Rational Design and Synthesis of Diverse Pyrimidine Molecules Bearing Sulfonamide Moiety as Novel ERK Inhibitors

 , and
, and
Abstract
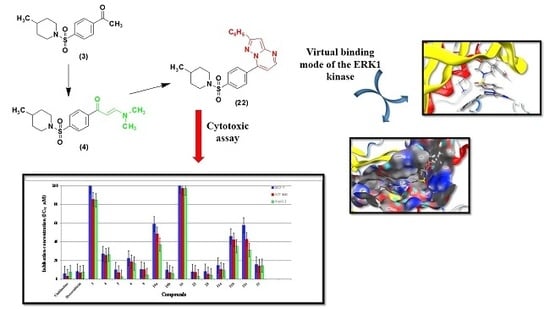
:
1. Introduction
2. Results and Discussion
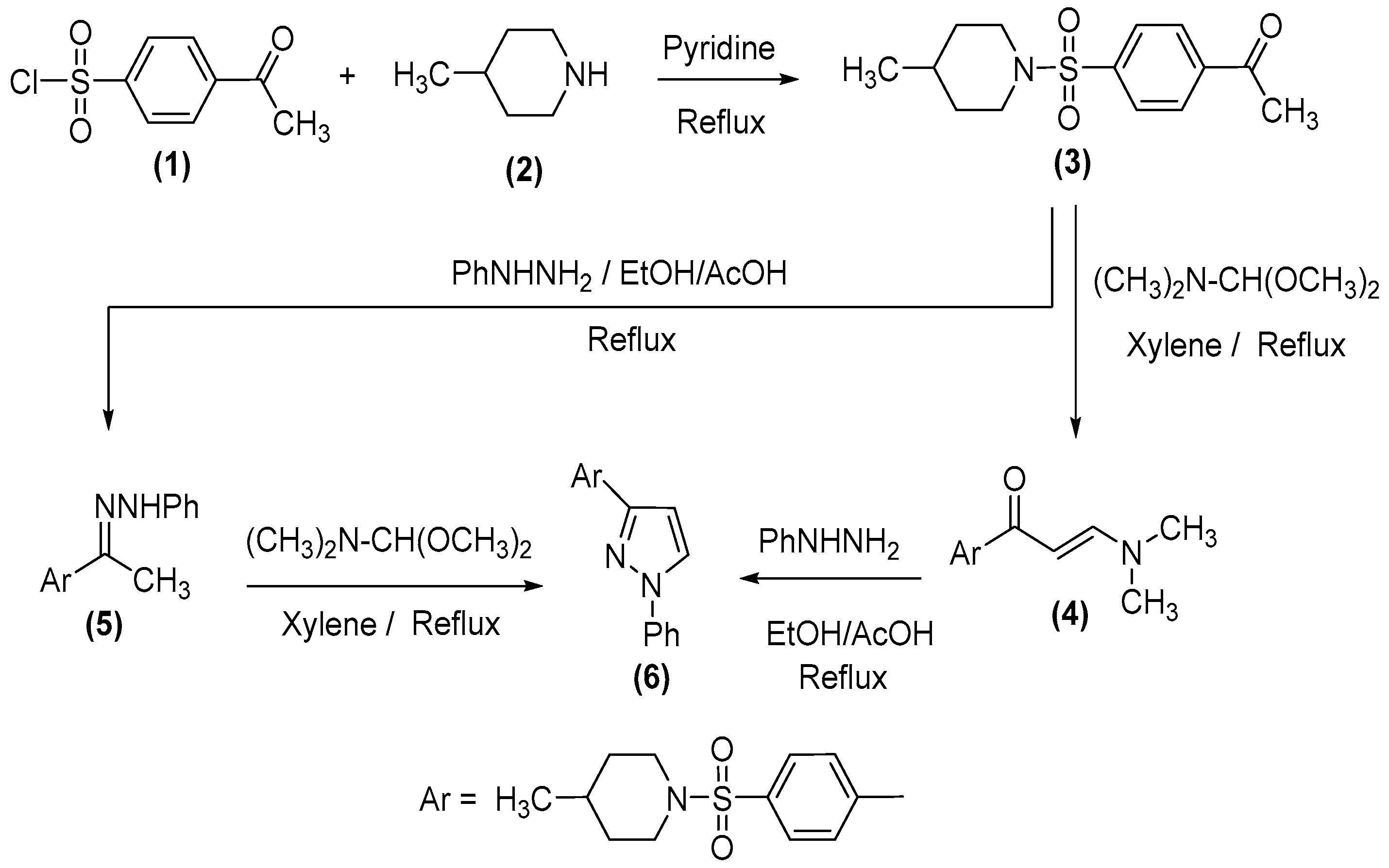
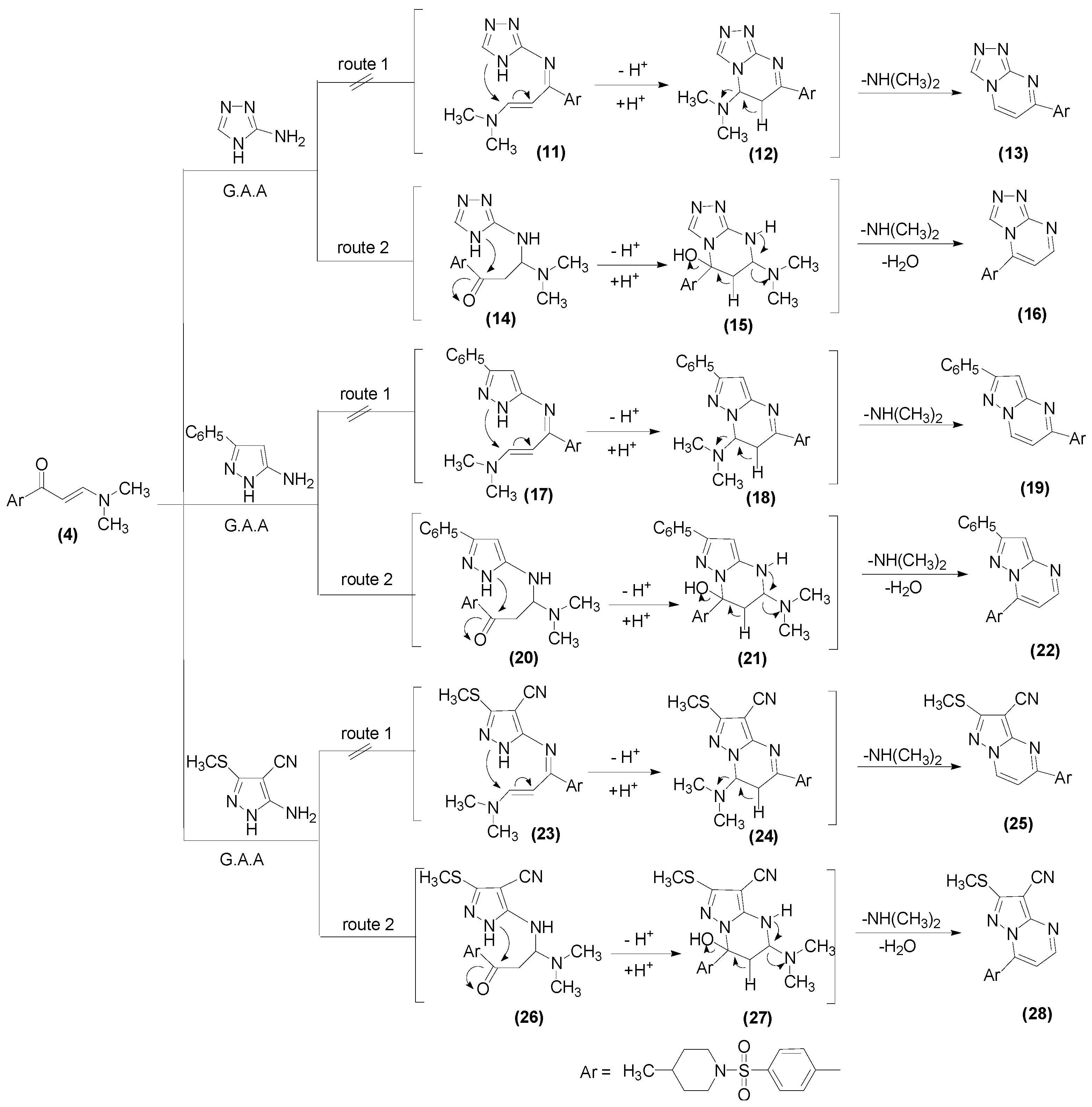
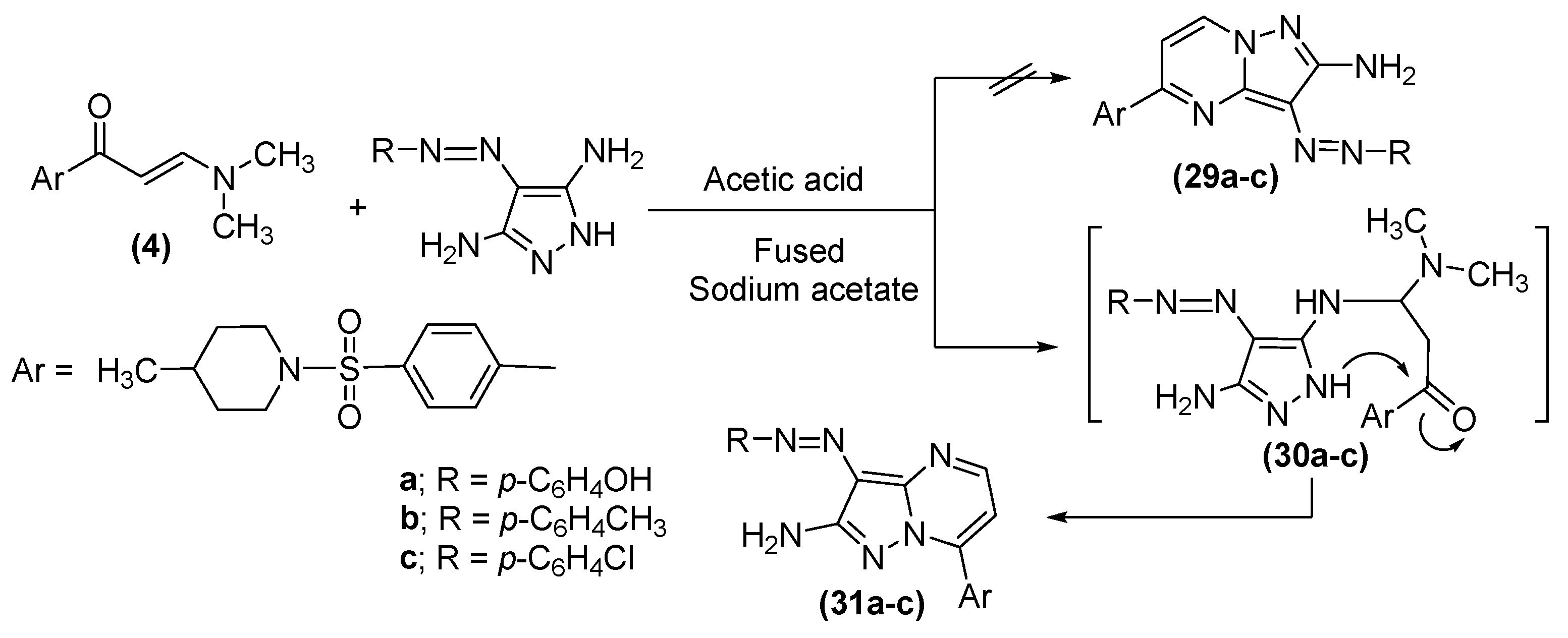
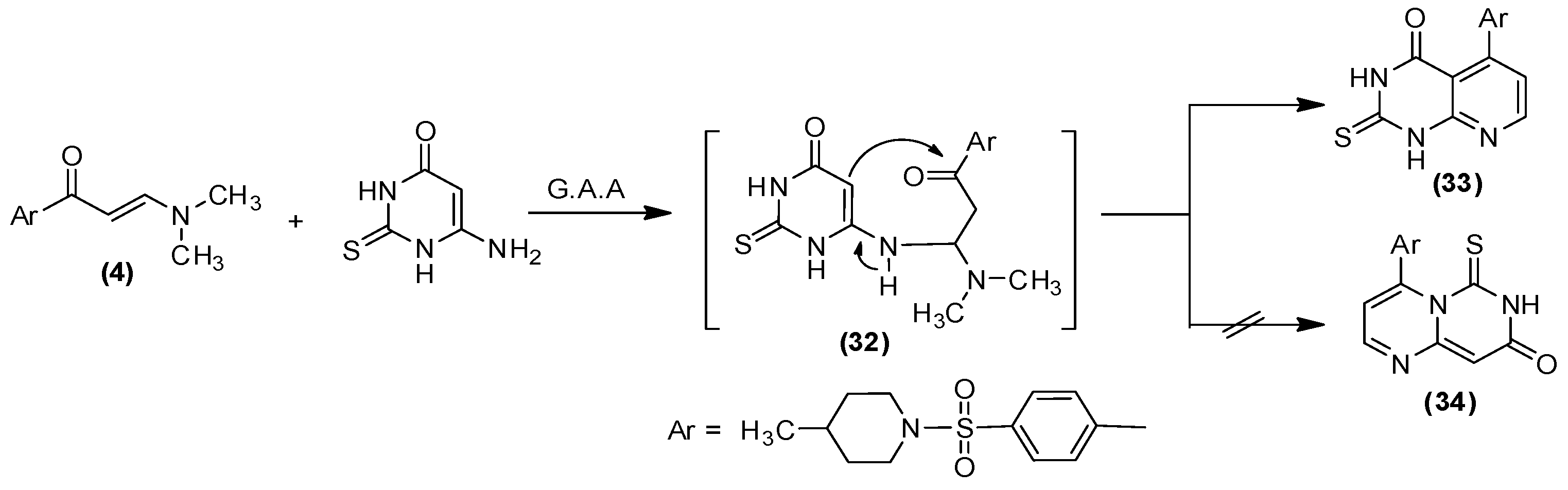
2.1. Chemistry
2.2. Biological Activity
In Vitro Anti-Proliferative Activity
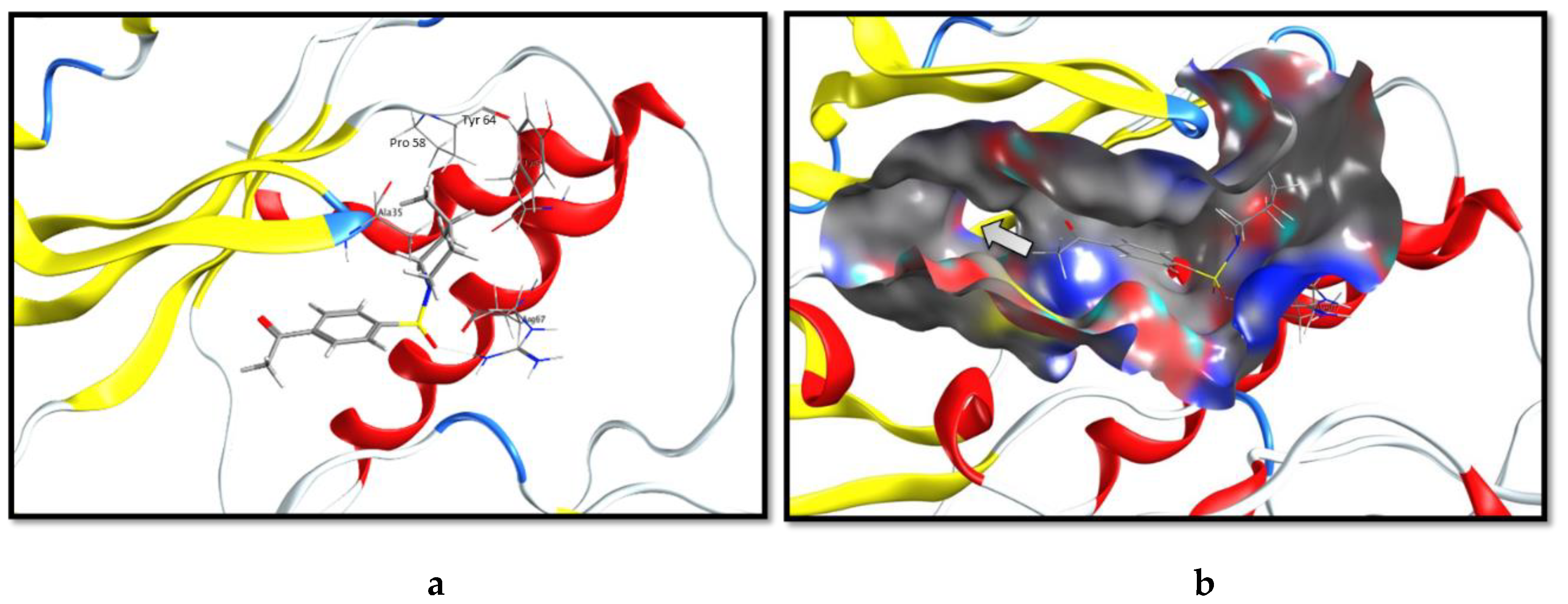
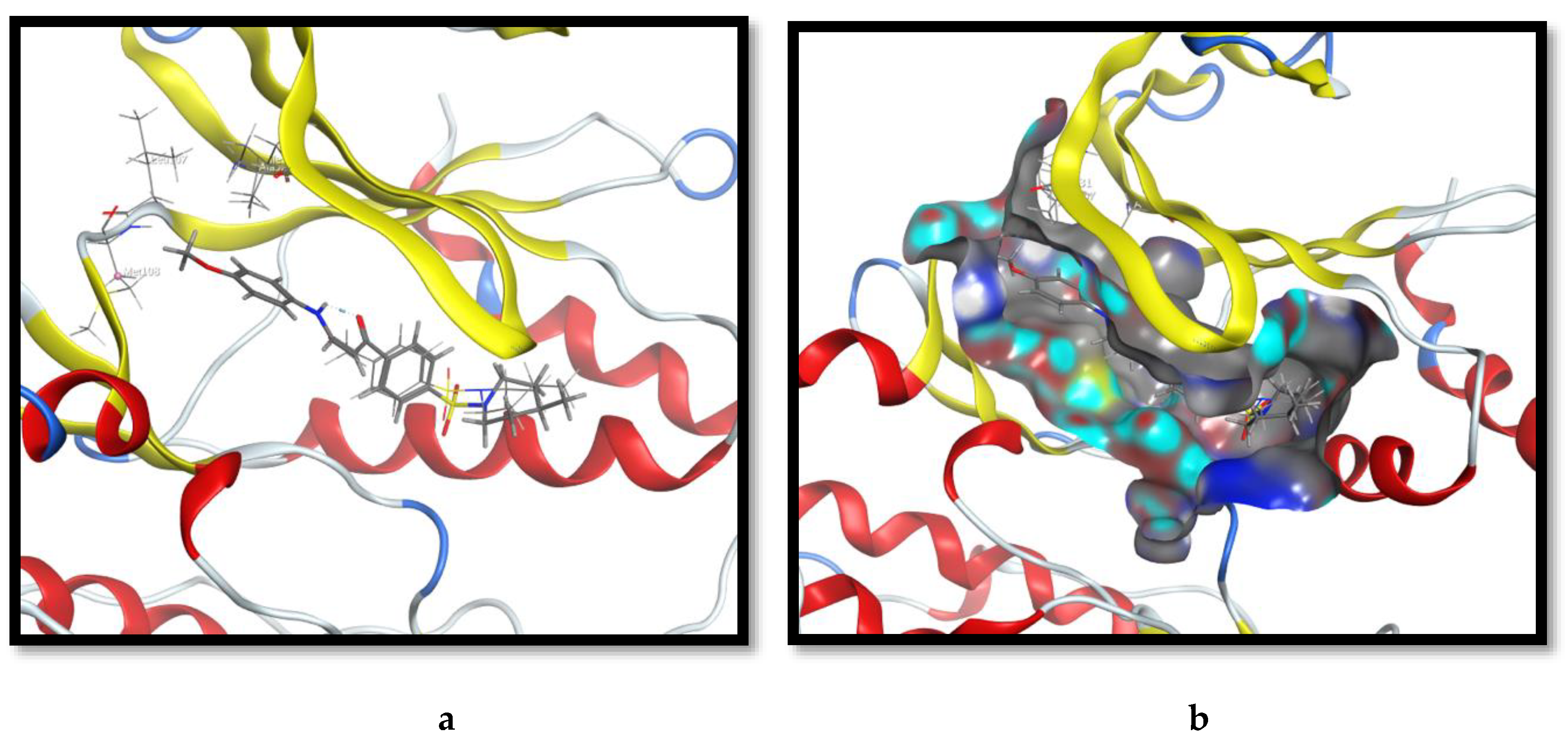
2.3. Molecular Docking and SAR Studies
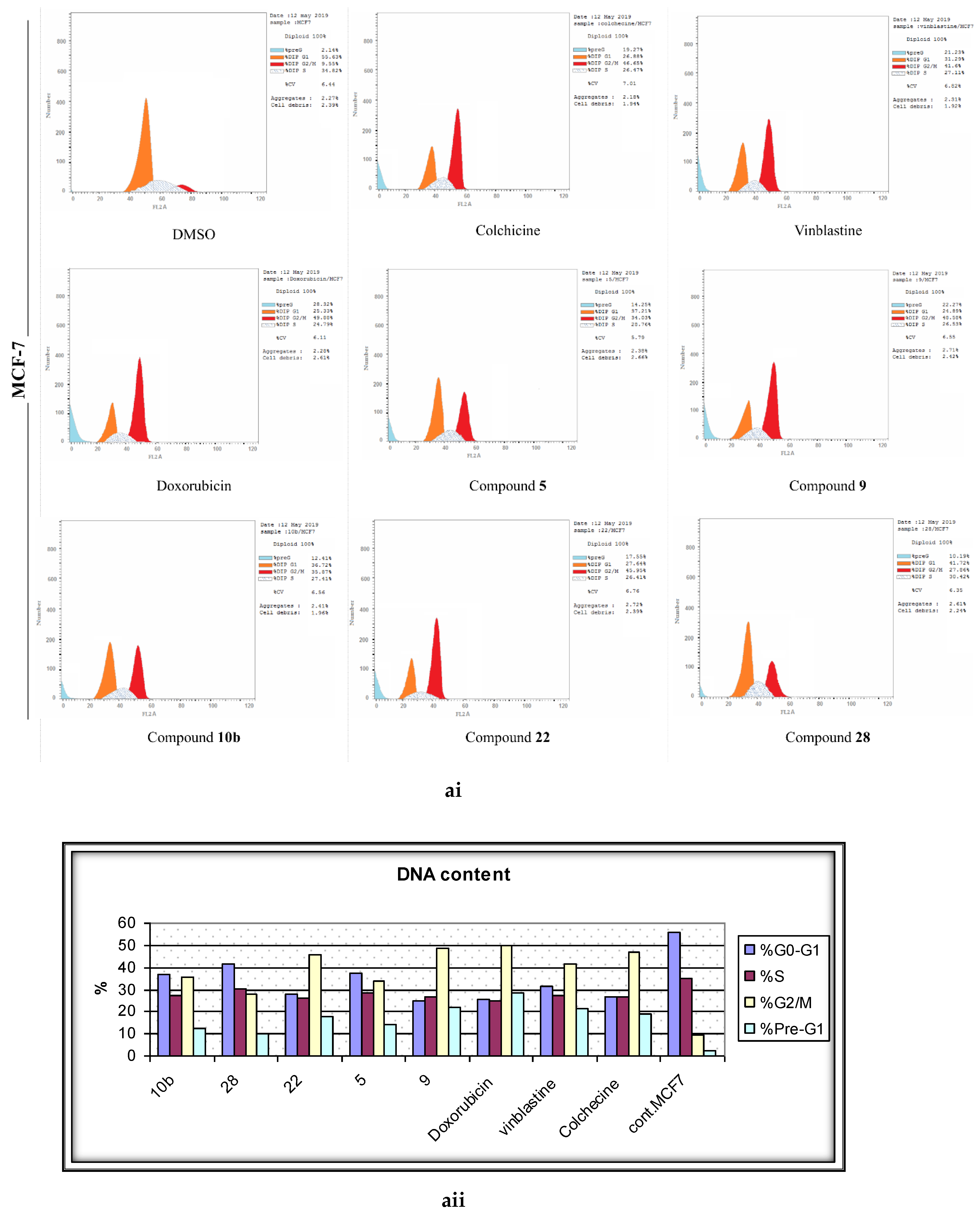
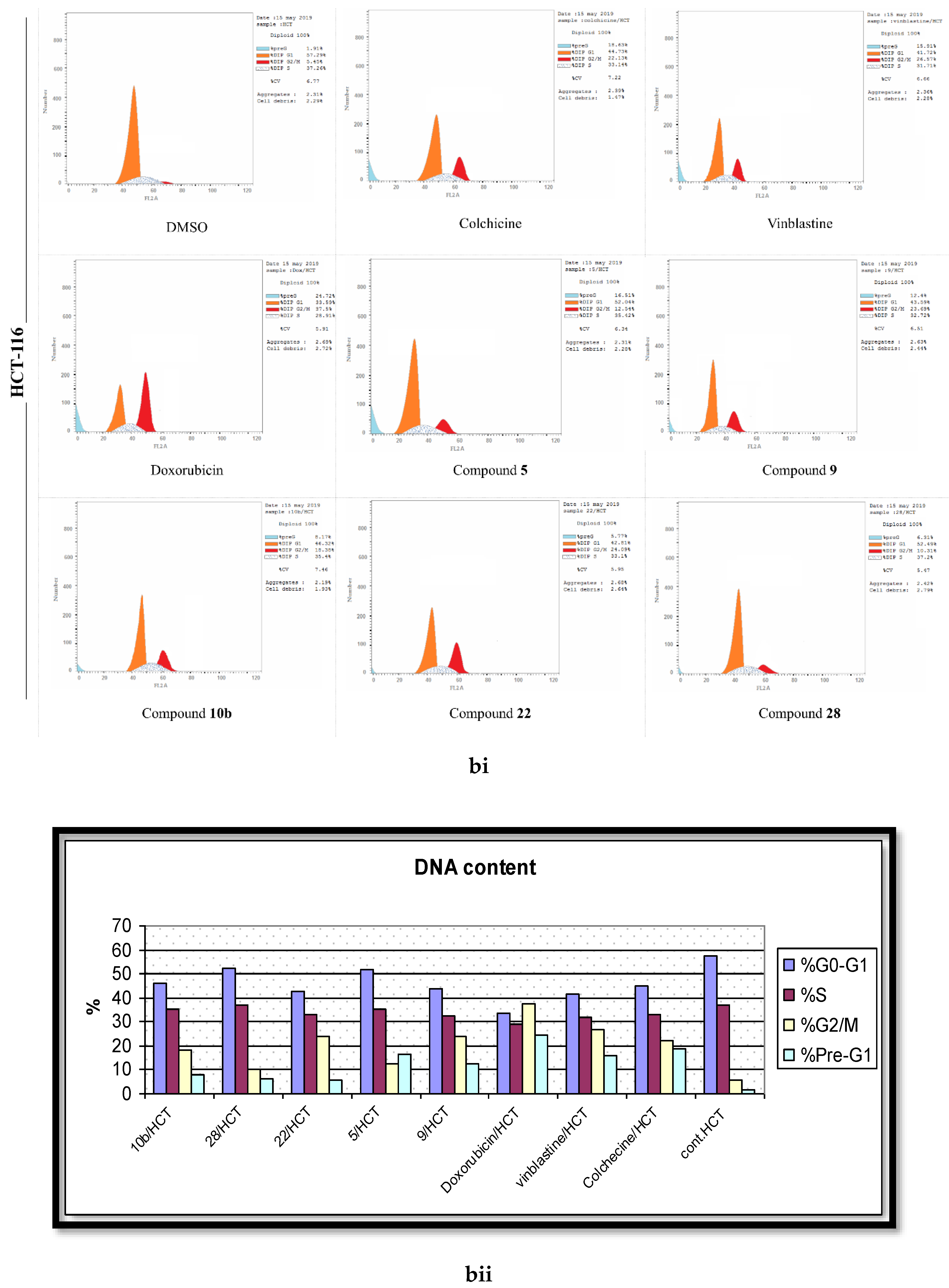
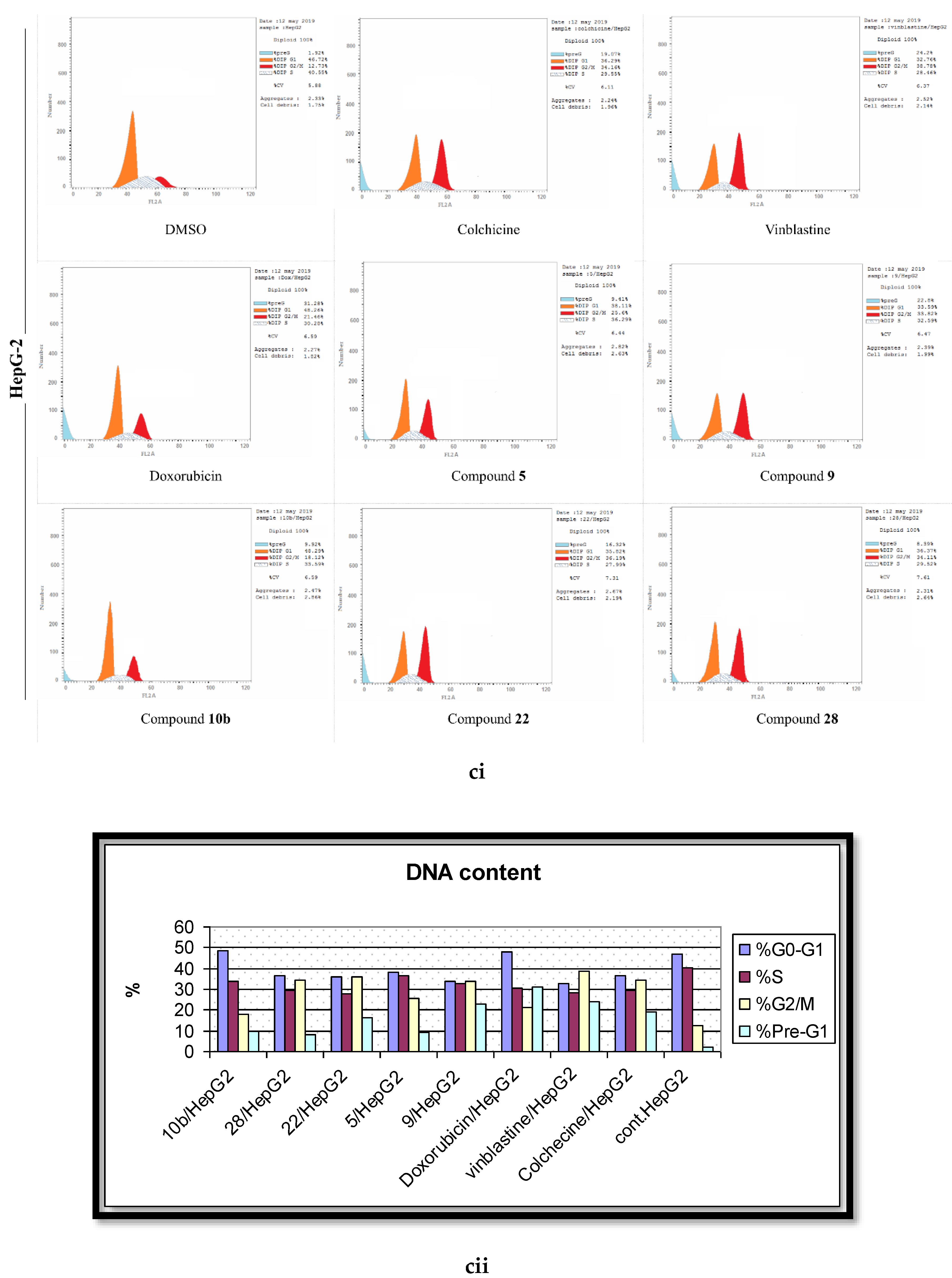
2.4. Analysis of Cell Cycle Distribution
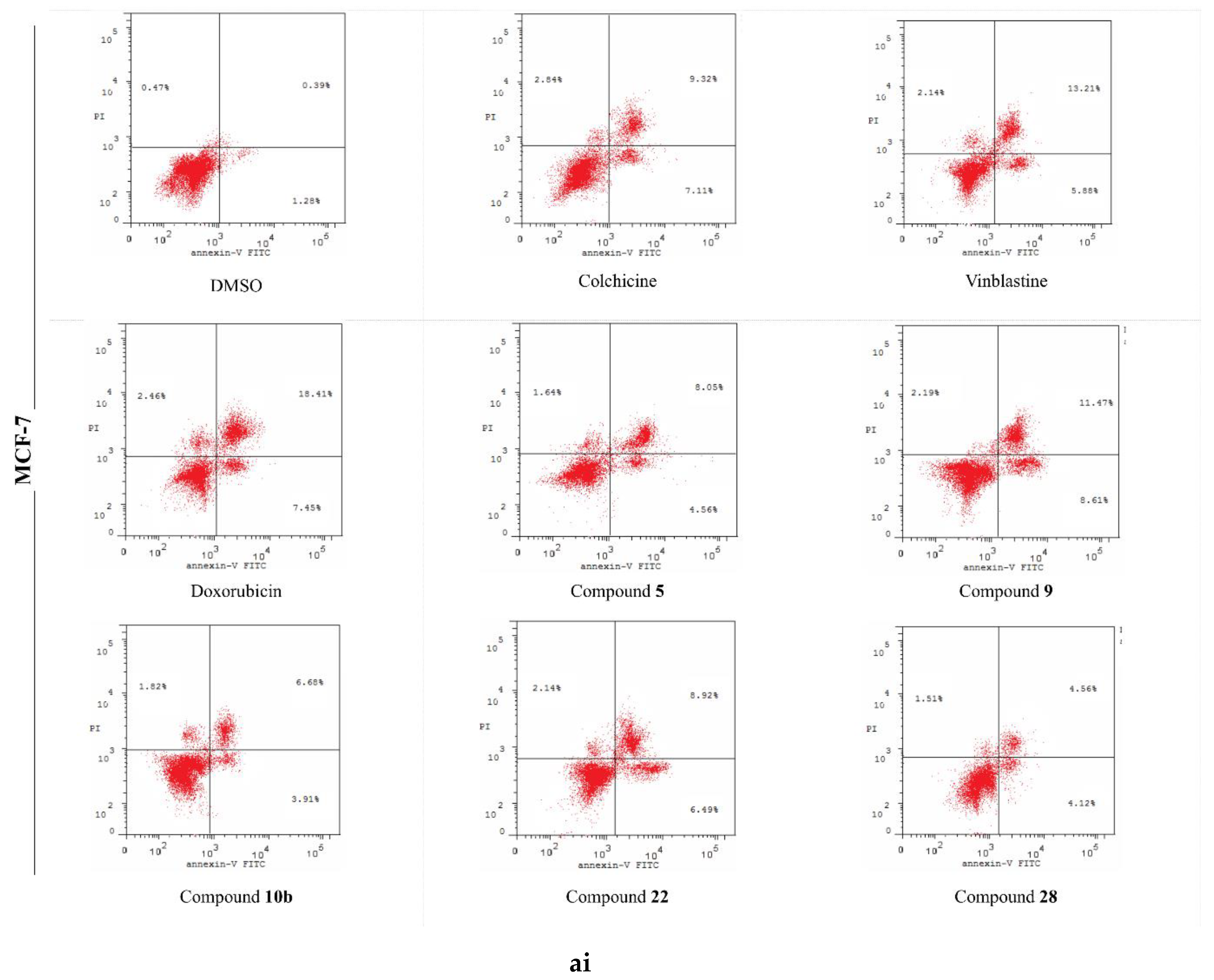
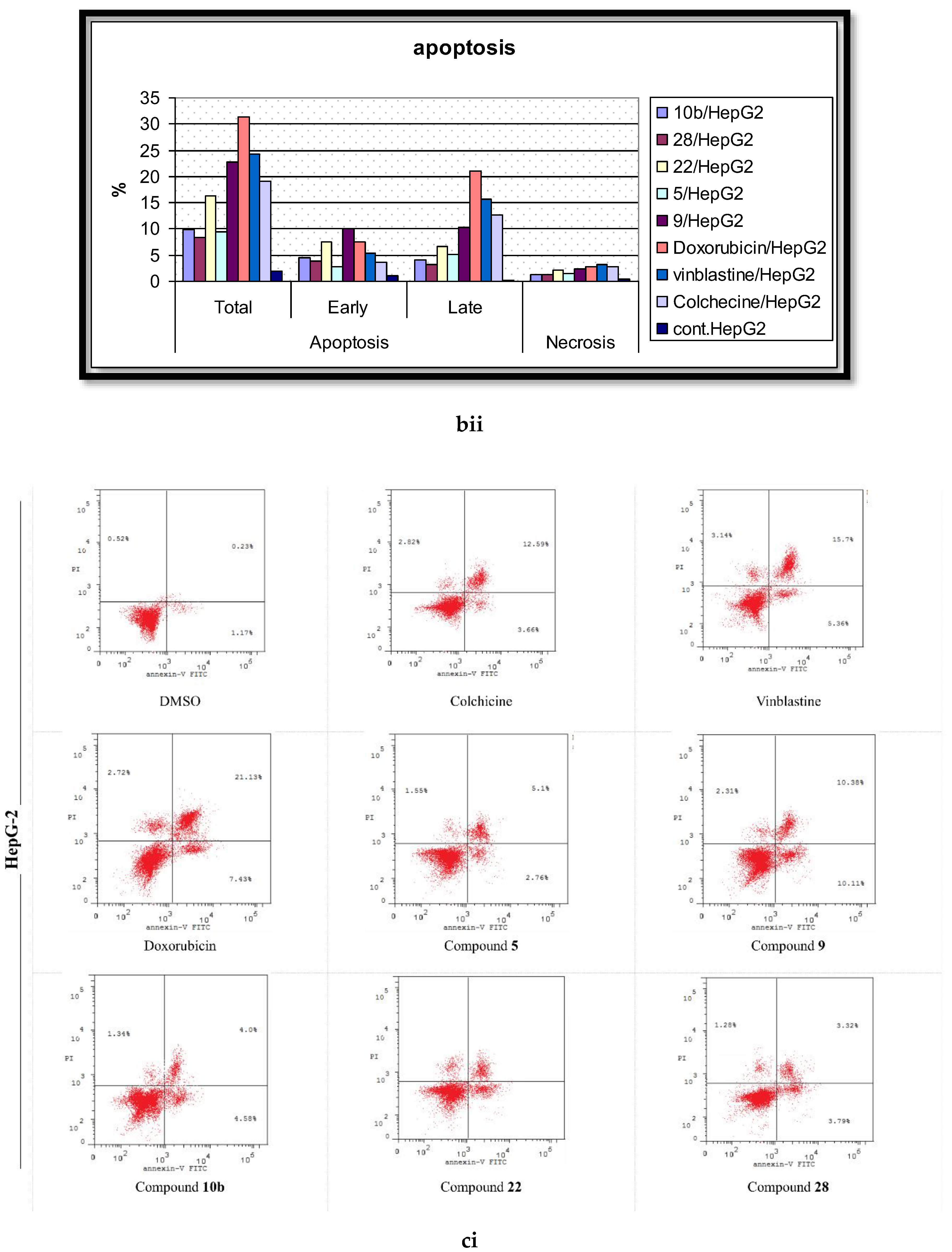
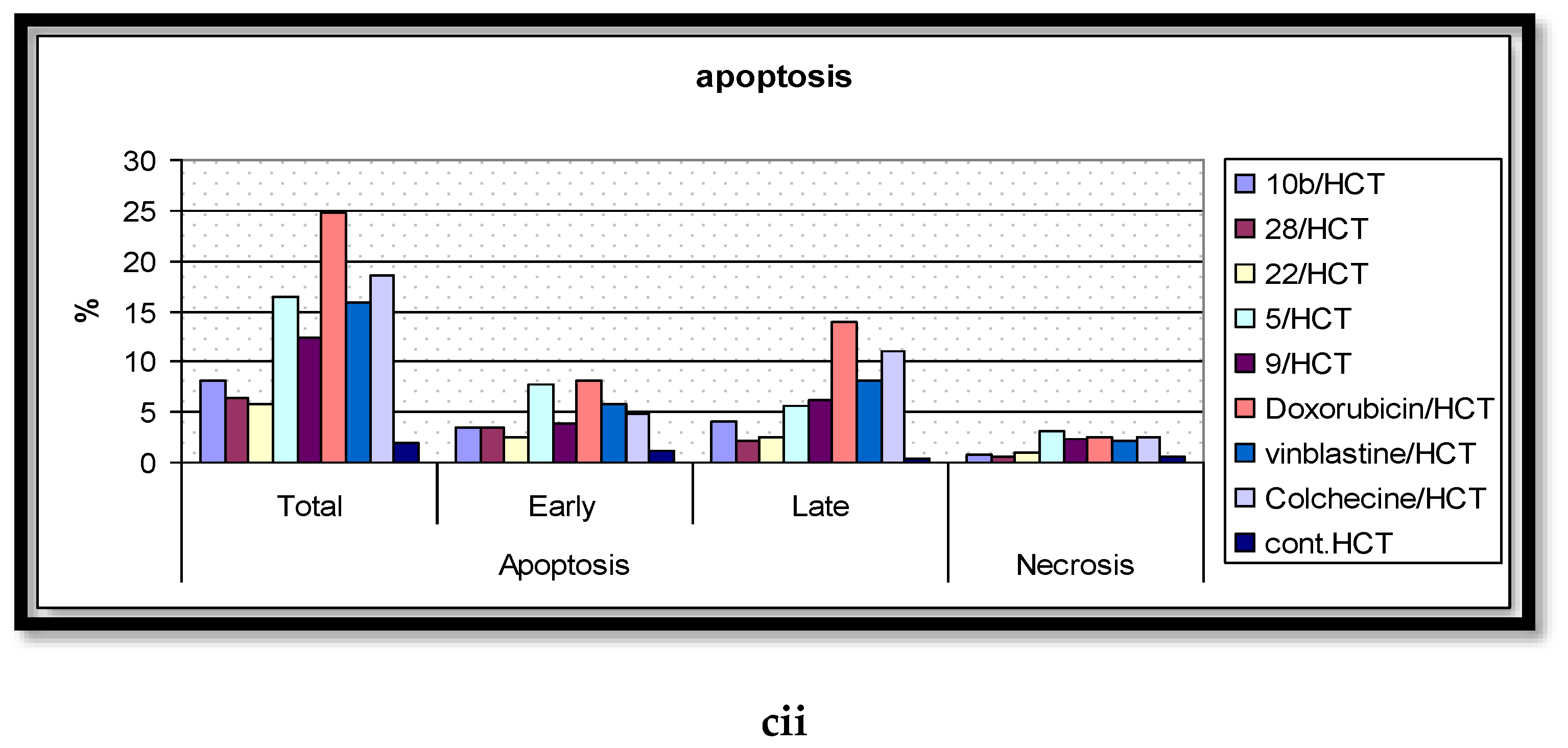
2.5. Analysis of Cell Apoptosis
3. Materials and Methods
3.1. General Methods
3.1.1. 1-[4-(4-Methyl-piperidine-1-sulfonyl)phenyl]ethanone (3)
3.1.2. (E)-3-(Dimethylamino)-1-(4-((4-methylpiperidin-1-yl)sulfonyl)phenyl)prop-2-en-1-one (4)
3.1.3. (E)-4-Methyl-1-((4-(1-(2-phenylhydrazineylidene)ethyl)phenyl)sulfonyl)piperidine (5)
3.1.4. 4-Methyl-1-[4-(1-phenyl-1H-pyrazol-3-yl)benzenesulfonyl]piperidine (6)
3.1.5. 4-[4-(4-Methyl-piperidine-1-sulfonyl)-phenyl]pyrimidin-2-ylamine (9)
3.1.6. General Procedure for Preparation of (10a, b)
3.1.7. (E)-1-(4-((4-Methylpiperidin-1-yl)sulfonyl)phenyl)-3-(p-tolylamino)prop-2-en-1-one (10a)
3.1.8. (E)-3-((4-Methoxyphenyl)amino)-1-(4-((4-methylpiperidin-1-yl)sulfon-yl)phenyl)prop-2-en-1-one (10b)
3.1.9. General Procedure for Preparation of Compounds 16, 22, 28
3.1.10. 5-[4-(4-Methyl-piperidine-1-sulfonyl)phenyl][1,2,4]triazolo[4,3-a]pyrimidine (16)
3.1.11. 7-(4-((4-Methylpiperidin-1-yl)sulfonyl)phenyl)-2-phenylpyrazolo[1,5-a]pyramidine (22)
3.1.12. 7-(4-((4-Methylpiperidin-1-yl)sulfonyl)phenyl)-2-(methylthio)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (28)
3.1.13. General Procedure for Preparation of (31a–c)
3.1.14. (E)-4-((2-Amino-7-(4-((4-methylpiperidin-1-yl)sulfonyl)phenyl)pyrazolo[1,5-a]pyrimidin-3-yl)diazenyl)phenol (31a)
3.1.15. (E)-7-(4-((4-Methylpiperidin-1-yl)sulfonyl)phenyl)-3-(p-tolyldiazenyl)pyrazolo[1,5-a]pyramidin-2-amine (31b)
3.1.16. (E)-3-((4-Chlorophenyl)diazenyl)-7-(4-((4-methylpiperidin-1-yl)sulfonyl)phenyl)pyrazolo[1,5-a]pyrimidin-2-amine (31c)
3.1.17. 5-(4-((4-Methylpiperidin-1-yl)sulfonyl)phenyl)-2-thioxo-2,3-dihydropyrido[2–d]pyrimidin-4(1H)-one (33)
3.2. Molecular Modeling
3.3. Protein Structure Preparation
3.4. Ligand Structure Preparation
3.5. Molecular Docking
3.6. Biological Screening
3.6.1. Cell Culture
3.6.2. Evaluation of Anti-Proliferative Activity
