Maintenance of Type 2 Response by CXCR6-Deficient ILC2 in Papain-Induced Lung Inflammation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
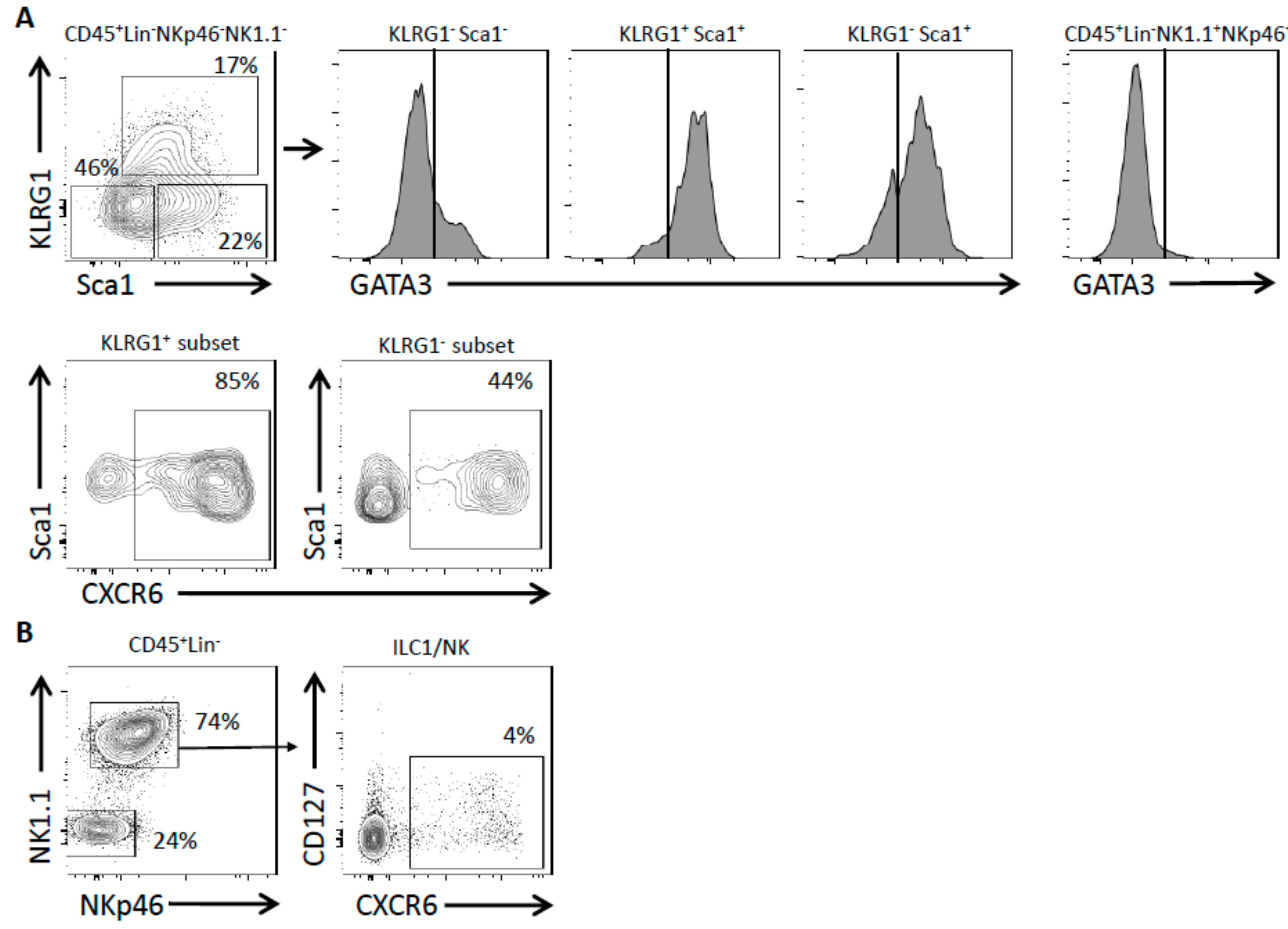
2.1. Lung ILC Subsets Differentially Express CXCR6
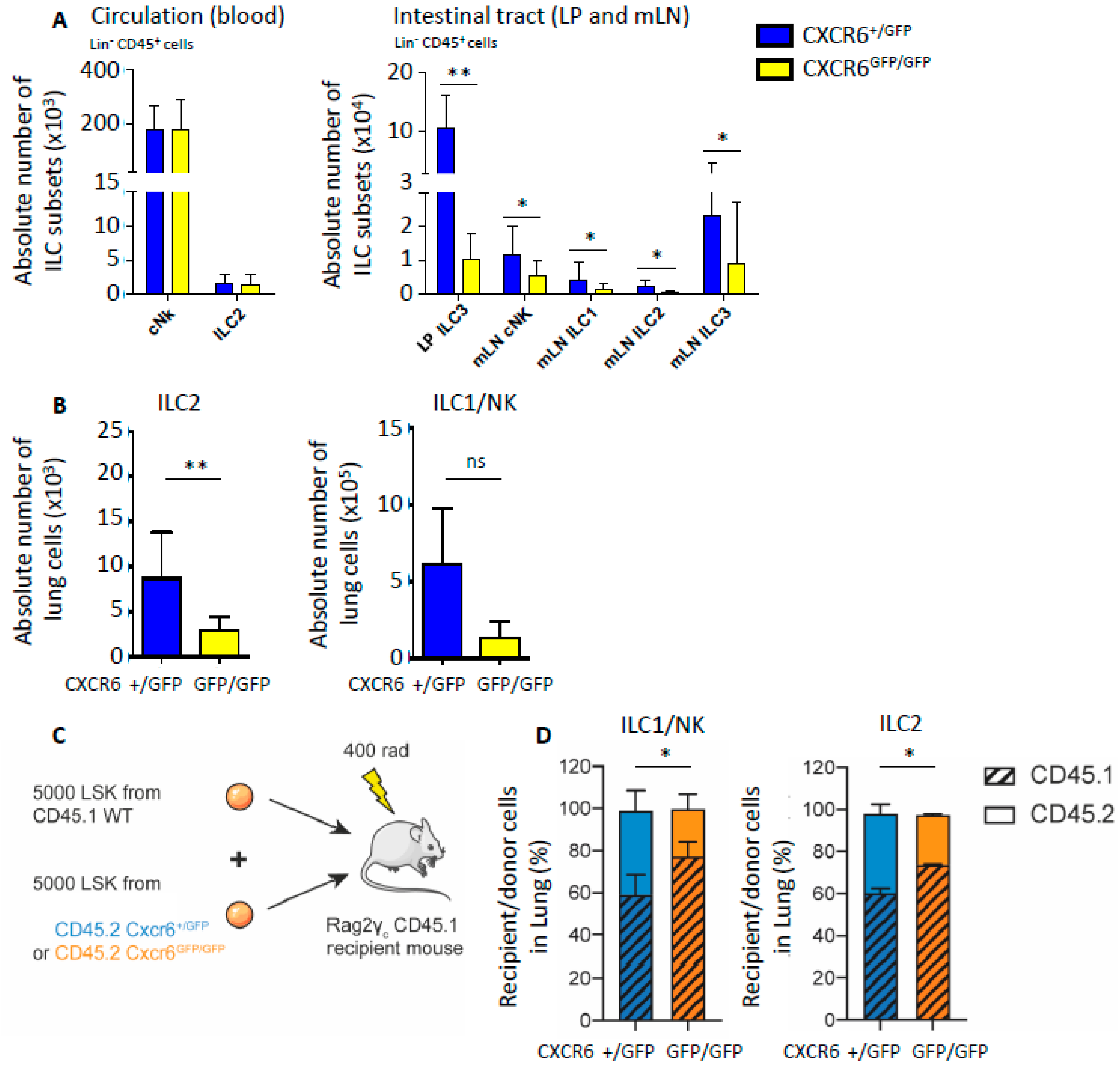
2.2. CXCR6 Deficiency Alters Lung ILC Subset Distribution at Homeostasis
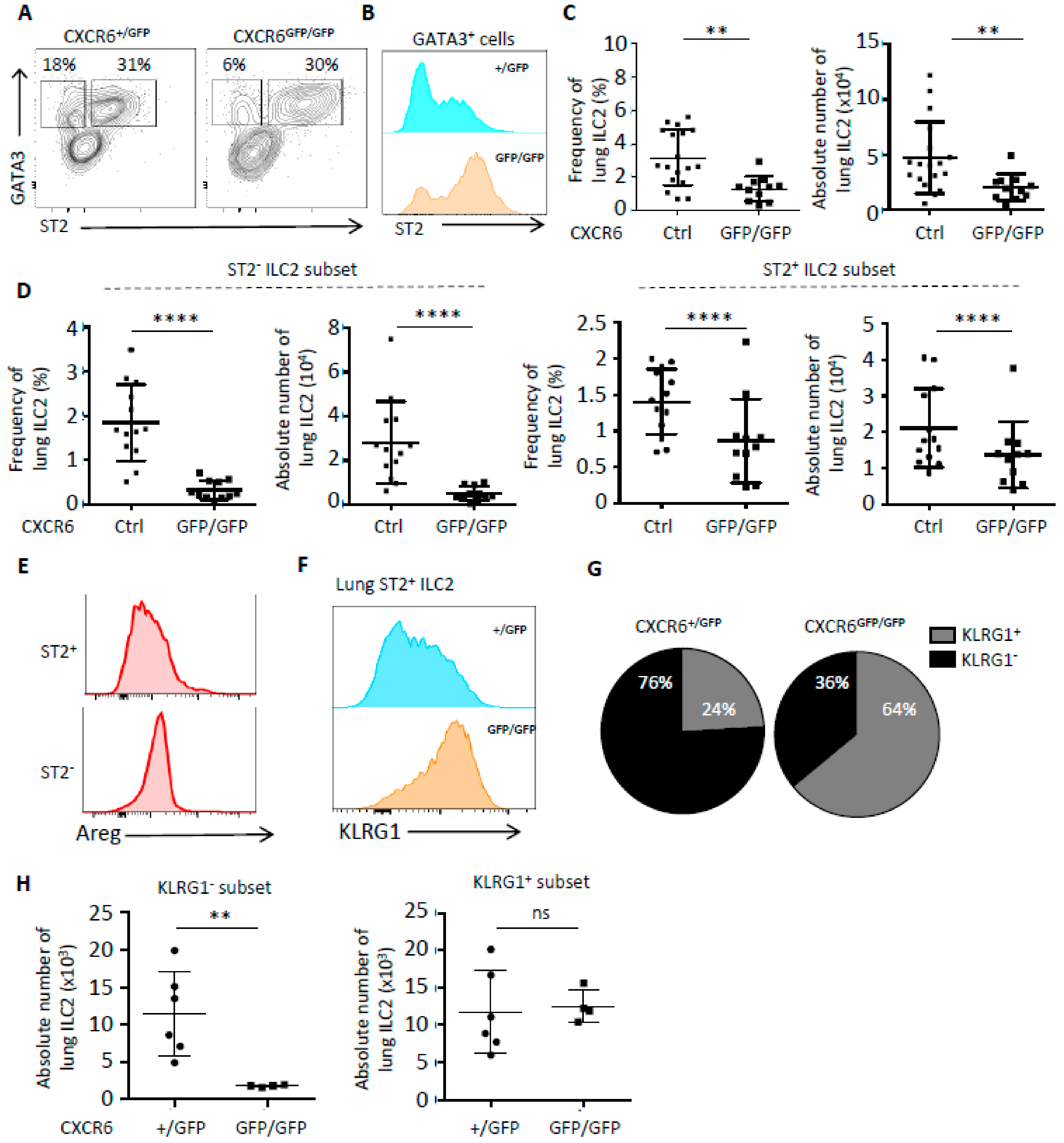
2.3. In Case of Lung Inflammation, CXCR6 Deficiency Impacts the Number of ILC2 and Their Phenotype
2.4. CXCR6 Does Not Influence the Capacity of ILC2 to Secrete IL-5 and IL-13 during Inflammation
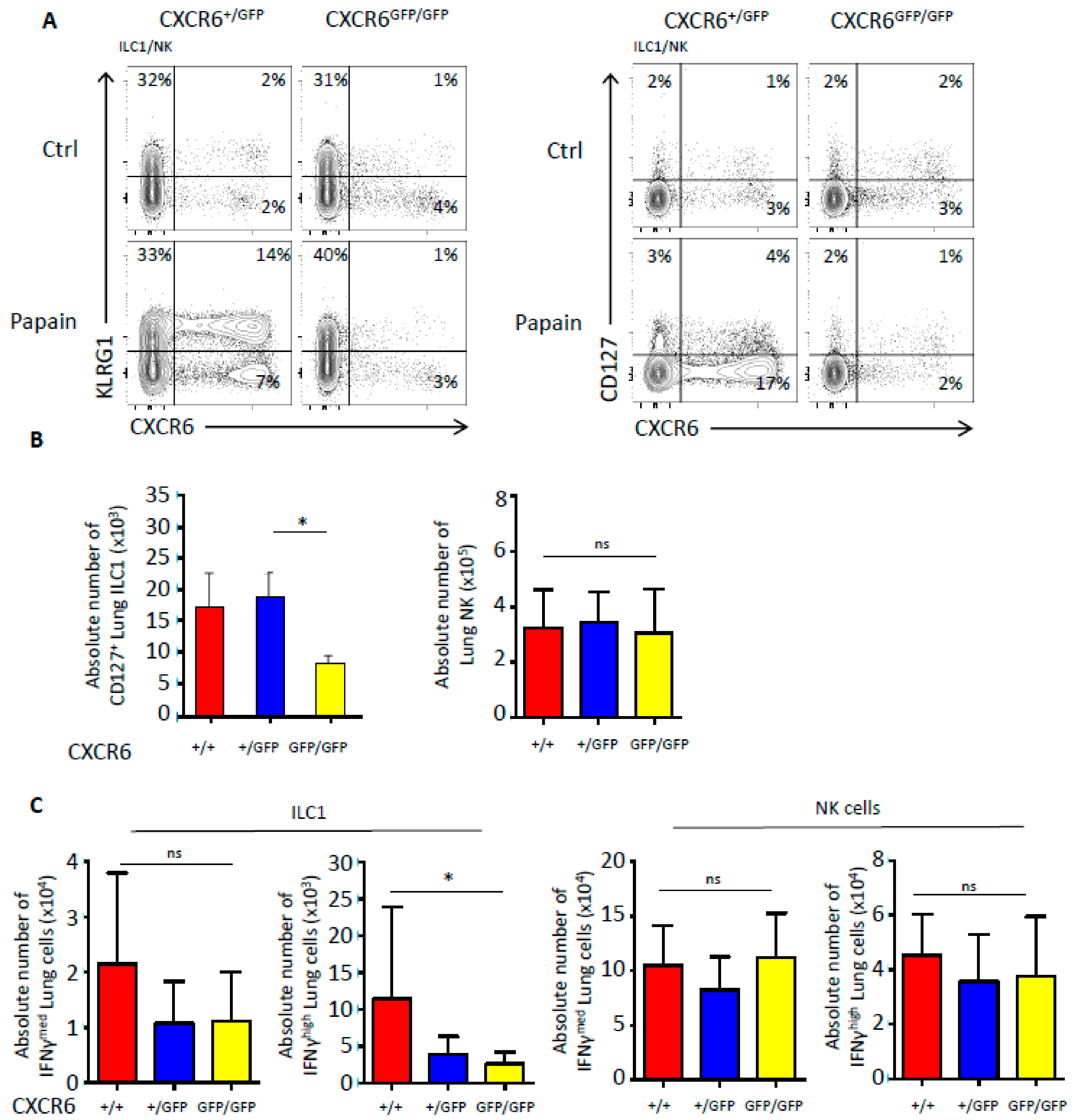
2.5. CXCR6 Deficiency Also Alters the Number and Functions of Type 1 Innate Lymphoid Cells
3. Discussion
4. Materials and Methods
4.1. Mice and Animal Facilities
4.2. Inflammation of Lung
4.3. Cell Preparation
4.4. Lymphocyte Activation
4.5. Flow Cytometry
4.6. In Vivo Reconstitution
4.7. RT-qPCR Analysis
4.8. Exsanguination Experiment
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ILC | Innate Lymphoid Cells |
| NK | Natural Killer |
| FCS | Fetal Calf Serum |
| mLN | Mesenteric Lymph Node |
| GFP | Green Fluorescent Protein |
References
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halim, T.Y.F.; Steer, C.A.; Mathä, L.; Gold, M.J.; Martinez-Gonzalez, I.; McNagny, K.M.; McKenzie, A.N.J.; Takei, F. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 2014, 40, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gonzalez, I.; Steer, C.A.; Takei, F. Lung ILC2s link innate and adaptive responses in allergic inflammation. Trends Immunol. 