A Promising Future for Stem-Cell-Based Therapies in Muscular Dystrophies—In Vitro and In Vivo Treatments to Boost Cellular Engraftment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Muscular Dystrophies
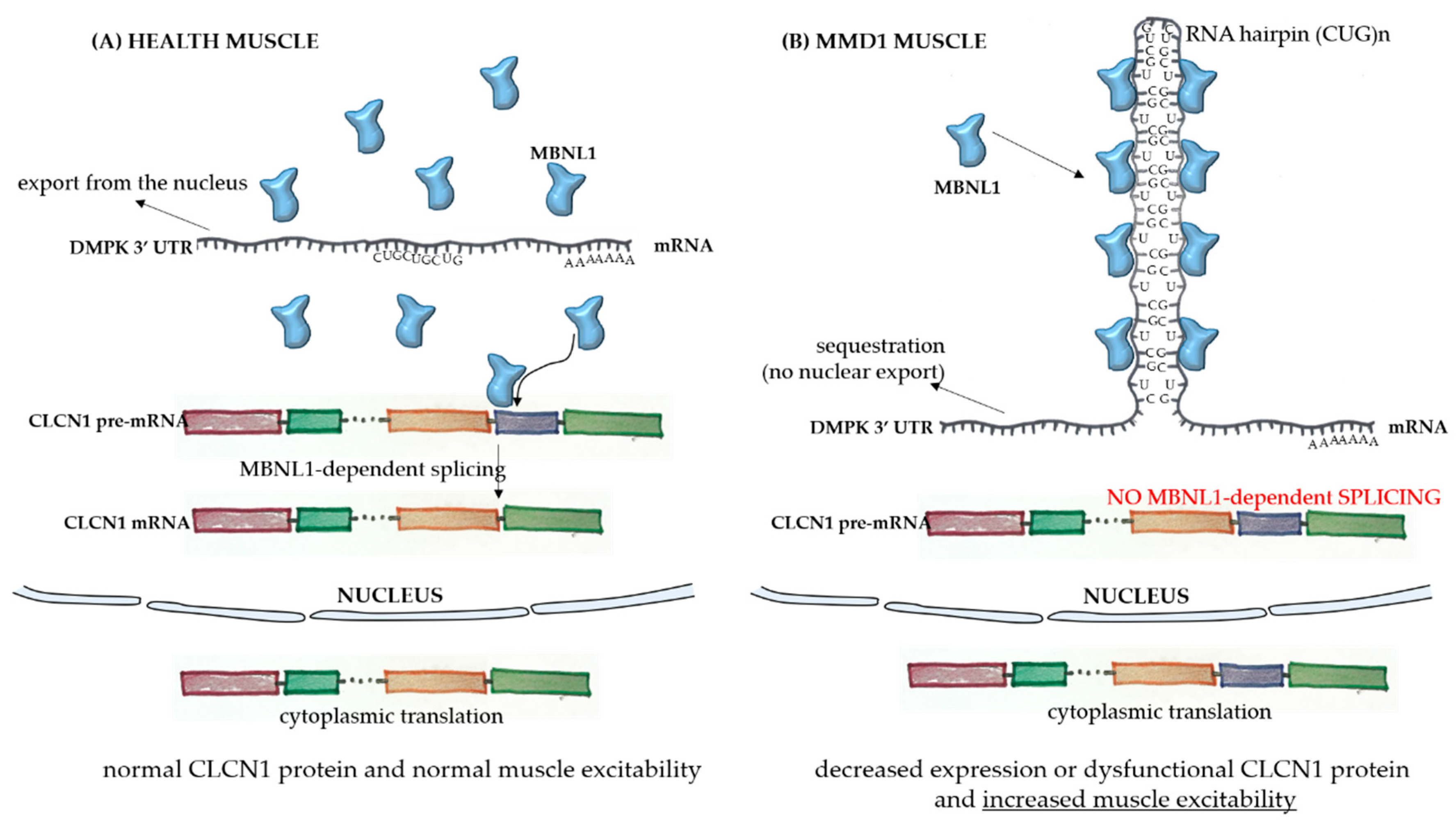
1.1. Myotonic Muscular Dystrophy (MMD)
1.2. Oculopharyngeal Muscular Dystrophy (OPMD)
1.3. Emery–Dreifuss Muscular Dystrophy (EDMD)
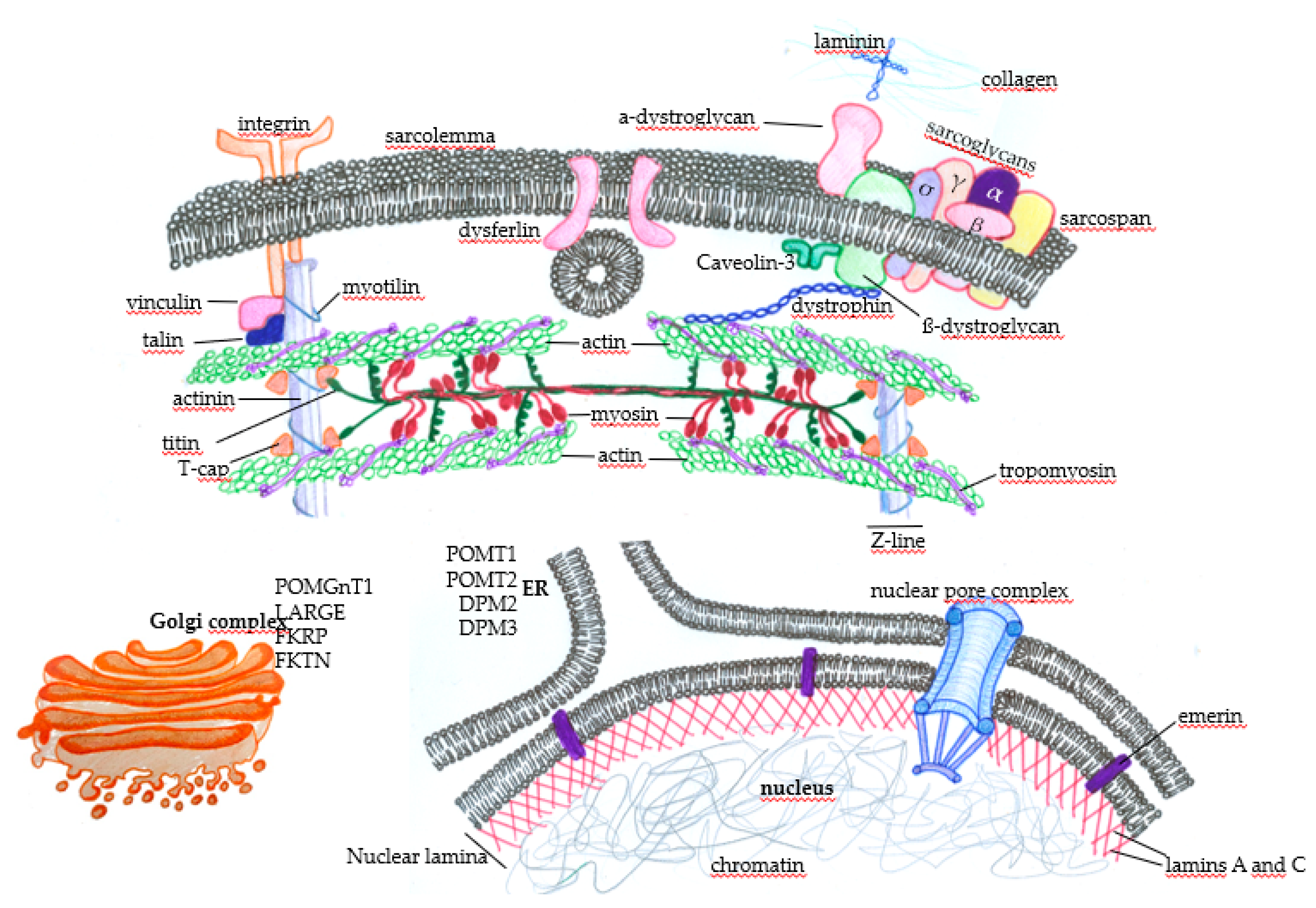
1.4. Limb-Girdle Muscular Dystrophy (LGMD)
1.5. Facioscapulohumeral Muscular Dystrophy (FSHD)
1.6. Congenital Muscular Dystrophy (CMD) and Distal Muscular Dystrophy
1.7. Duchenne Muscular Dystrophy (DMD) and Becker Muscular Dystrophy (BMD)
2. General Concepts for Stem-Cell-Based Thearapies
3. Progenitor Cells Found in Skeletal Muscles and Stem Cell Thearapies
4. Satellite Cells, the Main Stem Cell Target for Muscle Therapy
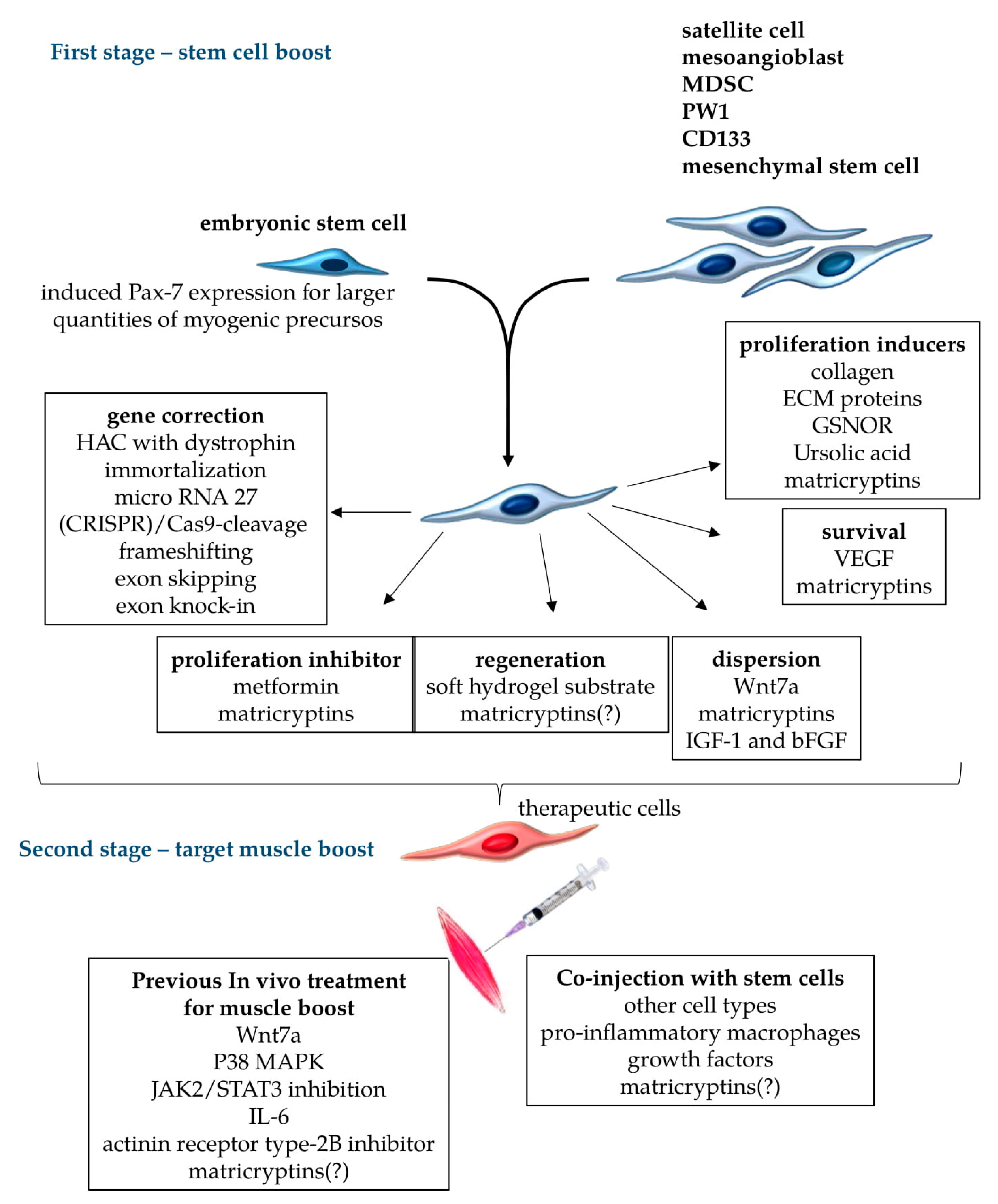
5. Proposals for Improvement of Stem-Cell-Based Therapies
5.1. Stem Cell Treatments
5.2. Pecipient Muscle Treatments
6. Matricryptins as Possible New Players in Stem Cell Therapy
7. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Theadom, A.; Rodrigues, M.; Roxburgh, R.; Balalla, S.; Higgins, C.; Bhattacharjee, R.; Jones, K.; Krishnamurthi, R.; Feigin, V. Prevalence of muscular dystrophies: A systematic literature review. Neuroepidemiology 2014, 43, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Udd, B.; Krahe, R. The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012, 11, 891–905. [Google Scholar] [CrossRef]
- Garrott, H.M.; Walland, M.J.; O’Day, J. Recurrent posterior capsular opacification and capsulorhexis contracture after cataract surgery in myotonic dystrophy. Clin. Exp. Ophthalmol. 2004, 32, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Winblad, S.; Lindberg, C.; Hansen, S. Temperament and character in patients with classical myotonic dystrophy type 1 (DM-1). Neuromuscul. Disord. 2005, 15, 287–292. [Google Scholar] [CrossRef]
- Rönnblom, A.; Forsberg, H.; Danielsson, A. Gastrointestinal symptoms in myotonic dystrophy. Scand. J. Gastroenterol. 1996, 31, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, K.; Ariyasu, H.; Ishibashi, T.; Kawai, S.; Uraki, S.; Koh, J.; Ito, H.; Akamizu, T. Myotonic dystrophy type 1 with diabetes mellitus, mixed hypogonadism and adrenal insufficiency. Endocrinol. Diabetes. Metab. Case Rep. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Ashizawa, T.; Sarkar, P.S. Myotonic dystrophy types 1 and 2. Handb. Clin. Neurol. 2011, 101, 193–237. [Google Scholar] [CrossRef]
- Angeard, N.; Huerta, E.; Jacquette, A.; Cohen, D.; Xavier, J.; Gargiulo, M.; Servais, L.; Eymard, B.; Héron, D. Childhood-onset form of myotonic dystrophy type 1 and autism spectrum disorder: Is there comorbidity? Neuromuscul. Disord. 2018, 28, 216–221. [Google Scholar] [CrossRef]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell 1992, 69, 385. [Google Scholar] [CrossRef]
- López-Morató, M.; Brook, J.D.; Wojciechowska, M. Small Molecules Which Improve Pathogenesis of Myotonic Dystrophy Type 1. Front. Neurol. 2018, 9, 349. [Google Scholar] [CrossRef]
- LoRusso, S.; Weiner, B.; Arnold, W.D. Myotonic Dystrophies: Targeting Therapies for Multisystem Disease. Neurotherapeutics 2018, 15, 872–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, J.W.; Ricker, K.; Jacobsen, J.F.; Rasmussen, L.J.; Dick, K.A.; Kress, W.; Schneider, C.; Koch, M.C.; Beilman, G.J.; Harrison, A.R.; et al. Myotonic dystrophy type 2: Molecular, diagnostic and clinical spectrum. Neurology 2003, 60, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Schoser, B.; Timchenko, L. Myotonic dystrophies 1 and 2: Complex diseases with complex mechanisms. Curr. Genomics. 2010, 11, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Ranum, L.P.; Rasmussen, P.F.; Benzow, K.A.; Koob, M.D.; Day, J.W. Genetic mapping of a second myotonic dystrophy locus. Nat. Genet. 1998, 19, 196–198. [Google Scholar] [CrossRef]
- Tomé, F.M.; Fardeau, M. Nuclear inclusions in oculopharyngeal dystrophy. Acta Neuropathol. 1980, 49, 85–87. [Google Scholar] [CrossRef]
- Brais, B.; Bouchard, J.P.; Xie, Y.G.; Rochefort, D.L.; Chrétien, N.; Tomé, F.M.; Lafrenière, R.G.; Rommens, J.M.; Uyama, E.; Nohira, O.; et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet. 1998, 18, 164–167. [Google Scholar] [CrossRef]
- Kerwitz, Y.; Kühn, U.; Lilie, H.; Knoth, A.; Scheuermann, T.; Friedrich, H.; Schwarz, E.; Wahle, E. Stimulation of poly(A) polymerase through a direct interaction with the nuclear poly(A) binding protein allosterically regulated by RNA. EMBO J. 2003, 22, 3705–3714. [Google Scholar] [CrossRef] [Green Version]
- Benoit, B.; Mitou, G.; Chartier, A.; Temme, C.; Zaessinger, S.; Wahle, E.; Busseau, I.; Simonelig, M. An essential cytoplasmic function for the nuclear poly(A) binding protein, PABP2, in poly(A) tail length control and early development in Drosophila. Dev. Cell 2005, 9, 511–522. [Google Scholar] [CrossRef]