3.7. Cell Cycle Analysis
3.8. Flow Cytometry by Annexin V-FITC
3.9. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hunter, T. Signaling—2000 and Beyond. Cell 2000, 100, 113–127. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samadani, R.; Zhang, J.; Brophy, A.; Oashi, T.; Priyakumar, U.D.; Raman, E.P.; St John, F.J.; Jung, K.-Y.; Fletcher, S.; Pozharski, E.; et al. Small-molecule inhibitors of ERK-mediated immediate early gene expression and proliferation of melanoma cells expressing mutated BRaf. Biochem. J. 2015, 467, 425–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danish, M.; Bibi, A.; Gilani, K.; Raza, M.A.; Ashfaq, M.; Arshad, M.N.; Asiri, A.M.; Ayub, K. Antiradical, Antimicrobial and Enzyme Inhibition Evaluation of Sulfonamide Derived Esters; Synthesis, X-Ray Analysis and DFT Studies. J. Mol. Struct. 2019, 1175, 379–388. [Google Scholar] [CrossRef]
- Shafieyoon, P.; Mehdipour, E.; Mary, Y.S. Synthesis, characterization and biological investigation of glycine-based sulfonamide derivative and its complex: Vibration assignment, HOMO – LUMO analysis, MEP and molecular docking. J. Mol. Struct. 2019, 1181, 244–252. [Google Scholar] [CrossRef]
- Rocha, M.; Piro, O.E.; Echeverría, G.A.; Pastoriza, A.C.; Sgariglia, M.A.; Soberón, J.R.; Gil, D.M. Co(II), Ni(II) and Cu(II) ternary complexes with sulfadiazine and dimethylformamide: Synthesis, spectroscopic characterization, crystallographic study and antibacterial activity. J. Mol. Struct. 2019, 1176, 605–613. [Google Scholar] [CrossRef]
- Eren, B.; Ünal, A.; Özdemir-Koçak, F. Combined experimental and theoretical studies on the chemical and spectroscopic properties of an antimicrobial n-(Phenyl)dimethyldisulfonimide. J. Mol. Struct. 2019, 1175, 542–550. [Google Scholar] [CrossRef]
- Sunil Kumar, A.; Kudva, J.; Madan Kumar, S.; Vishwanatha, U.; Kumar, V.; Naral, D. Synthesis, characterization, crystal structure, Hirshfeld interaction and bio-evaluation studies of 4-amino quinazoline sulfonamide derivatives. J. Mol. Struct. 2018, 1167, 142–153. [Google Scholar] [CrossRef]
- Scozzafava, A.; Owa, T.; Mastrolorenzo, A.; Supuran, C. Anticancer and Antiviral Sulfonamides. Curr. Med. Chem. 2005, 10, 925–953. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Phan, V.H.G.; Lee, D.S.; Thambi, T.; Huynh, D.P. Bioresorbable pH- and temperature-responsive injectable hydrogels-incorporating electrosprayed particles for the sustained release of insulin. Polym. Degrad. Stab. 2019, 162, 36–46. [Google Scholar] [CrossRef]