2015, 36, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Cui, L.; Yu, G.; Yang, X.; Chen, Y.; Wan, X. NK Cells Alleviate Lung Inflammation by Negatively Regulating Group 2 Innate Lymphoid Cells. J. Immunol. 2017, 198, 3336–3344. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, J.C.; Van Dyken, S.J.; Von Moltke, J.; Cheng, L.E.; Mohapatra, A.; Molofsky, A.B.; Thornton, E.E.; Krummel, M.F.; Chawla, A.; Liang, H.-E.; et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 2013, 502, 245–248. [Google Scholar] [CrossRef] [Green Version]
- Kuperman, D.A.; Huang, X.; Koth, L.L.; Chang, G.H.; Dolganov, G.M.; Zhu, Z.; Elias, J.A.; Sheppard, D.; Erle, D.J. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat. Med. 2002, 8, 885–889. [Google Scholar] [CrossRef]
- Halim, T.Y.F.; Krauss, R.H.; Sun, A.C.; Takei, F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity 2012, 36, 451–463. [Google Scholar] [CrossRef]
- Licona-Limón, P.; Kim, L.K.; Palm, N.W.; Flavell, R.A. TH2, allergy and group 2 innate lymphoid cells. Nat. Immunol. 2013, 14, 536–542. [Google Scholar] [CrossRef]
- Mathias, C.B.; Guernsey, L.A.; Zammit, D.; Brammer, C.; Wu, C.A.; Thrall, R.S.; Aguila, H.L. Pro-inflammatory role of natural killer cells in the development of allergic airway disease. Clin. Exp. Allergy 2014, 44, 589–601. [Google Scholar] [CrossRef]
