Riding the Wave: The SINE-Specific V Highly-Conserved Domain Spread into Mammalian Genomes Exploiting the Replication Burst of the MER6 DNA Transposon



Abstract
:
1. Introduction
2. Results
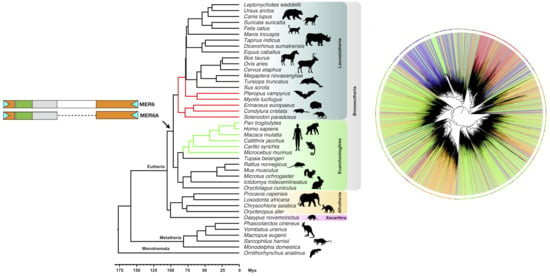
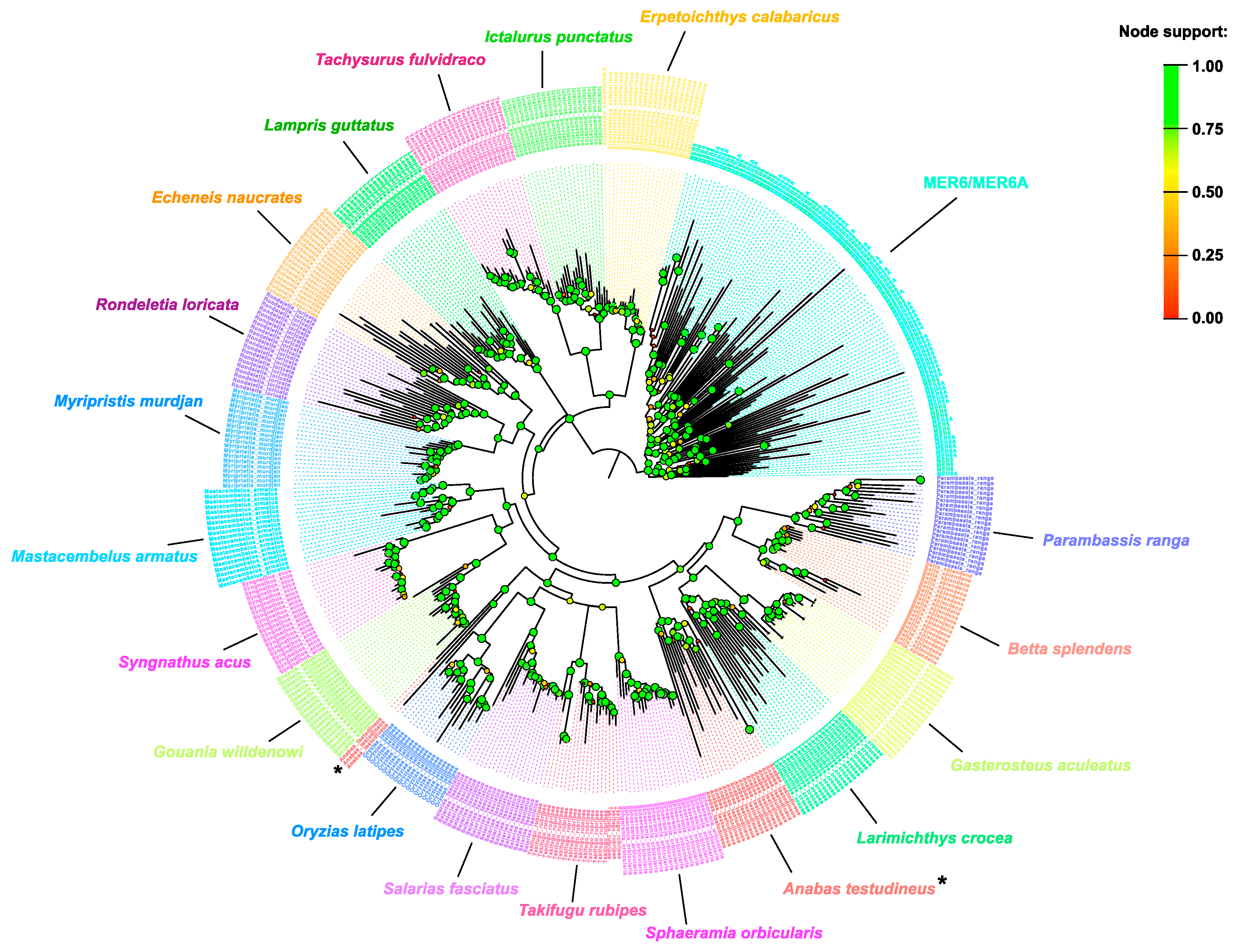
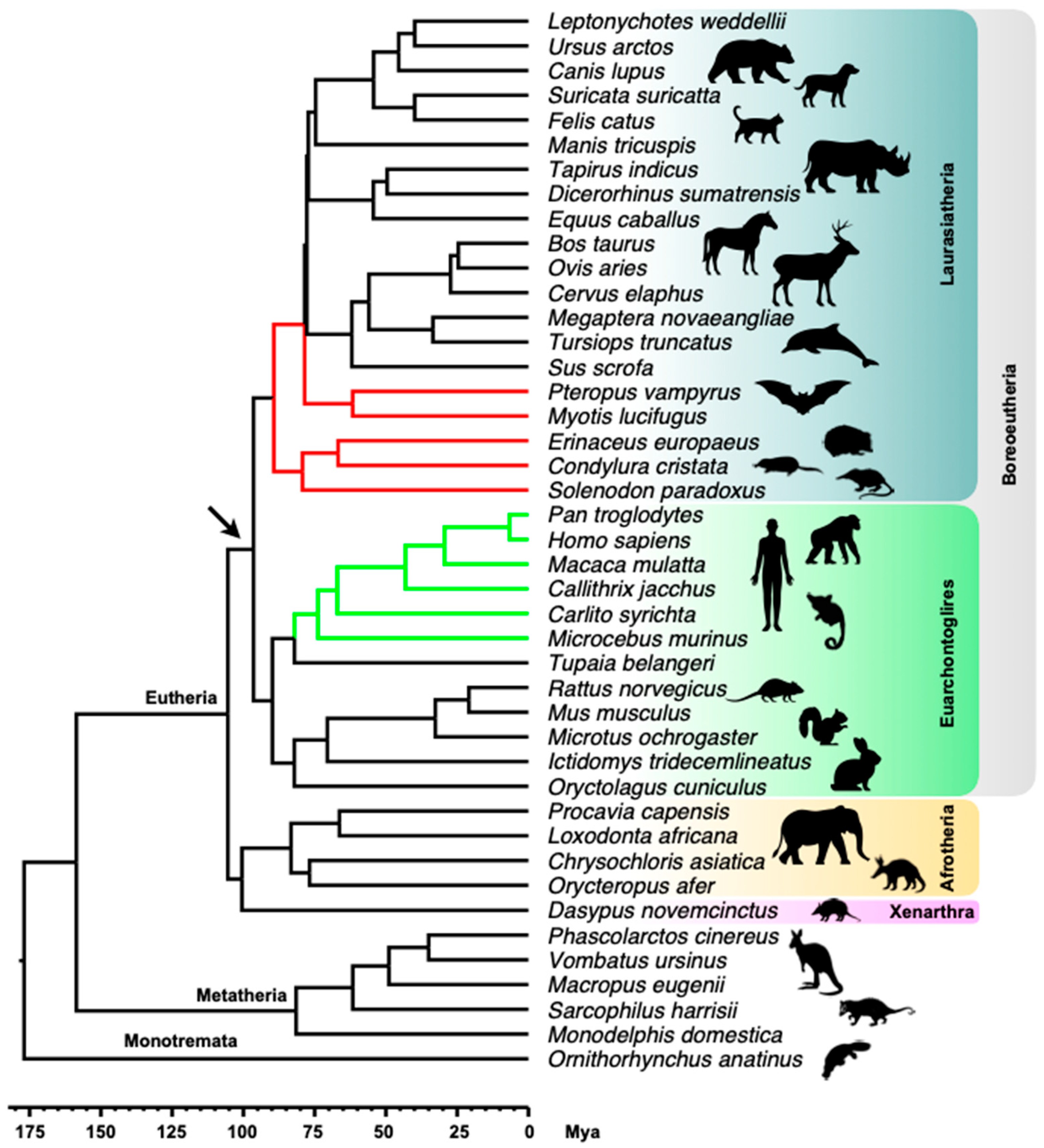
2.1. MER6/MER6A Taxonomic Distribution
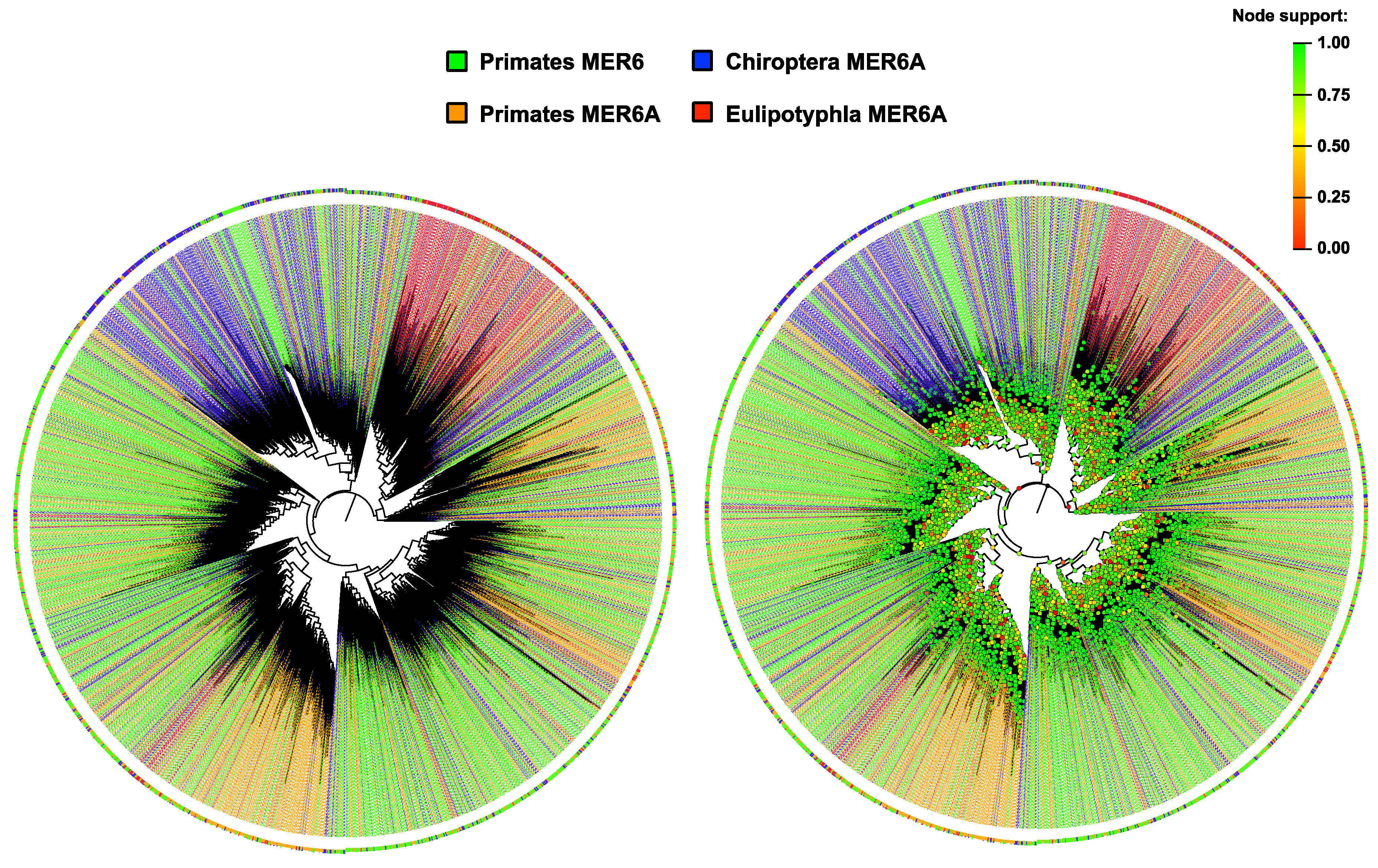
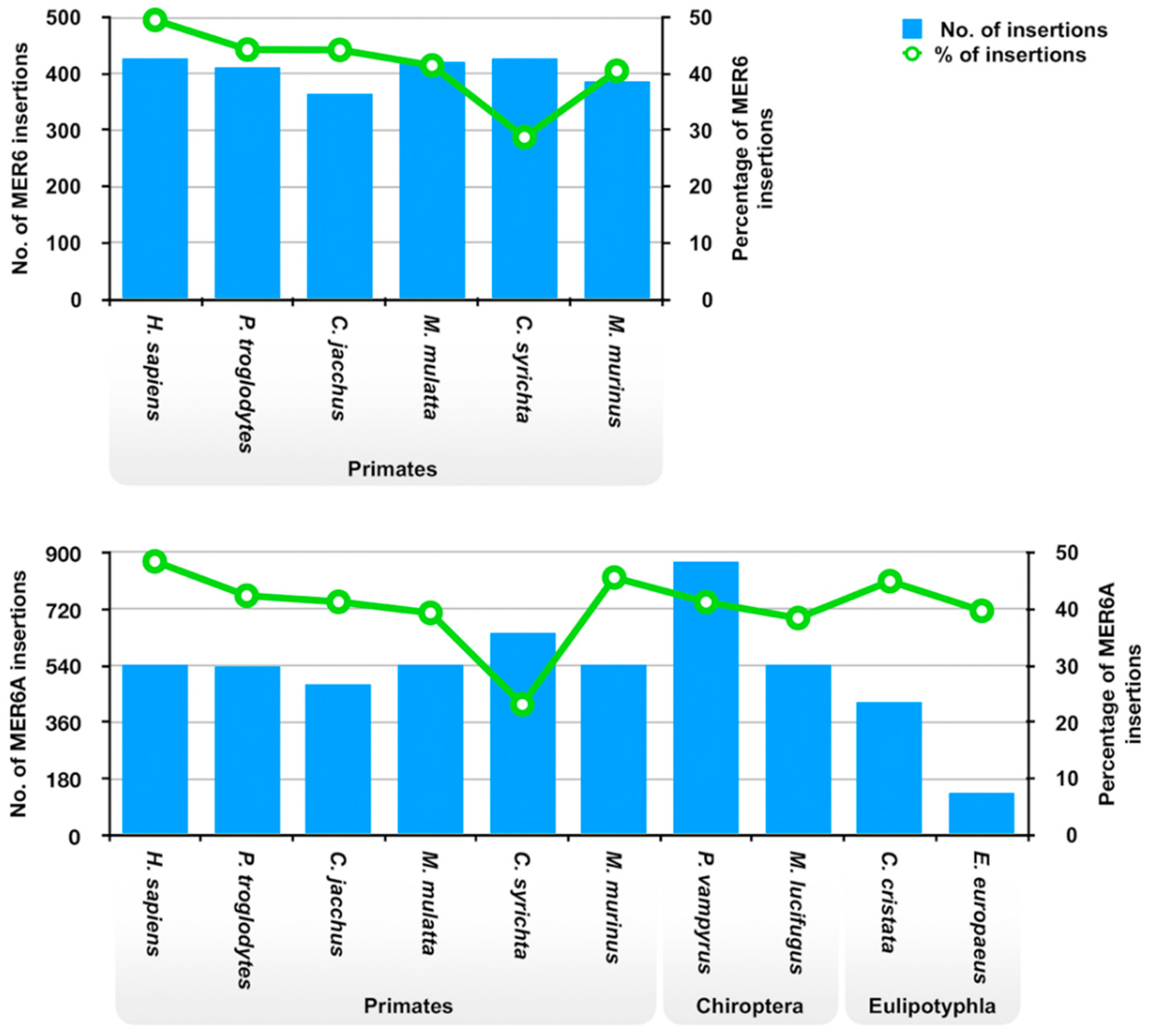
2.2. MER6 and MER6A Copy Number and Nucleotide Variability
2.3. MER6 Occurrence within Genic Regions
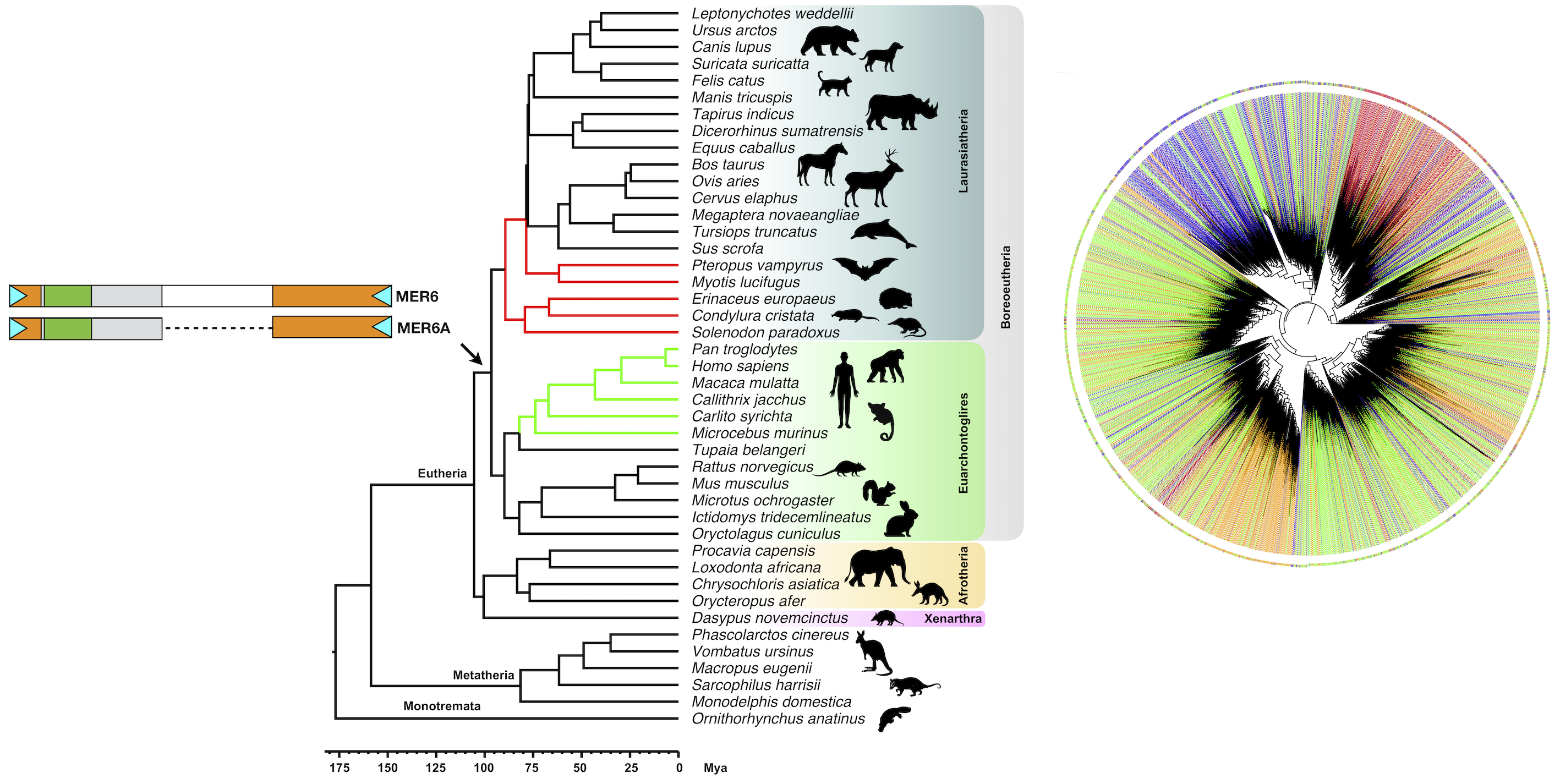
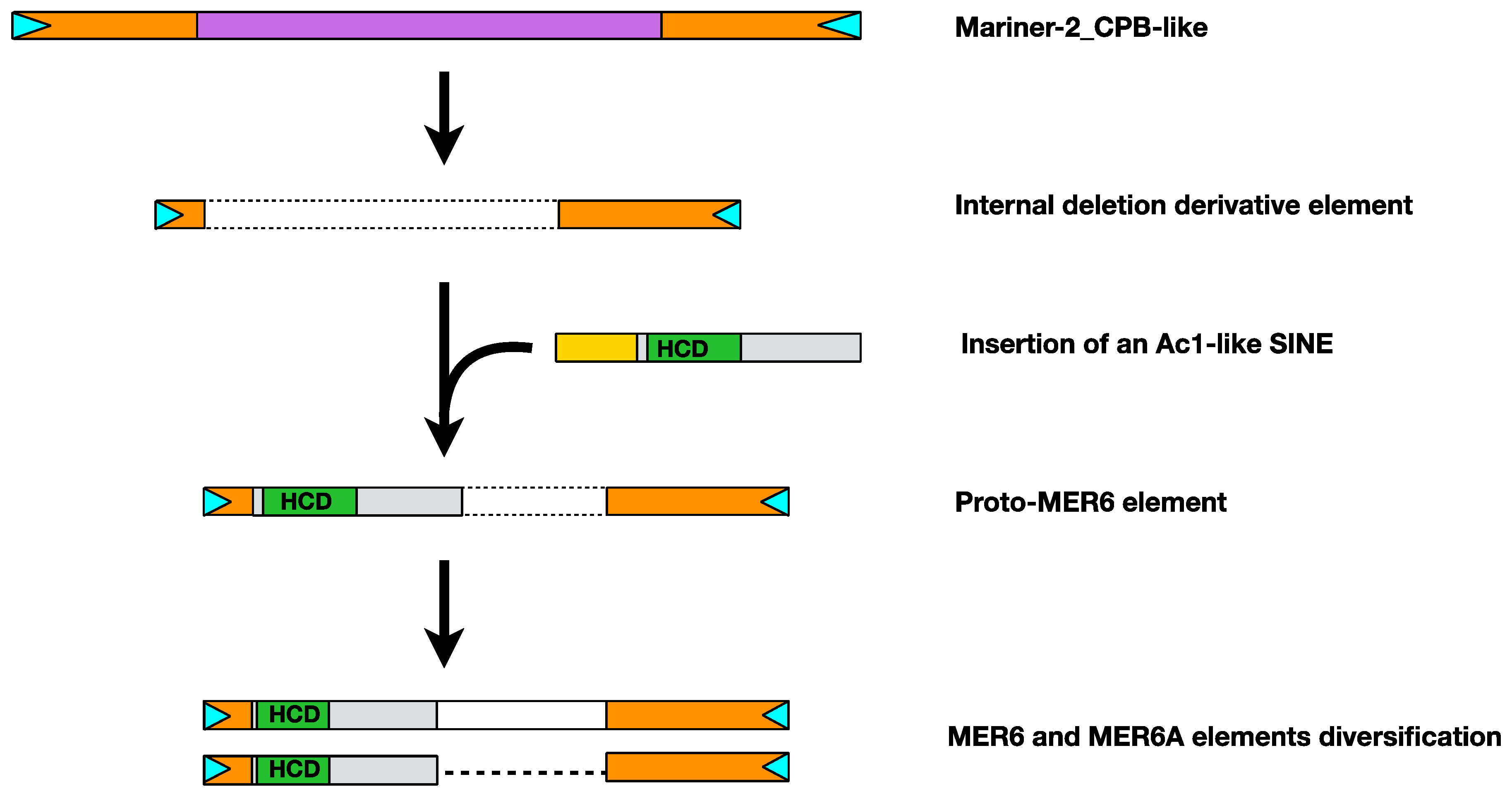