- Ghareb, N.; El-Sayed, N.M.; Abdelhameed, R.; Yamada, K.; Elgawish, M.S. Toward a treatment of diabesity: Rational design, synthesis and biological evaluation of benzene-sulfonamide derivatives as a new class of PTP-1B inhibitors. Bioorg. Chem. 2019, 86, 322–338. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, Z.; Morahan, G.; Arooj, M.; Sobolev, A.N.; Hameed, S. Synthesis of new arylsulfonylspiroimidazolidine-2′,4′-diones and study of their effect on stimulation of insulin release from MIN6 cell line, inhibition of human aldose reductase, sorbitol accumulations in various tissues and oxidative stress. Eur. J. Med. Chem. 2019, 168, 154–175. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Luo, G. Facile synthesis of acyl sulfonamides from carboxyic acids using the Mukaiyama reagent. Tetrahedron Lett. 2019, 60, 268–271. [Google Scholar] [CrossRef]
- Zhang, J.; Li, J.; Su, H.; Zhao, Y.; Zeng, X.; Hu, M.; Xiao, W.; Mao, X. H-bonding effect of oxyanions enhanced photocatalytic degradation of sulfonamides by g-C3N4 in aqueous solution. J. Hazard. Mater. 2019, 366, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Ozmen Ozgun, D.; Gul, H.I.; Yamali, C.; Sakagami, H.; Gulcin, I.; Sukuroglu, M.; Supuran, C.T. Synthesis and bioactivities of pyrazoline benzensulfonamides as carbonic anhydrase and acetylcholinesterase inhibitors with low cytotoxicity. Bioorg. Chem. 2019, 84, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Soltani, S.; Magri, P.; Rogalski, M.; Kadri, M. Charge-transfer complexes of hypoglycemic sulfonamide with π-acceptors: Experimental and DFT-TDDFT studies. J. Mol. Struct. 2019, 1175, 105–116. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.M.; Angeli, A.; El-Azab, A.S.; Hammouda, M.E.A.; El-Sherbeny, M.A.; Supuran, C.T. Synthesis and anti-inflammatory activity of sulfonamides and carboxylates incorporating trimellitimides: Dual cyclooxygenase/carbonic anhydrase inhibitory actions. Bioorg. Chem. 2019, 84, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, Z.; Sławiński, J.; Saczewski, F.; Innocenti, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis and inhibition of the human cytosolic isozymes I and II and transmembrane isozymes IX, XII (cancer-associated) and XIV with 4-substituted 3-pyridinesulfonamides. Eur. J. Med. Chem. 2010, 45, 2396–2404. [Google Scholar] [CrossRef] [PubMed]
- Renzi, G.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Topical sulfonamide antiglaucoma agents incorporating secondary amine moieties. Bioorg. Med. Chem. Lett. 2000, 10, 673–676. [Google Scholar] [CrossRef]
- Salehi, M.; Kubicki, M.; Galini, M.; Jafari, M.; Malekshah, R.E. Synthesis, characterization and crystal structures of two novel sulfa drug Schiff base ligands derived sulfonamide and molecular docking study. J. Mol. Struct. 2019, 1180, 595–602. [Google Scholar] [CrossRef]