- Haworth, O.; Cernadas, M.; Levy, B.D. NK cells are effectors for resolvin E1 in the timely resolution of allergic airway inflammation. J. Immunol. 2011, 186, 6129–6135. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Phipps, S.; Angkasekwinai, P.; Dong, C.; Foster, P.S. NK cell deficiency predisposes to viral-induced Th2-type allergic inflammation via epithelial-derived IL-25. J. Immunol. 2010, 185, 4681–4690. [Google Scholar] [CrossRef] [PubMed]
- Barnig, C.; Cernadas, M.; Dutile, S.; Liu, X.; Perrella, M.A.; Kazani, S.; Wechsler, M.E.; Israel, E.; Levy, B.D. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med. 2013, 5, 174ra26. [Google Scholar] [CrossRef] [PubMed]
- Rafei-Shamsabadi, D.A.; Van de Poel, S.; Dorn, B.; Kunz, S.; Martin, S.F.; Klose, C.S.N.; Arnold, S.J.; Tanriver, Y.; Ebert, K.; Diefenbach, A.; et al. Lack of Type 2 Innate Lymphoid Cells Promotes a Type I-Driven Enhanced Immune Response in Contact Hypersensitivity. J. Investig. Dermatol. 2018, 138, 1962–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Possot, C.; Schmutz, S.; Chea, S.; Boucontet, L.; Louise, A.; Cumano, A.; Golub, R. Notch signaling is necessary for adult, but not fetal, development of RORγt(+) innate lymphoid cells. Nat. Immunol. 2011, 12, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Chea, S.; Possot, C.; Perchet, T.; Petit, M.; Cumano, A.; Golub, R. CXCR6 Expression Is Important for Retention and Circulation of ILC Precursors. Mediators Inflamm. 2015, 2015, 368427. [Google Scholar] [CrossRef]