- Bresson, S.M.; Conrad, N.K. The human nuclear poly(a)-binding protein promotes RNA hyperadenylation and decay. PLoS Genet. 2013, 9, e1003893. [Google Scholar] [CrossRef]
- Malerba, A.; Klein, P.; Bachtarzi, H.; Jarmin, S.A.; Cordova, G.; Ferry, A.; Strings, V.; Espinoza, M.P.; Mamchaoui, K.; Blumen, S.C.; et al. PABPN1 gene therapy for oculopharyngeal muscular dystrophy. Nat. Commun. 2017, 8, 14848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, J.E.; Berger, Z.; Rubinsztein, D.C. Oculopharyngeal muscular dystrophy: Potential therapies for an aggregate-associated disorder. Int. J. Biochem. Cell Biol. 2006, 38, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Barbezier, N.; Chartier, A.; Bidet, Y.; Buttstedt, A.; Voisset, C.; Galons, H.; Blondel, M.; Schwarz, E.; Simonelig, M. Antiprion drugs 6-aminophenanthridine and guanabenz reduce PABPN1 toxicity and aggregation in oculopharyngeal muscular dystrophy. EMBO Mol. Med. 2011, 3, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E.; Dreifuss, F.E. Unusual type of benign x-linked muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 1966, 29, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, A.S.; Wagener, M.E.; Warren, S.T.; Hopkins, L.C. Emery-Dreifuss muscular dystrophy. Semin Neurol 1999, 19, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Helbling-Leclerc, A.; Bonne, G.; Schwartz, K. Emery-Dreifuss muscular dystrophy. Eur. J. Hum. Genet. 2002, 10, 157–161. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Muntoni, F. Muscular dystrophies. Lancet 2013, 381, 845–860. [Google Scholar] [CrossRef]
- Madej-Pilarczyk, A.; Kochański, A. Emery-Dreifuss muscular dystrophy: The most recognizable laminopathy. Folia Neuropathol. 2016, 54, 1–8. [Google Scholar] [CrossRef]
- Bione, S.; Maestrini, E.; Rivella, S.; Mancini, M.; Regis, S.; Romeo, G.; Toniolo, D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat. Genet. 1994, 8, 323–327. [Google Scholar] [CrossRef]
- Manilal, S.; Recan, D.; Sewry, C.A.; Hoeltzenbein, M.; Llense, S.; Leturcq, F.; Deburgrave, N.; Barbot, J.; Man, N.; Muntoni, F.; et al. Mutations in Emery-Dreifuss muscular dystrophy and their effects on emerin protein expression. Hum. Mol. Genet. 1998, 7, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtizberea, A.; Bécane, H.M.; Recan, D.; Merlini, L.; Wehnert, M.; Boor, R.; Reuner, U.; et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 2000, 48, 170–180. [Google Scholar] [CrossRef]
- Angori, S.; Capanni, C.; Faulkner, G.; Bean, C.; Boriani, G.; Lattanzi, G.; Cenni, V. Emery-Dreifuss Muscular Dystrophy-Associated Mutant Forms of Lamin A Recruit the Stress Responsive Protein Ankrd2 into the Nucleus, Affecting the Cellular Response to Oxidative Stress. Cell Physiol. Biochem. 2017, 42, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, S.; Menditto, I.; Degano, M.; Rodolico, C.; Merlini, L.; D’Amico, A.; Palmucci, L.; Berardinelli, A.; Pegoraro, E.; Trevisan, C.P.; et al. Phenotypic clustering of lamin A/C mutations in neuromuscular patients. Neurology 2007, 69, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bethmann, C.; Worth, N.F.; Davies, J.D.; Wasner, C.; Feuer, A.; Ragnauth, C.D.; Yi, Q.; Mellad, J.A.; Warren, D.T.; et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum. Mol. Genet. 2007, 16, 2816–2833. [Google Scholar] [CrossRef]
- Gesson, K.; Vidak, S.; Foisner, R. Lamina-associated polypeptide (LAP)2α and nucleoplasmic lamins in adult stem cell regulation and disease. Semin. Cell Dev. Biol. 2014, 29, 116–124. [Google Scholar] [CrossRef]
- Gueneau, L.; Bertrand, A.T.; Jais, J.P.; Salih, M.A.; Stojkovic, T.; Wehnert, M.; Hoeltzenbein, M.; Spuler, S.; Saitoh, S.; Verschueren, A.; et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 2009, 85, 338–353. [Google Scholar] [CrossRef]
- Wahbi, K.; Meune, C.; Hamouda, E.H.; Stojkovic, T.; Laforêt, P.; Bécane, H.M.; Eymard, B.; Duboc, D. Cardiac assessment of limb-girdle muscular dystrophy 2I patients: An echography, Holter ECG and magnetic resonance imaging study. Neuromuscul. Disord. 2008, 18, 650–655. [Google Scholar] [CrossRef]
- Bönnemann, C.G.; Wang, C.H.; Quijano-Roy, S.; Deconinck, N.; Bertini, E.; Ferreiro, A.; Muntoni, F.; Sewry, C.; Béroud, C.; Mathews, K.D.; et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul. Disord. 2014, 24, 289–311. [Google Scholar] [CrossRef]
- Vissing, J. Limb girdle muscular dystrophies: Classification, clinical spectrum and emerging therapies. Curr. Opin. Neurol. 2016, 29, 635–641. [Google Scholar] [CrossRef]
- Guglieri, M.; Magri, F.; D’Angelo, M.G.; Prelle, A.; Morandi, L.; Rodolico, C.; Cagliani, R.; Mora, M.; Fortunato, F.; Bordoni, A.; et al. Clinical, molecular, and protein correlations in a large sample of genetically diagnosed Italian limb girdle muscular dystrophy patients. Hum. Mutat. 2008, 29, 258–266. [Google Scholar] [CrossRef]