- Bashandy, M.S.; Alsaid, M.S.; Arafa, R.K.; Ghorab, M.M. Design, synthesis and molecular docking of novel n,n-dimethylbenzenesulfonamide derivatives as potential antiproliferative agents. J. Enzyme Inhib. Med. Chem. 2014, 29, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Ghorab, M.M.; Ceruso, M.; Alsaid, M.S.; Nissan, Y.M.; Arafa, R.K.; Supuran, C.T. Novel sulfonamides bearing pyrrole and pyrrolopyrimidine moieties as carbonic anhydrase inhibitors: Synthesis, cytotoxic activity and molecular modeling. Eur. J. Med. Chem. 2014, 87, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Bhavanarushi, S.; Kanakaiah, V.; Bharath, G.; Gangagnirao, A.; Vatsala Rani, J. Synthesis and antibacterial activity of 4,4′-(aryl or alkyl methylene)-bis(1H-pyrazol-5-ol) derivatives. Med. Chem. Res. 2014, 23, 158–167. [Google Scholar] [CrossRef]
- Nimbarte, V.D.; Murtuza, H.; Phaniraj, S.; Shrivastava, S.; Naidu, V.G.M.; Satheesh Kumar, N.; Atcha, K.R. Design, synthesis and biological evaluation of 4-(1- (4(sulphanilamide) phenyl)-3-(methyl)-1H-pyrazol-5-yl)dine urea and n-acyl derivatives as a soluble epoxide hydrolase inhibitors. Med. Chem. Res. 2014, 23, 2178–2197. [Google Scholar] [CrossRef]
- Rizk, H.F.; Ibrahim, S.A.; El-Borai, M.A. Synthesis, fastness properties, color assessment and antimicrobial activity of some azo reactive dyes having pyrazole moiety. Dye. Pigment. 2015, 112, 86–92. [Google Scholar] [CrossRef]
- Surendra Kumar, R.; Arif, I.A.; Ahamed, A.; Idhayadhulla, A. Anti-inflammatory and antimicrobial activities of novel pyrazole analogues. Saudi. J. Biol. Sci. 2016, 23, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Sribalan, R.; Banuppriya, G.; Kirubavathi, M.; Jayachitra, A.; Padmini, V. Multiple biological activities and molecular docking studies of newly synthesized 3-(pyridin-4-yl)-1H-pyrazole-5-carboxamide chalcone hybrids. Bioorg. Med. Chem. Lett. 2016, 26, 5624–5630. [Google Scholar] [CrossRef] [PubMed]
- B’Bhatt, H.; Sharma, S. 2-(5-Chlorobenzo[d]thiazol-2-ylimino)thiazolidin-4-one derivatives as an antimicrobial agent. Arab. J. Chem. 2017, 10, S531–S538. [Google Scholar] [CrossRef] [Green Version]
- Nagamallu, R.; Srinivasan, B.; Ningappa, M.B.; Kariyappa, A.K. Synthesis of novel coumarin appended bis(formylpyrazole) derivatives: Studies on their antimicrobial and antioxidant activities. Bioorg. Med. Chem. Lett. 2016, 26, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Saddik, A.A.; Kamal El-Dean, A.M.; El-Said, W.A.; Hassan, K.M.; Abbady, M.S. Synthesis, Antimicrobial, and Anticancer Activities of a New Series of Thieno[2,3–d] Pyrimidine Derivatives. J. Heterocycl. Chem. 2018, 55, 2111–2122. [Google Scholar] [CrossRef]
- Anupama, B.; Dinda, S.C.; Prasad, Y.R.; Rao, A.V. Synthesis and antimicrobial activity of some new 1,3,5-trisubstituted-2-pyrazolines. Int. J. Res. Pharm. Chem. 2012, 2, 249–253. [Google Scholar]