- Robinette, M.L.; Fuchs, A.; Cortez, V.S.; Lee, J.S.; Wang, Y.; Durum, S.K.; Gilfillan, S.; Colonna, M. Immunological Genome Consortium Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat. Immunol. 2015, 16, 306–317. [Google Scholar] [CrossRef]
- Satoh-Takayama, N.; Vosshenrich, C.A.J.; Lesjean-Pottier, S.; Sawa, S.; Lochner, M.; Rattis, F.; Mention, J.-J.; Thiam, K.; Cerf-Bensussan, N.; Mandelboim, O.; et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 2008, 29, 958–970. [Google Scholar] [CrossRef]
- Stegmann, K.A.; Robertson, F.; Hansi, N.; Gill, U.; Pallant, C.; Christophides, T.; Pallett, L.J.; Peppa, D.; Dunn, C.; Fusai, G.; et al. CXCR6 marks a novel subset of T-bet(lo)Eomes(hi) natural killer cells residing in human liver. Sci Rep. 2016, 6, 26157. [Google Scholar] [CrossRef]
- Cong, J.; Wei, H. Natural Killer Cells in the Lungs. Front. Immunol. 2019, 10, 1416. [Google Scholar] [CrossRef] [Green Version]
- Robinson, B.W.; Pinkston, P.; Crystal, R.G. Natural killer cells are present in the normal human lung but are functionally impotent. J. Clin. Investig. 1984, 74, 942–950. [Google Scholar] [CrossRef]
- Sawa, S.; Lochner, M.; Satoh-Takayama, N.; Dulauroy, S.; Bérard, M.; Kleinschek, M.; Cua, D.; Di Santo, J.P.; Eberl, G. RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat. Immunol. 2011, 12, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, F.; Molofsky, A.B.; Morar, M.M.; Rosenzwajg, M.; Liang, H.-E.; Klatzmann, D.; Locksley, R.M.; Bluestone, J.A. Interleukin-5-producing group 2 innate lymphoid cells control eosinophilia induced by interleukin-2 therapy. Blood 2014, 124, 3572–3576. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Guo, L.; Qiu, J.; Chen, X.; Hu-Li, J.; Siebenlist, U.; Williamson, P.R.; Urban, J.F.; Paul, W.E. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential “inflammatory” type 2 innate lymphoid cells. Nat. Immunol. 2015, 16, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mao, K.; Chen, X.; Sun, M.-A.; Kawabe, T.; Li, W.; Usher, N.; Zhu, J.; Urban, J.F.; Paul, W.E.; et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science 2018, 359, 114–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geissmann, F.; Cameron, T.O.; Sidobre, S.; Manlongat, N.; Kronenberg, M.; Briskin, M.J.; Dustin, M.L.; Littman, D.R. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 2005, 3, e113. [Google Scholar] [CrossRef]
- Schneider, C.; Lee, J.; Koga, S.; Ricardo-Gonzalez, R.R.; Nussbaum, J.C.; Smith, L.K.; Villeda, S.A.; Liang, H.-E.; Locksley, R.M. Tissue-Resident Group 2 Innate Lymphoid Cells Differentiate by Layered Ontogeny and In Situ Perinatal Priming. Immunity 2019, 50, 1425–1438. [Google Scholar] [CrossRef]
- Trinchieri, G. Biology of natural killer cells. Adv. Immunol. 1989, 47, 187–376. [Google Scholar]
- Huntington, N.D.; Tabarias, H.; Fairfax, K.; Brady, J.; Hayakawa, Y.; Degli-Esposti, M.A.; Smyth, M.J.; Tarlinton, D.M.; Nutt, S.L. NK cell maturation and peripheral homeostasis is associated with KLRG1 up-regulation. J. Immunol. 2007, 178, 4764–4770. [Google Scholar] [CrossRef]