- Liewluck, T.; Milone, M. Untangling the complexity of limb-girdle muscular dystrophies. Muscle Nerve. 2018, 58, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, E.; Rezaee, M.; Barreto, G.E.; Sahebkar, A. Prevalence, pathological mechanisms, and genetic basis of limb-girdle muscular dystrophies: A review. J. Cell Physiol. 2019, 234, 7874–7884. [Google Scholar] [CrossRef] [PubMed]
- Padberg, G.W.; Lunt, P.W.; Koch, M.; Fardeau, M. Diagnostic criteria for facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 1991, 1, 231–234. [Google Scholar] [CrossRef]
- Statland, J.M.; Tawil, R. Facioscapulohumeral Muscular Dystrophy. Continuum. Minneap. Minn. 2016, 22, 1916–1931. [Google Scholar] [CrossRef] [Green Version]
- van Deutekom, J.C.; Wijmenga, C.; van Tienhoven, E.A.; Gruter, A.M.; Hewitt, J.E.; Padberg, G.W.; van Ommen, G.J.; Hofker, M.H.; Frants, R.R. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 1993, 2, 2037–2042. [Google Scholar] [CrossRef]
- Hamel, J.; Tawil, R. Facioscapulohumeral Muscular Dystrophy: Update on Pathogenesis and Future Treatments. Neurotherapeutics 2018, 15, 863–871. [Google Scholar] [CrossRef] [Green Version]
- Lemmers, R.J.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.; Amell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012, 44, 1370–1374. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.N.; Xiong, H. Genetic and Clinical Advances of Congenital Muscular Dystrophy. Chin. Med. J. Engl. 2017, 130, 2624–2631. [Google Scholar] [CrossRef]
- Mercuri, E.; Muntoni, F. The ever-expanding spectrum of congenital muscular dystrophies. Ann. Neurol. 2012, 72, 9–17. [Google Scholar] [CrossRef]
- Falsaperla, R.; Praticò, A.D.; Ruggieri, M.; Parano, E.; Rizzo, R.; Corsello, G.; Vitaliti, G.; Pavone, P. Congenital muscular dystrophy: From muscle to brain. Ital. J. Pediatr. 2016, 42, 78. [Google Scholar] [CrossRef]
- Kang, P.B.; Morrison, L.; Iannaccone, S.T.; Graham, R.J.; Bönnemann, C.G.; Rutkowski, A.; Hornyak, J.; Wang, C.H.; North, K.; Oskoui, M.; et al. Evidence-based guideline summary: Evaluation, diagnosis, and management of congenital muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology 2015, 84, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Pénisson-Besnier, I. Distal myopathies. Rev. Neurol. Paris 2013, 169, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.; Liewluck, T. The unfolding spectrum of inherited distal myopathies. Muscle Ner. 2019, 59, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef]
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; de Kock, S.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Phys. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef]
- Yiu, E.M.; Kornberg, A.J. Duchenne muscular dystrophy. J. Paediatr. Child. Health 2015, 51, 759–764. [Google Scholar] [CrossRef]
- Nakamura, A. Mutation-Based Therapeutic Strategies for Duchenne Muscular Dystrophy: From Genetic Diagnosis to Therapy. J. Pers. Med. 2019, 9. [Google Scholar] [CrossRef]
- Okada, T.; Takeda, S. Current Challenges and Future Directions in Recombinant AAV-Mediated Gene Therapy of Duchenne Muscular Dystrophy. Pharm. Basel 2013, 6, 813–836. [Google Scholar] [CrossRef] [Green Version]
- Fairclough, R.J.; Wood, M.J.; Davies, K.E. Therapy for Duchenne muscular dystrophy: Renewed optimism from genetic approaches. Nat. Rev. Genet. 2013, 14, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Mah, J.K. Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr. Dis. Treat. 2016, 12, 1795–1807. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M. Duchenne and Becker muscular dystrophies. Neurol Clin. 2014, 32, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M. The muscular dystrophies. Semin Neurol 2012, 32, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Mao, A.S.; Mooney, D.J. Regenerative medicine: Current therapies and future directions. Proc. Natl. Acad. Sci. USA 2015, 112, 14452–14459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrovidov, S.; Shi, X.; Sadeghian, R.B.; Salehi, S.; Fujie, T.; Bae, H.; Ramalingam, M.; Khademhosseini, A. Stem Cell Differentiation Toward the Myogenic Lineage for Muscle Tissue Regeneration: A Focus on Muscular Dystrophy. Stem. Cell Rev. 2015, 11, 866–884. [Google Scholar] [CrossRef]
- Sampaolesi, M.; Biressi, S.; Tonlorenzi, R.; Innocenzi, A.; Draghici, E.; Cusella de Angelis, M.G.; Cossu, G. Cell therapy of primary myopathies. Arch. Ital. De Biol. 2005, 143, 235–242. [Google Scholar]
- Negroni, E.; Bigot, A.; Butler-Browne, G.S.; Trollet, C.; Mouly, V. Cellular Therapies for Muscular Dystrophies: Frustrations and Clinical Successes. Hum. Gene Ther. 2016, 27, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Skuk, D.; Caron, N.J.; Goulet, M.; Roy, B.; Tremblay, J.P. Resetting the problem of cell death following muscle-derived cell transplantation: Detection, dynamics and mechanisms. J. Neuropathol. Exp. Neurol. 2003, 62, 951–967. [Google Scholar] [CrossRef]
