen, J.; Barthel, S.; Schelosky, L.; Cervos-Navarro, J.; DeArmond, S.J. Ischemic stress induces deposition of amyloid beta immunoreactivity in human brain. Acta Neuropathol. 1995, 90, 461–466. [Google Scholar] [CrossRef]
- Launer, L.J.; Ross, G.W.; Petrovitch, H.; Masaki, K.; Foley, D.; White, L.R.; Havlik, R.J. Midlife blood pressure and dementia: The Honolulu-Asia aging study. Neurobiol. Aging 2000, 21, 49–55. [Google Scholar] [CrossRef]
- Leibson, C.L.; Rocca, W.A.; Hanson, V.A.; Cha, R.; Kokmen, E.; O’Brien, P.C.; Palumbo, P.J. Risk of dementia among persons with diabetes mellitus: A population-based cohort study. Am. J. Epidemiol. 1997, 145, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Dagogo-Jack, S.E.; Wiethop, B.V.; Murphy, C.; Nevins, R.T.; Fleischman, S.; Rice, V.; Newcomer, J.W.; Cryer, P.E. Effects of hyperglycemia on memory and hormone levels in dementia of the Alzheimer type: A longitudinal study. Behav. Neurosci. 1993, 107, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Meneilly, G.S.; Hill, A. Alterations in glucose metabolism in patients with Alzheimer’s disease. J. Am. Geriatr. Soc. 1993, 41, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Stolk, R.P.; Hofman, A.; van Harskamp, F.; Grobbee, D.E.; Breteler, M.M. Association of diabetes mellitus and dementia: The Rotterdam Study. Diabetologia 1996, 39, 1392–1397. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.S.; Seshadri, S.; Beiser, A.; Zhang, Y.; Felson, D.; Hannan, M.T.; Au, R.; Wolf, P.A.; Kiel, D.P. Bone mineral density and the risk of Alzheimer disease. Arch. Neurol. 2005, 62, 107–111. [Google Scholar] [CrossRef]
- Kwok, M.K.; Lin, S.L.; Schooling, C.M. Re-thinking Alzheimer’s disease therapeutic targets using gene-based tests. EBioMedicine 2018, 37, 461–470. [Google Scholar] [CrossRef]
- Kupershmidt, I.; Su, Q.J.; Grewal, A.; Sundaresh, S.; Halperin, I.; Flynn, J.; Shekar, M.; Wang, H.; Park, J.; Cui, W.; et al. Ontology-based meta-analysis of global collections of high-throughput public data. PLoS ONE 2010, 5, e13066. [Google Scholar] [CrossRef]
- Lex, A.; Gehlenborg, N.; Strobelt, H.; Vuillemot, R.; Pfister, H. UpSet: Visualization of Intersecting Sets. IEEE Trans. Vis. Comput Graph. 2014, 20, 1983–1992. [Google Scholar] [CrossRef]
- Xia, J.; Gill, E.E.; Hancock, R.E. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823–844. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- ENCODE_Project_Consortium. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011, 9, e1001046. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, A.; Xu, H.; Krishnan, J.; Berger, S.I.; Mazloom, A.R.; Ma’ayan, A. ChEA: Transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 2010, 26, 2438–2444. [Google Scholar] [CrossRef]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Cheneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D260–D266. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef]
- Sethupathy, P.; Corda, B.; Hatzigeorgiou, A.G. TarBase: A comprehensive database of experimentally supported animal microRNA targets. RNA 2006, 12, 192–197. [Google Scholar] [CrossRef]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef]
- Pinero, J.; Bravo, A.; Queralt-Rosinach, N.; Gutierrez-Sacristan, A.; Deu-Pons, J.; Centeno, E.; Garcia-Garcia, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Datasets | Platform | Cases | Controls | References |
|---|---|---|---|---|---|
| MCI | GSE63063 (GSE63060, GSE63061) | Illumina GPL6947 | 80 | 104 | [43,44,45] |
| MCI | GSE63063 (GSE63060, GSE63061) | Illumina GPL10558 | 109 | 136 | [43,44,45] |