- Robbins, S.H.; Nguyen, K.B.; Takahashi, N.; Mikayama, T.; Biron, C.A.; Brossay, L. Cutting edge: Inhibitory functions of the killer cell lectin-like receptor G1 molecule during the activation of mouse NK cells. J. Immunol. 2002, 168, 2585–2589. [Google Scholar] [CrossRef]
- Wijaya, R.S.; Read, S.A.; Schibeci, S.; Eslam, M.; Azardaryany, M.K.; El-Khobar, K.; Van der Poorten, D.; Lin, R.; Yuen, L.; Lam, V.; et al. KLRG1+ natural killer cells exert a novel antifibrotic function in chronic hepatitis B. J. Hepatol. 2019, 71, 252–264. [Google Scholar] [CrossRef]
- Martinez-Gonzalez, I.; Mathä, L.; Steer, C.A.; Ghaedi, M.; Poon, G.F.T.; Takei, F. Allergen-Experienced Group 2 Innate Lymphoid Cells Acquire Memory-like Properties and Enhance Allergic Lung Inflammation. Immunity 2016, 45, 198–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knipfer, L.; Schulz-Kuhnt, A.; Kindermann, M.; Greif, V.; Symowski, C.; Voehringer, D.; Neurath, M.F.; Atreya, I.; Wirtz, S. A CCL1/CCR8-dependent feed-forward mechanism drives ILC2 functions in type 2-mediated inflammation. J. Exp. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Puttur, F.; Denney, L.; Gregory, L.G.; Vuononvirta, J.; Oliver, R.; Entwistle, L.J.; Walker, S.A.; Headley, M.B.; McGhee, E.J.; Pease, J.E.; et al. Pulmonary environmental cues drive group 2 innate lymphoid cell dynamics in mice and humans. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duerr, C.U.; McCarthy, C.D.A.; Mindt, B.C.; Rubio, M.; Meli, A.P.; Pothlichet, J.; Eva, M.M.; Gauchat, J.-F.; Qureshi, S.T.; Mazer, B.D.; et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat. Immunol. 2016, 17, 65–75. [Google Scholar] [CrossRef]