- Ho, P.P.; Lahey, L.J.; Mourkioti, F.; Kraft, P.E.; Filareto, A.; Brandt, M.; Magnusson, K.E.G.; Finn, E.E.; Chamberlain, J.S.; Robinson, W.H.; et al. Engineered DNA plasmid reduces immunity to dystrophin while improving muscle force in a model of gene therapy of Duchenne dystrophy. Proc. Natl. Acad. Sci. USA 2018, 115, E9182–E9191. [Google Scholar] [CrossRef] [Green Version]
- Danisovic, L.; Culenova, M.; Csobonyeiova, M. Induced Pluripotent Stem Cells for Duchenne Muscular Dystrophy Modeling and Therapy. Cells 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Mouly, V.; Aamiri, A.; Perie, S.; Mamchaoui, K.; Barani, A.; Bigot, A.; Bouazza, B.; Francois, V.; Furling, D.; Jacquemin, V.; et al. Myoblast transfer therapy: Is there any light at the end of the tunnel? Acta Myol. Myopathies Cardiomyopathies J. Mediterr. Soc. Myol. 2005, 24, 128–133. [Google Scholar]
- Negroni, E.; Butler-Browne, G.S.; Mouly, V. Myogenic stem cells: Regeneration and cell therapy in human skeletal muscle. Pathol. Biol. 2006, 54, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.M.; Harting, M.T.; Jimenez, F.; Monzon-Posadas, W.O.; Xue, H.; Savitz, S.I.; Laine, G.A.; Cox, C.S., Jr. Pulmonary passage is a major obstacle for intravenous stem cell delivery: The pulmonary first-pass effect. Stem Cells Dev. 2009, 18, 683–692. [Google Scholar] [CrossRef]
- Ortiz-Vitali, J.L.; Darabi, R. iPSCs as a Platform for Disease Modeling, Drug Screening, and Personalized Therapy in Muscular Dystrophies. Cells 2019, 8. [Google Scholar] [CrossRef]
- Grounds, M.D.; Garrett, K.L.; Lai, M.C.; Wright, W.E.; Beilharz, M.W. Identification of skeletal muscle precursor cells in vivo by use of MyoD1 and myogenin probes. Cell Tissue Res. 1992, 267, 99–104. [Google Scholar] [CrossRef]
- Meregalli, M.; Farini, A.; Belicchi, M.; Parolini, D.; Cassinelli, L.; Razini, P.; Sitzia, C.; Torrente, Y. Perspectives of stem cell therapy in Duchenne muscular dystrophy. FEBS J. 2013, 280, 4251–4262. [Google Scholar] [CrossRef]
- Tamaki, T.; Okada, Y.; Uchiyama, Y.; Tono, K.; Masuda, M.; Wada, M.; Hoshi, A.; Ishikawa, T.; Akatsuka, A. Clonal multipotency of skeletal muscle-derived stem cells between mesodermal and ectodermal lineage. Stem Cells Dayton. Ohio. 2007, 25, 2283–2290. [Google Scholar] [CrossRef]
- Cossu, G.; Bianco, P. Mesoangioblasts--vascular progenitors for extravascular mesodermal tissues. Curr. Opin. Genet. Dev. 2003, 13, 537–542. [Google Scholar] [CrossRef]
- Benchaouir, R.; Meregalli, M.; Farini, A.; D’Antona, G.; Belicchi, M.; Goyenvalle, A.; Battistelli, M.; Bresolin, N.; Bottinelli, R.; Garcia, L.; et al. Restoration of human dystrophin following transplantation of exon-skipping-engineered DMD patient stem cells into dystrophic mice. Cell Stem Cell 2007, 1, 646–657. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.J.; Pannerec, A.; Cadot, B.; Parlakian, A.; Besson, V.; Gomes, E.R.; Marazzi, G.; Sassoon, D.A. Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat. Cell Biol. 2010, 12, 257–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouger, K.; Larcher, T.; Dubreil, L.; Deschamps, J.Y.; Le Guiner, C.; Jouvion, G.; Delorme, B.; Lieubeau, B.; Carlus, M.; Fornasari, B.; et al. Systemic delivery of allogenic muscle stem cells induces long-term muscle repair and clinical efficacy in duchenne muscular dystrophy dogs. Am. J. Pathol. 2011, 179, 2501–2518. [Google Scholar] [CrossRef] [PubMed]
- Minasi, M.G.; Riminucci, M.; De Angelis, L.; Borello, U.; Berarducci, B.; Innocenzi, A.; Caprioli, A.; Sirabella, D.; Baiocchi, M.; De Maria, R.; et al. The meso-angioblast: A multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Dev. Camb. Engl. 2002, 129, 2773–2783. [Google Scholar]
- Sampaolesi, M.; Blot, S.; D’Antona, G.; Granger, N.; Tonlorenzi, R.; Innocenzi, A.; Mognol, P.; Thibaud, J.L.; Galvez, B.G.; Barthelemy, I.; et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 2006, 444, 574–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedesco, F.S.; Gerli, M.F.; Perani, L.; Benedetti, S.; Ungaro, F.; Cassano, M.; Antonini, S.; Tagliafico, E.; Artusi, V.; Longa, E.; et al. Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci. Trans. Med. 2012, 4, 140ra189. [Google Scholar] [CrossRef]
- Tedesco, F.S.; Cossu, G. Stem cell therapies for muscle disorders. Curr. Opin. Neurol. 2012, 25, 597–603. [Google Scholar] [CrossRef]
- Miraglia, S.; Godfrey, W.; Yin, A.H.; Atkins, K.; Warnke, R.; Holden, J.T.; Bray, R.A.; Waller, E.K.; Buck, D.W. A novel five-transmembrane hematopoietic stem cell antigen: Isolation, characterization, and molecular cloning. Blood 1997, 90, 5013–5021. [Google Scholar] [CrossRef]
- Torrente, Y.; Belicchi, M.; Sampaolesi, M.; Pisati, F.; Meregalli, M.; D’Antona, G.; Tonlorenzi, R.; Porretti, L.; Gavina, M.; Mamchaoui, K.; et al. Human circulating AC133(+) stem cells restore dystrophin expression and ameliorate function in dystrophic skeletal muscle. J. Clin. Investig. 2004, 114, 182–195. [Google Scholar] [CrossRef]