- Hemdan, M.M.; Abd El-Mawgoude, H.K. Synthesis and Antimicrobial Evaluation of Thieno[2,3–d]pyrimidine, Thieno[2′,3′:4,5]pyrimido[1,2–a][1,3,5]triazine, Thieno[2,3–d]-1,3-thiazine and 1, 2,4-Triazole Systems. Chem. Pharm. 2015, 63, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Khalafallah, A.K.; Ahmed, M.A. Synthesis and antimicrobial activity of some novel S-glucosides of 4-amino- pyrimidine-2-(1H )-thione derivatives. Curr. Sci. Perspect. 2018, 4, 30–36. [Google Scholar]
- Sirakanyan, S.N.; Hakobyan, E.K.; Hovakimyan, A.A.; Geronikaki, A.; Petrou, A.; Spinelli, D.; Kartsev, V.G. Synthesis and antimicrobial activity of new amino derivatives of pyrano[4′’,3′’:4′,5′]pyrido[3′,2′:4,5]- [3′,2′:4,5]thieno[3,2-d]pyrimidine. An. Acad. Bras. Cienc. 2018, 90, 1043–1057. [Google Scholar] [CrossRef] [PubMed]
- Sankappa Rai, U.; Isloor, A.M.; Shetty, P.; Pai, K.S.R.; Fun, H.K. Synthesis and in vitro biological evaluation of new pyrazole chalcones and heterocyclic diamides as potential anticancer agents. Arab. J. Chem. 2015, 8, 317–321. [Google Scholar] [CrossRef]
- Mohareb, R.M.; Abdallah, A.E.M.; Abdelaziz, M.A. New approaches for the synthesis of pyrazole, thiophene, thieno[2–b]pyridine, and thiazole derivatives together with their anti-tumor evaluations. Med. Chem. Res. 2014, 23, 564–579. [Google Scholar] [CrossRef]
- Nakao, S.; Mabuchi, M.; Shimizu, T.; Itoh, Y.; Takeuchi, Y.; Ueda, M.; Mizuno, H.; Shigi, N.; Ohshio, I.; Jinguji, K.; et al. Design and synthesis of prostate cancer antigen-1 (PCA-1/ALKBH3) inhibitors as anti-prostate cancer drugs. Bioorg. Med. Chem. Lett. 2014, 24, 1071–1074. [Google Scholar] [CrossRef] [PubMed]
- Aydın, S.; Kaushik-Basu, N.; Özbaş-Turan, S.; Akbuğa, J.; Mega Tiber, P.; Orun, O.; Gurukumar, K.R.; Basu, A.; Güniz Küçükgüzel, Ş. Synthesis of 1-aroyl-3,5-dimethyl-1H-pyrazoles as Anti-HCV and Anticancer Agents. Lett. Drug Des. Discov. 2014, 11, 121–131. [Google Scholar] [CrossRef]
- Ren, J.; Wang, S.; Ni, H.; Yao, R.; Liao, C.; Ruan, B. Synthesis, Characterization and Antitumor Activity of Novel Ferrocene-Based Amides Bearing Pyrazolyl Moiety. J. Inorg. Organomet. Polym. Mater. 2015, 25, 419–426. [Google Scholar] [CrossRef]
- Kamal, A.; Shaik, A.B.; Polepalli, S.; Santosh Reddy, V.; Bharath Kumar, G.; Gupta, S.; Rama Krishna, K.V.S.; Nagabhushana, A.; Mishra, R.K.; Jain, N. Pyrazole-oxadiazole conjugates: Synthesis, antiproliferative activity and inhibition of tubulin polymerization. Org. Biomol. Chem. 2014, 12, 7993–8007. [Google Scholar] [CrossRef] [PubMed]