- Zaiss, D.M.; Yang, L.; Shah, P.R.; Kobie, J.J.; Urban, J.F.; Mosmann, T.R. Amphiregulin, a TH2 cytokine enhancing resistance to nematodes. Science 2006, 314, 1746. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.K.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meunier, S.; Chea, S.; Garrido, D.; Perchet, T.; Petit, M.; Cumano, A.; Golub, R. Maintenance of Type 2 Response by CXCR6-Deficient ILC2 in Papain-Induced Lung Inflammation. Int. J. Mol. Sci. 2019, 20, 5493. https://doi.org/10.3390/ijms20215493
Meunier S, Chea S, Garrido D, Perchet T, Petit M, Cumano A, Golub R. Maintenance of Type 2 Response by CXCR6-Deficient ILC2 in Papain-Induced Lung Inflammation. International Journal of Molecular Sciences. 2019; 20(21):5493. https://doi.org/10.3390/ijms20215493
Chicago/Turabian StyleMeunier, Sylvain, Sylvestre Chea, Damien Garrido, Thibaut Perchet, Maxime Petit, Ana Cumano, and Rachel Golub. 2019. "Maintenance of Type 2 Response by CXCR6-Deficient ILC2 in Papain-Induced Lung Inflammation" International Journal of Molecular Sciences 20, no. 21: 5493. https://doi.org/10.3390/ijms20215493
APA StyleMeunier, S., Chea, S., Garrido, D., Perchet, T., Petit, M., Cumano, A., & Golub, R. (2019). Maintenance of Type 2 Response by CXCR6-Deficient ILC2 in Papain-Induced Lung Inflammation. International Journal of Molecular Sciences, 20(21), 5493. https://doi.org/10.3390/ijms20215493