- Uchida, N.; Buck, D.W.; He, D.; Reitsma, M.J.; Masek, M.; Phan, T.V.; Tsukamoto, A.S.; Gage, F.H.; Weissman, I.L. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14720–14725. [Google Scholar] [CrossRef] [Green Version]
- Negroni, E.; Riederer, I.; Chaouch, S.; Belicchi, M.; Razini, P.; Di Santo, J.; Torrente, Y.; Butler-Browne, G.S.; Mouly, V. In vivo myogenic potential of human CD133+ muscle-derived stem cells: A quantitative study. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- Gavina, M.; Belicchi, M.; Rossi, B.; Ottoboni, L.; Colombo, F.; Meregalli, M.; Battistelli, M.; Forzenigo, L.; Biondetti, P.; Pisati, F.; et al. VCAM-1 expression on dystrophic muscle vessels has a critical role in the recruitment of human blood-derived CD133+ stem cells after intra-arterial transplantation. Blood 2006, 108, 2857–2866. [Google Scholar] [CrossRef] [Green Version]
- Jackson, W.M.; Nesti, L.J.; Tuan, R.S. Potential therapeutic applications of muscle-derived mesenchymal stem and progenitor cells. Expert Opin. Biol. Ther. 2010, 10, 505–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gang, E.J.; Jeong, J.A.; Hong, S.H.; Hwang, S.H.; Kim, S.W.; Yang, I.H.; Ahn, C.; Han, H.; Kim, H. Skeletal myogenic differentiation of mesenchymal stem cells isolated from human umbilical cord blood. Stem Cells Dayt. Ohio 2004, 22, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, K.; Leelahavanichkul, A.; Yuen, P.S.; Mayer, B.; Parmelee, A.; Doi, K.; Robey, P.G.; Leelahavanichkul, K.; Koller, B.H.; Brown, J.M.; et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 2009, 15, 42–49. [Google Scholar] [CrossRef]
- Ichim, T.E.; Alexandrescu, D.T.; Solano, F.; Lara, F.; Campion Rde, N.; Paris, E.; Woods, E.J.; Murphy, M.P.; Dasanu, C.A.; Patel, A.N.; et al. Mesenchymal stem cells as anti-inflammatories: Implications for treatment of Duchenne muscular dystrophy. Cell. Immunol. 2010, 260, 75–82. [Google Scholar] [CrossRef]
- De Bari, C.; Dell’Accio, F.; Vandenabeele, F.; Vermeesch, J.R.; Raymackers, J.M.; Luyten, F.P. Skeletal muscle repair by adult human mesenchymal stem cells from synovial membrane. J. Cell Biol. 2003, 160, 909–918. [Google Scholar] [CrossRef] [Green Version]
- Montarras, D.; L’Honore, A.; Buckingham, M. Lying low but ready for action: The quiescent muscle satellite cell. FEBS J. 2013, 280, 4036–4050. [Google Scholar] [CrossRef]
- Shi, X.; Garry, D.J. Muscle stem cells in development, regeneration, and disease. Genes Dev. 2006, 20, 1692–1708. [Google Scholar] [CrossRef]
- Seale, P.; Sabourin, L.A.; Girgis-Gabardo, A.; Mansouri, A.; Gruss, P.; Rudnicki, M.A. Pax7 is required for the specification of myogenic satellite cells. Cell 2000, 102, 777–786. [Google Scholar] [CrossRef]
- Kuang, S.; Charge, S.B.; Seale, P.; Huh, M.; Rudnicki, M.A. Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J. Cell Biol. 2006, 172, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Relaix, F.; Montarras, D.; Zaffran, S.; Gayraud-Morel, B.; Rocancourt, D.; Tajbakhsh, S.; Mansouri, A.; Cumano, A.; Buckingham, M. Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J. Cell Biol 2006, 172, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Sabourin, L.A.; Rudnicki, M.A. The molecular regulation of myogenesis. Clin. Genet. 2000, 57, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.; Doyonnas, R.; Kraft, P.; Vitorovic, S.; Blau, H.M. Self-renewal and expansion of single transplanted muscle stem cells. Nature 2008, 456, 502–506. [Google Scholar] [CrossRef] [Green Version]
- Irintchev, A.; Zeschnigk, M.; Starzinski-Powitz, A.; Wernig, A. Expression pattern of M-cadherin in normal, denervated, and regenerating mouse muscles. Dev. Dynam. Off. Publ. Am. Assoc. Anatom. 1994, 199, 326–337. [Google Scholar] [CrossRef]
- Beauchamp, J.R.; Heslop, L.; Yu, D.S.; Tajbakhsh, S.; Kelly, R.G.; Wernig, A.; Buckingham, M.E.; Partridge, T.A.; Zammit, P.S. Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. J. Cell Biol. 2000, 151, 1221–1234. [Google Scholar] [CrossRef]
- Gnocchi, V.F.; White, R.B.; Ono, Y.; Ellis, J.A.; Zammit, P.S. Further characterisation of the molecular signature of quiescent and activated mouse muscle satellite cells. PLoS ONE 2009, 4, e5205. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Majka, M.; Kucia, M.; Drukala, J.; Pietrzkowski, Z.; Peiper, S.; Janowska-Wieczorek, A. Expression of functional CXCR4 by muscle satellite cells and secretion of SDF-1 by muscle-derived fibroblasts is associated with the presence of both muscle progenitors in bone marrow and hematopoietic stem/progenitor cells in muscles. Stem Cells Dayt. Ohio. 2003, 21, 363–371. [Google Scholar] [CrossRef]
- Meech, R.; Gonzalez, K.N.; Barro, M.; Gromova, A.; Zhuang, L.; Hulin, J.A.; Makarenkova, H.P. Barx2 is expressed in satellite cells and is required for normal muscle growth and regeneration. Stem Cells Dayt. Ohio. 2012, 30, 253–265. [Google Scholar] [CrossRef]