- Halawa, A.H.; Fouda, A.M.; Al-Dies, A.M.; El-Agrody, A.M. Synthesis, Biological Evaluation and Molecular Docking Studies of 4H-benzo[h]chromenes, 7H-benzo[h]chromeno[2,3–d]pyrimidines as Antitumor. Lett. Drug Des. Discov. 2016, 13, 77–88. [Google Scholar] [CrossRef]
- El-Agrody, A.M.; Halawa, A.H.; Fouda, A.M.; Al-Dies, A.A.M. The anti-proliferative activity of novel 4H-benzo[h]chromenes, 7H-benzo[h]chromeno[2,3–d]pyrimidines and the structure–activity relationships of the 2-, 3-positions and fused rings at the 2, 3-positions. J. Saudi Chem. Soc. 2017, 21, 82–90. [Google Scholar] [CrossRef]
- El-Agrody, A.M.; Fouda, A.M.; Al-Dies, A.A.M. Studies on the synthesis, in vitro antitumor activity of 4H-benzo[h]chromene, 7H-benzo[h]chromene[2,3–d]pyrimidine derivatives and structure-Activity relationships of the 2-,3- and 2,3-positions. Med. Chem. Res. 2014, 23, 3187–3199. [Google Scholar] [CrossRef]
- Okasha, R.M.; Alblewi, F.F.; Afifi, T.H.; Naqvi, A.; Fouda, A.M.; Al-Dies, A.A.M.; El-Agrody, A.M.; Belmont, P.; Bunce, R.A. Design of new Benzo[h]chromene derivatives: Antitumor activities and structure-activity relationships of the 2,3-Positions and fused rings at the 2,3-positions. Molecules 2017, 22, 479. [Google Scholar] [CrossRef] [PubMed]
- El-Sehemi, A.G.; Bondock, S.; Ammar, Y.A. Transformations of naproxen into pyrazolecarboxamides: Search for potent anti-inflammatory, analgesic and ulcerogenic agents. Med. Chem. Res. 2014, 23, 827–838. [Google Scholar] [CrossRef]
- Antre, R.V.; Cendilkumar, A.; Goli, D.; Andhale, G.S.; Oswal, R.J. Microwave assisted synthesis of novel pyrazolone derivatives attached to a pyrimidine moiety and evaluation of their anti-inflammatory, analgesic and antipyretic activities. Saudi Pharm. J. 2011, 19, 233–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagle, A.S.; Khare, S.; Kumar, A.B.; Supek, F.; Buchynskyy, A.; Mathison, C.J.N.; Chennamaneni, N.K.; Pendem, N.; Buckner, F.S.; Gelb, M.H.; et al. Recent developments in drug discovery for leishmaniasis and human african trypanosomiasis. Chem. Rev. 2014, 114, 11305–11347. [Google Scholar] [CrossRef] [PubMed]
- Bekhit, A.A.; Hassan, A.M.M.; Abd El Razik, H.A.; El-Miligy, M.M.M.; El-Agroudy, E.J.; Bekhit, A.E.D.A. New heterocyclic hybrids of pyrazole and its bioisosteres: Design, synthesis and biological evaluation as dual acting antimalarial-antileishmanial agents. Eur. J. Med. Chem. 2015, 94, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Suryawanshi, S.N.; Kumar, S.; Shivahare, R.; Pandey, S.; Tiwari, A.; Gupta, S. Design, synthesis and biological evaluation of aryl pyrimidine derivatives as potential leishmanicidal agents. Bioorg. Med. Chem. Lett. 2013, 23, 5235–5238. [Google Scholar] [CrossRef] [PubMed]