2.4. MER6 Composite Structure
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TE | Transposable elements |
| SINE | Short interspersed elements |
| MITE | Miniature inverted-repeat transposable element |
| HCD | Highly conserved domain |
| mRNA | Messenger RNA |
| miRNA | Micro-RNA |
| lncRNA | Long non-coding RNA |
| tRNA | Transfer RNA |
| UTR | Untranslated region |
| GTR | General Time Reversible |
References
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Goburnova, V.; Seluanov, A.; Hammel, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef]
- McClintock, B. A Cytological and Genetical Study of Triploid Maize. Genetics 1929, 14, 180–222. [Google Scholar]
- Kramerov, D.A.; Vassetzky, N.S. Origin and evolution of SINEs in eukaryotic genomes. Heredity 2011, 107, 487–495. [Google Scholar] [CrossRef]
- Feschotte, C.; Pritham, E.J. DNA Transposons and the evolution of eukaryotic genomes. Annu. Rev. Genet. 2007, 41, 331–368. [Google Scholar] [CrossRef]
- Chalopin, D.; Naville, M.; Plard, F.; Galiana, D.; Volff, J.-N. Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates. Genome Biol. Evol. 2015, 7, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.K. LINEs Contribute to the origins of middle bodies of SINEs besides 3′ tails. Genome Biol. Evol. 2018, 10, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, A.; Mantovani, B. Conserved domains and SINE diversity during animal evolution. Genomics 2013, 102, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, N.; Labuda, D. CORE-SINEs: Eukaryotic short interspersed retroposing elements with common sequence motifs. Proc. Natl. Acad. Sci. USA 1999, 96, 2869–2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogiwara, I.; Miya, M.; Ohshima, K.; Okada, N. V-SINEs: A new superfamily of vertebrate SINEs that are widespread in vertebrate genomes and retain a strongly