- Collins, C.A.; Olsen, I.; Zammit, P.S.; Heslop, L.; Petrie, A.; Partridge, T.A.; Morgan, J.E. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 2005, 122, 289–301. [Google Scholar] [CrossRef]
- Tremblay, J.P.; Malouin, F.; Roy, R.; Huard, J.; Bouchard, J.P.; Satoh, A.; Richards, C.L. Results of a triple blind clinical study of myoblast transplantations without immunosuppressive treatment in young boys with Duchenne muscular dystrophy. Cell Trans. 1993, 2, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Kissel, J.T.; Amato, A.A.; King, W.; Signore, L.; Prior, T.W.; Sahenk, Z.; Benson, S.; McAndrew, P.E.; Rice, R.; et al. Myoblast transfer in the treatment of Duchenne’s muscular dystrophy. N. Engl. J. Med. 1995, 333, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Gussoni, E.; Blau, H.M.; Kunkel, L.M. The fate of individual myoblasts after transplantation into muscles of DMD patients. Nat. Med. 1997, 3, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Partridge, T.A.; Morgan, J.E.; Coulton, G.R.; Hoffman, E.P.; Kunkel, L.M. Conversion of mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature 1989, 337, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Cerletti, M.; Jurga, S.; Witczak, C.A.; Hirshman, M.F.; Shadrach, J.L.; Goodyear, L.J.; Wagers, A.J. Highly efficient, functional engraftment of skeletal muscle stem cells in dystrophic muscles. Cell 2008, 134, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, F.S.; Dellavalle, A.; Diaz-Manera, J.; Messina, G.; Cossu, G. Repairing skeletal muscle: Regenerative potential of skeletal muscle stem cells. J. Clin. Inv. 2010, 120, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.C.; Rudnicki, M.A. Satellite Cells and Skeletal Muscle Regeneration. Compr. Phys. 2015, 5, 1027–1059. [Google Scholar] [CrossRef]
- Darabi, R.; Arpke, R.W.; Irion, S.; Dimos, J.T.; Grskovic, M.; Kyba, M.; Perlingeiro, R.C. Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell 2012, 10, 610–619. [Google Scholar] [CrossRef]
- Charville, G.W.; Cheung, T.H.; Yoo, B.; Santos, P.J.; Lee, G.K.; Shrager, J.B.; Rando, T.A. Ex Vivo Expansion and In Vivo Self-Renewal of Human Muscle Stem Cells. Stem Cell Rep. 2015, 5, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Garcia, S.M.; Tamaki, S.; Lee, S.; Wong, A.; Jose, A.; Dreux, J.; Kouklis, G.; Sbitany, H.; Seth, R.; Knott, P.D.; et al. High-Yield Purification, Preservation, and Serial Transplantation of Human Satellite Cells. Stem Cell Rep. 2018, 10, 1160–1174. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, P.M.; Havenstrite, K.L.; Magnusson, K.E.; Sacco, A.; Leonardi, N.A.; Kraft, P.; Nguyen, N.K.; Thrun, S.; Lutolf, M.P.; Blau, H.M. Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science 2010, 329, 1078–1081. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Wang, Y.X.; von Maltzahn, J.; Soleimani, V.D.; Yin, H.; Rudnicki, M.A. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell Stem Cell 2013, 12, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Urciuolo, A.; Quarta, M.; Morbidoni, V.; Gattazzo, F.; Molon, S.; Grumati, P.; Montemurro, F.; Tedesco, F.S.; Blaauw, B.; Cossu, G.; et al. Collagen VI regulates satellite cell self-renewal and muscle regeneration. Nat. Commun. 2013, 4, 1964. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.M.S.; Ancillotti, M.T.C.; Rangel, L.P.; Fontenele, M.; Figueiredo-Freitas, C.; Possidonio, A.C.; Soares, C.P.; Sorenson, M.M.; Mermelstein, C.; Nogueira, L. Balance between S-nitrosylation and denitrosylation modulates myoblast proliferation independently of soluble guanylyl cyclase activation. Am. J. Phys. Cell Phys. 2017, 313, C11–C26. [Google Scholar] [CrossRef] [PubMed]
- Pavlidou, T.; Rosina, M.; Fuoco, C.; Gerini, G.; Gargioli, C.; Castagnoli, L.; Cesareni, G. Regulation of myoblast differentiation by metabolic perturbations induced by metformin. PLoS ONE 2017, 12, e0182475. [Google Scholar] [CrossRef] [PubMed]
- Bakhtiari, N.; Hosseinkhani, S.; Soleimani, M.; Hemmati, R.; Noori-Zadeh, A.; Javan, M.; Tashakor, A. Short-term ursolic acid promotes skeletal muscle rejuvenation through enhancing of SIRT1 expression and satellite cells proliferation. Biomed. Pharmacother. Biomed. Pharmacother. 2016, 78, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Auwerx, J. Transcriptional targets of sirtuins in the coordination of mammalian physiology. Curr. Opin. Cell Biol. 2008, 20, 303–309. [Google Scholar] [CrossRef] [Green Version]
- Rathbone, C.R.; Booth, F.W.; Lees, S.J. Sirt1 increases skeletal muscle precursor cell proliferation. Eur. J. Cell Biol. 2009, 88, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Bouchentouf, M.; Benabdallah, B.F.; Bigey, P.; Yau, T.M.; Scherman, D.; Tremblay, J.P. Vascular endothelial growth factor reduced hypoxia-induced death of human myoblasts and improved their engraftment in mouse muscles. Gene Ther. 2008, 15, 404–414. [Google Scholar] [CrossRef]