- Desai, n.C.; Kotadiya, G.M.; Trivedi, A.R. Studies on molecular properties prediction, antitubercular and antimicrobial activities of novel quinoline based pyrimidine motifs. Bioorg. Med. Chem. Lett. 2014, 24, 3126–3130. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.B. Preparation of 2H-1,2,3-benzothiadiazine 1,1-dioxides, 11H-11,11a-dihydrobenzimidazo[1–b][1,2]benzisothiazole 5,5-dioxides, 6H-dibenzo[c,g][1,2,5]thiadiazocine 5,5-dioxides and 5H-dibenzo[c,g][1,2,6]thiadiazocine 6,6-dioxides. J. Heterocycl. Chem. 1968, 5, 453–459. [Google Scholar] [CrossRef]
- Dawood, K.M. Synthesis of Spiro-pyrazole-3,3′-thiopyrano[2,3–b]pyridines and Azolo[a]pyrido[2′,3′:5,6]- thiopyrano[3,4–d]pyrimidines as New Ring Systems with Antifungal and Antibacterial Activities. J. Heterocycl. Chem. 2005, 42, 221–225. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Y.; Chen, S.; Chen, B.; Jia, Y.; Zeng, Z. Synthesis and Biological Activities of Schiff Bases of 3-amino-1H-1,2,4-triazole. Chin. J. Chem. 2009, 27, 949–952. [Google Scholar] [CrossRef]
- Cocconcelli, G.; Ghiron, C.; Haydar, S.; Micco, I.; Zanaletti, R. Synthesis of novel4-fluoro-2H-pyrazol- 3-ylamines. Synth. Commun. 2010, 40, 2547–2555. [Google Scholar] [CrossRef]
- Maftei, C.V.; Fodor, E.; Jones, P.G.; Daniliuc, C.G.; Franz, M.H.; Kelter, G.; Fiebig, H.H.; Tamm, M.; Neda, I. Novel 1,2,4-Oxadiazoles and Trifluoromethylpyridines Related to Natural Products: Synthesis, Structural Analysis and Investigation of their Antitumor Activity. Tetrahedron 2016, 72, 1185–1199. [Google Scholar] [CrossRef]
- Ho, Y.W. 5-(1-Pyrrolyl)-2-phenylthieno[2,3–d]pyrimidine as Building Block in Heterocyclic Synthesis: Novel Synthesis of Some Pyrazoles, Pyrimidines, Imidazo[1,5–a]pyrimidines, Pyrazolo[1,5-a]pyrimidines, Pyrido(pyrimido)pyrazolo[1,5-a]pyrimidines, 1,2,4-Triazolo[1,5-a]pyrimidine and a 1,2,3,4-Tetrazolo[1,5-a] 1,2,3,4-Tetrazolo[1,5-a]pyrimidine Derivative. J. Chin. Chem. Soc. 2007, 54, 1075–1085. [Google Scholar] [CrossRef]
- Wen, L.R.; Wang, S.W.; Li, M.; Yang, H.Z. Reaction of enaminones with aminopyrazoles: Synthesis, structures and bioactivities of 7-aryl-3-cyano-2-substituted pyrazolo[1,5-a]pyrimidines. Chin. J. Chem. 2005, 23, 1231–1235. [Google Scholar] [CrossRef]
- Rangnekar, D.W.; Puro, S.S. Synthesis and dyeing characteristics of new2-methyl-3-arylazo-6-phenyl- 7-amino- and 7-acetamido-pyrazolo- [1,5-a]pyrimidines. Indian, J. Fibre Text. 1990, 15, 37–38. [Google Scholar]
- Vajrabhaya, L.; Korsuwannawong, S. Cytotoxicity evaluation of a Thai he rb using tetrazolium (MTT) and sulforhodamine B (SRB) assays. J. Anal. Sci. Technol. 2018, 9, 1–6. [Google Scholar] [CrossRef]