| ADD | GSE63063 (GSE63060, GSE63061) | Illumina GPL6947 | 142 | 104 | [43,44,45] |
| ADD | GSE63063 (GSE63060, GSE63061) | Illumina GPL10558 | 139 | 136 | [43,44,45] |
| ADD | E-MTAB-6094 | Agilent G4845A | 22 | 13 | Not published |
| AdvADD | GSE97760 | Agilent GPL16699 | 9 | 10 | [46] |
| Gene | miRNA | Degree | Betweenness |
|---|---|---|---|
| MCI | hsa-mir-16-5p | 15 | 29305 |
| hsa-mir-92a-3p | 11 | 13618 | |
| hsa-mir-26b-5p | 10 | 16008 | |
| hsa-mir-106b-5p | 7 | 8335 | |
| hsa-mir-93-5p | 7 | 7370 | |
| hsa-mir-20a-5p | 7 | 7297 | |
| hsa-mir-320a | 7 | 3580 | |
| hsa-let-7a-5p | 6 | 11229 | |
| hsa-mir-484 | 6 | 3618 | |
| hsa-mir-615-3p | 6 | 3342 | |
| hsa-mir-18a-3p | 5 | 8997 | |
| hsa-mir-7977 | 5 | 6593 | |
| hsa-mir-17-5p | 5 | 4484 | |
| hsa-mir-155-5p | 5 | 4269 | |
| hsa-mir-193b-3p | 5 | 3279 | |
| hsa-mir-450a-1-3p | 5 | 3266 | |
| hsa-mir-887-5p | 5 | 1840 | |
| ADD | hsa-mir-16-5p | 51 | 101307 |
| hsa-mir-26b-5p | 48 | 69947 | |
| hsa-mir-92a-3p | 33 | 53149 | |
| hsa-mir-193b-3p | 30 | 65562 | |
| hsa-mir-17-5p | 28 | 45790 | |
| hsa-mir-93-5p | 28 | 32553 | |
| hsa-mir-106b-5p | 28 | 32245 | |
| hsa-mir-20a-5p | 26 | 27726 | |
| hsa-mir-192-5p | 26 | 22623 | |
| hsa-mir-335-5p | 25 | 40587 | |
| hsa-mir-186-5p | 24 | 40049 | |
| hsa-mir-615-3p | 24 | 26450 | |
| hsa-let-7b-5p | 23 | 44950 | |
| hsa-mir-155-5p | 23 | 33976 | |
| hsa-mir-215-5p | 23 | 16736 | |
| hsa-mir-20b-5p | 21 | 16266 | |
| hsa-mir-519d-3p | 21 | 16266 | |
| hsa-let-7a-5p | 20 | 22235 |
| Gene | Disease Associated | Degree | Betweenness |
|---|---|---|---|
| MCI | Autosomal recessive predisposition | 5 | 1795 |
| Failure to gain weight | 3 | 306 | |
| Pediatric failure to thrive | 3 | 306 | |
| Muscle degeneration | 3 | 296 | |
| Neurogenic muscular atrophy | 3 | 296 | |
| Skeletal muscle atrophy | 3 | 296 | |
| Neurogenic muscle atrophy, especially in the lower limbs | 3 | 296 | |
| Cerebellar ataxia | 3 | 161 | |
| Muscle hypotonia | 3 | 161 | |
| Hyperreflexia | 3 | 161 | |
| Global developmental delay | 3 | 161 | |
| Cognitive delay | 3 | 161 | |
| Mental and motor retardation | 3 | 161 | |
| Hypertrophic cardiomyopathy | 3 | 123 | |
| ADD | Autosomal recessive predisposition | 11 | 25695 |
| Liver cirrhosis experimental | 9 | 7088 | |
| Schizophrenia | 8 | 11805 | |
| Strabismus | 7 | 16263 | |
| Failure to gain weight | 6 | 6147 | |
| Pediatric failure to thrive | 6 | 6147 | |
| Hyperreflexia | 6 | 4072 | |
| Muscle hypotonia | 6 | 3821 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bottero, V.; Potashkin, J.A. Meta-Analysis of Gene Expression Changes in the Blood of Patients with Mild Cognitive Impairment and Alzheimer’s Disease Dementia. Int. J. Mol. Sci. 2019, 20, 5403. https://doi.org/10.3390/ijms20215403
Bottero V, Potashkin JA. Meta-Analysis of Gene Expression Changes in the Blood of Patients with Mild Cognitive Impairment and Alzheimer’s Disease Dementia. International Journal of Molecular Sciences. 2019; 20(21):5403. https://doi.org/10.3390/ijms20215403
Chicago/Turabian StyleBottero, Virginie, and Judith A. Potashkin. 2019. "Meta-Analysis of Gene Expression Changes in the Blood of Patients with Mild Cognitive Impairment and Alzheimer’s Disease Dementia" International Journal of Molecular Sciences 20, no. 21: 5403. https://doi.org/10.3390/ijms20215403
APA StyleBottero, V., & Potashkin, J. A. (2019). Meta-Analysis of Gene Expression Changes in the Blood of Patients with Mild Cognitive Impairment and Alzheimer’s Disease Dementia. International Journal of Molecular Sciences, 20(21), 5403. https://doi.org/10.3390/ijms20215403