- Lafreniere, J.F.; Caron, M.C.; Skuk, D.; Goulet, M.; Cheikh, A.R.; Tremblay, J.P. Growth factor coinjection improves the migration potential of monkey myogenic precursors without affecting cell transplantation success. Cell Trans. 2009, 18, 719–730. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; von Maltzahn, J.; Dumont, N.A.; Stark, D.A.; Wang, Y.X.; Nhan, K.; Frenette, J.; Cornelison, D.D.; Rudnicki, M.A. Wnt7a stimulates myogenic stem cell motility and engraftment resulting in improved muscle strength. J. Cell Biol. 2014, 205, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Bencze, M.; Negroni, E.; Vallese, D.; Yacoub-Youssef, H.; Chaouch, S.; Wolff, A.; Aamiri, A.; Di Santo, J.P.; Chazaud, B.; Butler-Browne, G.; et al. Proinflammatory macrophages enhance the regenerative capacity of human myoblasts by modifying their kinetics of proliferation and differentiation. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 2168–2179. [Google Scholar] [CrossRef] [PubMed]
- Lesault, P.F.; Theret, M.; Magnan, M.; Cuvellier, S.; Niu, Y.; Gherardi, R.K.; Tremblay, J.P.; Hittinger, L.; Chazaud, B. Macrophages improve survival, proliferation and migration of engrafted myogenic precursor cells into MDX skeletal muscle. PLoS ONE 2012, 7, e46698. [Google Scholar] [CrossRef] [PubMed]
- von Maltzahn, J.; Renaud, J.M.; Parise, G.; Rudnicki, M.A. Wnt7a treatment ameliorates muscular dystrophy. Proc. Natl. Acad. Sci. USA 2012, 109, 20614–20619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernet, J.D.; Doles, J.D.; Hall, J.K.; Kelly Tanaka, K.; Carter, T.A.; Olwin, B.B. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat. Med. 2014, 20, 265–271. [Google Scholar] [CrossRef]
- Price, F.D.; von Maltzahn, J.; Bentzinger, C.F.; Dumont, N.A.; Yin, H.; Chang, N.C.; Wilson, D.H.; Frenette, J.; Rudnicki, M.A. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med. 2014, 20, 1174–1181. [Google Scholar] [CrossRef] [Green Version]
- Kurosaka, M.; Machida, S. Interleukin-6-induced satellite cell proliferation is regulated by induction of the JAK2/STAT3 signalling pathway through cyclin D1 targeting. Cell Prol. 2013, 46, 365–373. [Google Scholar] [CrossRef]
- Alves, A.N.; Ribeiro, B.G.; Fernandes, K.P.; Souza, N.H.; Rocha, L.A.; Nunes, F.D.; Bussadori, S.K.; Mesquita-Ferrari, R.A. Comparative effects of low-level laser therapy pre- and post-injury on mRNA expression of MyoD, myogenin, and IL-6 during the skeletal muscle repair. Lasers Med. Sci. 2016, 31, 679–685. [Google Scholar] [CrossRef]
- Formicola, L.; Pannerec, A.; Correra, R.M.; Gayraud-Morel, B.; Ollitrault, D.; Besson, V.; Tajbakhsh, S.; Lachey, J.; Seehra, J.S.; Marazzi, G.; et al. Inhibition of the Activin Receptor Type-2B Pathway Restores Regenerative Capacity in Satellite Cell-Depleted Skeletal Muscle. Front. Phys. 2018, 9, 515. [Google Scholar] [CrossRef]
- Tedesco, F.S.; Hoshiya, H.; D’Antona, G.; Gerli, M.F.; Messina, G.; Antonini, S.; Tonlorenzi, R.; Benedetti, S.; Berghella, L.; Torrente, Y.; et al. Stem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophy. Sci. Trans. Med. 2011, 3, 96ra78. [Google Scholar] [CrossRef]
- Benedetti, S.; Uno, N.; Hoshiya, H.; Ragazzi, M.; Ferrari, G.; Kazuki, Y.; Moyle, L.A.; Tonlorenzi, R.; Lombardo, A.; Chaouch, S.; et al. Reversible immortalisation enables genetic correction of human muscle progenitors and engineering of next-generation human artificial chromosomes for Duchenne muscular dystrophy. EMBO Mol. Med. 2018, 10, 254–275. [Google Scholar] [CrossRef] [PubMed]
- Crist, C.G.; Montarras, D.; Pallafacchina, G.; Rocancourt, D.; Cumano, A.; Conway, S.J.; Buckingham, M. Muscle stem cell behavior is modified by microRNA-27 regulation of Pax3 expression. Proc. Natl. Acad. Sci. USA 2009, 106, 13383–13387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, L.; Xu, J.; Jiao, Y.; Li, H.; Pan, Z.; Duan, J.; Gu, T.; Hu, C.; Wang, C. MiR-27b Promotes Muscle Development by Inhibiting MDFI Expression. Cell. Physiol. Biochem. Int. J. Exp. Cell. Phys. Biochem. Pharmacol. 2018, 46, 2271–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; Jan, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [PubMed]
- van Agtmaal, E.L.; André, L.M.; Willemse, M.; Cumming, S.A.; van Kessel, I.D.G.; van den Broek, W.J.A.A.; Gourdon, G.; Furling, D.; Mouly, V.; Monckton, D.G.; et al. CRISPR/Cas9-Induced (CTG⋅CAG). Mol. Ther. 2017, 25, 24–43. [Google Scholar] [CrossRef]
- Perie, S.; Trollet, C.; Mouly, V.; Vanneaux, V.; Mamchaoui, K.; Bouazza, B.; Marolleau, J.P.; Laforet, P.; Chapon, F.; Eymard, B.; et al. Autologous myoblast transplantation for oculopharyngeal muscular dystrophy: A phase I/IIa clinical study. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 219–225. [Google Scholar] [CrossRef]
- Sivaraman, K.; Shanthi, C. Matrikines for therapeutic and biomedical applications. Life Sci. 2018, 214, 22–33. [Google Scholar] [CrossRef]
- Ricard-Blum, S.; Salza, R. Matricryptins and matrikines: Biologically active fragments of the extracellular matrix. Exp. Dermatol. 2014, 23, 457–463. [Google Scholar] [CrossRef]