- Roskoski, R. Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2019, 142, 151–168. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munshi, A.; Ramesh, R. Mitogen-Activated Protein Kinases and Their Role in Radiation Response. Genes & Cancer 2013, 4, 401–408. [Google Scholar] [CrossRef]
- Salaroglio, I.C.; Mungo, E.; Gazzano, E.; Kopecka, J.; Riganti, C. ERK is a Pivotal Player of Chemo-Immune-Resistance in Cancer. Int. J. Mol. Sci. 2019, 20, 2505. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) a | ||
|---|---|---|---|
| MCF-7 | HCT-116 | HepG-2 | |
| 3 | 100 ± 0.12 | 85.4 ± 0.03 | 84.52 ± 0.12 |
| 4 | 27.05 ± 0.35 | 25.44 ± 0.16 | 26.27 ± 0.5 |
| 5 | 9.96 ± 0.2 | 6.92 ± 0.11 | 2.56 ± 0.14 |
| 6 | 22.28 ± 0.13 | 18.56 ± 0.12 | 16.78 ± 0.11 |
| 9 | 10.43 ± 0.12 | 10.14 ± 0.05 | 4.36 ± 0.02 |
| 10a | 58.97 ± 0.3 | 48.48 ± 0.6 | 36.94 ± 0.1 |
| 10b | 9.89 ± 0.4 | 7.09 ± 0.1 | 5.72 ± 0.12 |
| 16 | 100 ± 0.13 | 97.22 ± 0.12 | 97.05 ± 0.11 |
| 22 | 7.84 ± 0.02 | 7.28 ± 0.1 | 2.96 ± 0.12 |
| 28 | 8.19 ± 0.03 | 5.38 ± 0.2 | 4.21 ± 0.01 |
| 31a | 14.69 ± 0.06 | 10.37 ± 0.18 | 9.36 ± 0.16 |
| 31b | 45.75 ± 0.1 | 42.08 ± 0.04 | 35.23 ± 0.02 |
| 31c | 57.61 ± 0.11 | 42.58 ± 0.13 | 31.19 ± 0.6 |
| 33 | 15.62 ± 0.12 | 14.07 ± 0.13 | 14.41 ± 0.11 |
| Vinblastine | 5.83 ± 0.13 | 3.2 ± 0.09 | 7.35 ± 0.42 |
| Doxorubicin | 8.19 ± 0.72 | 6.74 ± 0.68 | 7.52 ± 0.51 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halawa, A.H.; Eskandrani, A.A.; Elgammal, W.E.; Hassan, S.M.; Hassan, A.H.; Ebrahim, H.Y.; Mehany, A.B.M.; El-Agrody, A.M.; Okasha, R.M. Rational Design and Synthesis of Diverse Pyrimidine Molecules Bearing Sulfonamide Moiety as Novel ERK Inhibitors. Int. J. Mol. Sci. 2019, 20, 5592. https://doi.org/10.3390/ijms20225592
Halawa AH, Eskandrani AA, Elgammal WE, Hassan SM, Hassan AH, Ebrahim HY, Mehany ABM, El-Agrody AM, Okasha RM. Rational Design and Synthesis of Diverse Pyrimidine Molecules Bearing Sulfonamide Moiety as Novel ERK Inhibitors. International Journal of Molecular Sciences. 2019; 20(22):5592. https://doi.org/10.3390/ijms20225592
Chicago/Turabian StyleHalawa, Ahmed H., Areej A. Eskandrani, Walid E. Elgammal, Saber M. Hassan, Ahmed H. Hassan, Hassan Y. Ebrahim, Ahmed B. M. Mehany, Ahmed M. El-Agrody, and Rawda M. Okasha. 2019. "Rational Design and Synthesis of Diverse Pyrimidine Molecules Bearing Sulfonamide Moiety as Novel ERK Inhibitors" International Journal of Molecular Sciences 20, no. 22: 5592. https://doi.org/10.3390/ijms20225592
APA StyleHalawa, A. H., Eskandrani, A. A., Elgammal, W. E., Hassan, S. M., Hassan, A. H., Ebrahim, H. Y., Mehany, A. B. M., El-Agrody, A. M., & Okasha, R. M. (2019). Rational Design and Synthesis of Diverse Pyrimidine Molecules Bearing Sulfonamide Moiety as Novel ERK Inhibitors. International Journal of Molecular Sciences, 20(22), 5592. https://doi.org/10.3390/ijms20225592