- Davis, G.E.; Bayless, K.J.; Davis, M.J.; Meininger, G.A. Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am. J. Pathol. 2000, 156, 1489–1498. [Google Scholar] [CrossRef]
- Schor, S.L.; Schor, A.M. Phenotypic and genetic alterations in mammary stroma: Implications for tumour progression. Breast Cancer Res. BCR 2001, 3, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S.; Vallet, S.D. Fragments generated upon extracellular matrix remodeling: Biological regulators and potential drugs. Matrix Biol. J. Int. Soc. Matrix Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Guo, L.; Cui, X.; Liu, H.; Liu, L. Rh-endostatin Concomitant with Chemotherapy Versus Single Agent Chemotherapy for Treating Soft Tissue and Bone Sarcomas: A Systematic Review and Meta-Analysis. J. Pharm. Pharm. Sci. 2018, 21, 386–397. [Google Scholar] [CrossRef]
- Kikkawa, Y.; Hozumi, K.; Katagiri, F.; Nomizu, M.; Kleinman, H.K.; Koblinski, J.E. Laminin-111-derived peptides and cancer. Cell Adhes. Migr. 2013, 7, 150–256. [Google Scholar] [CrossRef] [PubMed]
- Nomizu, M.; Kim, W.H.; Yamamura, K.; Utani, A.; Song, S.Y.; Otaka, A.; Roller, P.P.; Kleinman, H.K.; Yamada, Y. Identification of cell binding sites in the laminin alpha 1 chain carboxyl-terminal globular domain by systematic screening of synthetic peptides. J. Biol. Chem. 1995, 270, 20583–20590. [Google Scholar] [CrossRef] [PubMed]
- Richard, B.L.; Nomizu, M.; Yamada, Y.; Kleinman, H.K. Identification of synthetic peptides derived from laminin alpha1 and alpha2 chains with cell type specificity for neurite outgrowth. Exp. Cell Res. 1996, 228, 98–105. [Google Scholar] [CrossRef]
- Mochizuki, M.; Philp, D.; Hozumi, K.; Suzuki, N.; Yamada, Y.; Kleinman, H.K.; Nomizu, M. Angiogenic activity of syndecan-binding laminin peptide AG73 (RKRLQVQLSIRT). Arch. Biochem. Biophys. 2007, 459, 249–255. [Google Scholar] [CrossRef]
- Mochizuki, M.; Kadoya, Y.; Wakabayashi, Y.; Kato, K.; Okazaki, I.; Yamada, M.; Sato, T.; Sakairi, N.; Nishi, N.; Nomizu, M. Laminin-1 peptide-conjugated chitosan membranes as a novel approach for cell engineering. FASEB J. Off. Publ. Feder. Am. Soc. Exp. Biol. 2003, 17, 875–877. [Google Scholar] [CrossRef]
- Ponce, M.L.; Kleinman, H.K. Identification of redundant angiogenic sites in laminin alpha1 and gamma1 chains. Exp. Cell Res. 2003, 285, 189–195. [Google Scholar] [CrossRef]
- Malinda, K.M.; Wysocki, A.B.; Koblinski, J.E.; Kleinman, H.K.; Ponce, M.L. Angiogenic laminin-derived peptides stimulate wound healing. Int. J. Biochem. Cell Biol. 2008, 40, 2771–2780. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, T.M.; Kibbey, M.C.; Zain, M.; Fridman, R.; Kleinman, H.K. Basement membrane and the SIKVAV laminin-derived peptide promote tumor growth and metastases. Cancer Metastasis Rev. 1991, 10, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, K.; Sephel, G.C.; Weeks, B.; Sasaki, M.; Martin, G.R.; Kleinman, H.K.; Yamada, Y. A synthetic peptide containing the IKVAV sequence from the A chain of laminin mediates cell attachment, migration, and neurite outgrowth. J. Biol. Chem. 1989, 264, 16174–16182. [Google Scholar] [PubMed]
- Kim, W.H.; Nomizu, M.; Song, S.Y.; Tanaka, K.; Kuratomi, Y.; Kleinman, H.K.; Yamada, Y. Laminin-alpha1-chain sequence Leu-Gln-Val-Gln-Leu-Ser-Ile-Arg (LQVQLSIR) enhances murine melanoma cell metastases. Int. J. Cancer 1998, 77, 632–639. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gois Beghini, D.; Iwao Horita, S.; Monteiro da Fonseca Cardoso, L.; Anastacio Alves, L.; Nagaraju, K.; Henriques-Pons, A. A Promising Future for Stem-Cell-Based Therapies in Muscular Dystrophies—In Vitro and In Vivo Treatments to Boost Cellular Engraftment. Int. J. Mol. Sci. 2019, 20, 5433. https://doi.org/10.3390/ijms20215433
Gois Beghini D, Iwao Horita S, Monteiro da Fonseca Cardoso L, Anastacio Alves L, Nagaraju K, Henriques-Pons A. A Promising Future for Stem-Cell-Based Therapies in Muscular Dystrophies—In Vitro and In Vivo Treatments to Boost Cellular Engraftment. International Journal of Molecular Sciences. 2019; 20(21):5433. https://doi.org/10.3390/ijms20215433
Chicago/Turabian StyleGois Beghini, Daniela, Samuel Iwao Horita, Liana Monteiro da Fonseca Cardoso, Luiz Anastacio Alves, Kanneboyina Nagaraju, and Andrea Henriques-Pons. 2019. "A Promising Future for Stem-Cell-Based Therapies in Muscular Dystrophies—In Vitro and In Vivo Treatments to Boost Cellular Engraftment" International Journal of Molecular Sciences 20, no. 21: 5433. https://doi.org/10.3390/ijms20215433
APA StyleGois Beghini, D., Iwao Horita, S., Monteiro da Fonseca Cardoso, L., Anastacio Alves, L., Nagaraju, K., & Henriques-Pons, A. (2019). A Promising Future for Stem-Cell-Based Therapies in Muscular Dystrophies—In Vitro and In Vivo Treatments to Boost Cellular Engraftment. International Journal of Molecular Sciences, 20(21), 5433. https://doi.org/10.3390/ijms20215433