conserved segment within each repetitive unit. Genome Res. 2002, 12, 316–324. [Google Scholar] [CrossRef]
- Nishihara, H.; Smit, A.F.; Okada, N. Functional noncoding sequences derived from SINEs in the mammalian genome. Genome Res. 2006, 16, 864–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishihara, H.; Plazzi, F.; Passamonti, M.; Okada, N. MetaSINEs: Broad distribution of a novel SINE superfamily in animals. Genome Biol. Evol. 2016, 8, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Akasaki, T.; Nikaido, M.; Nishihara, H.; Tsuchiya, K.; Segawa, S.; Okada, N. Characterization of a novel SINE superfamily from invertebrates: “Ceph-SINEs” from the genomes of squids and cuttlefish. Gene 2010, 454, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Piskurek, O.; Jackson, D.J. Tracking the ancestry of a deeply conserved eumetazoan SINE domain. Mol. Biol. Evol. 2011, 28, 2727–2730. [Google Scholar] [CrossRef]
- Matetovici, I.; Sajgo, S.; Ianc, B.; Ochis, C.; Bulzu, P.; Popescu, O.; Damert, A. Mobile element evolution playing jigsaw-SINEs in gastropod and bivalve mollusks. Genome Biol. Evol. 2016, 8, 253–270. [Google Scholar] [CrossRef]
- Luchetti, A.; Mantovani, B. Rare horizontal transmission does not hide long-term inheritance of SINE highly conserved domains in the metazoan evolution. Curr. Zool. 2016, 62, 667–674. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, N.; Labuda, D. Evolutionary inventions and continuity of CORE-SINEs in mammals. J. Mol. Biol. 2000, 298, 365–377. [Google Scholar] [CrossRef]
- Deragon, J.-M. SINE exaptation as cellular regulators occurred numerous times during eukaryote evolution. In Plant Transposable Elements; Grandbastien, M.-A., Casacuberta, J.M., Eds.; Springer: Berlin, Germany, 2012; pp. 253–271. [Google Scholar]
- Bejerano, G.; Lowe, C.B.; Ahituv, N.; King, B.; Siepel, A.; Salama, S.R.; Rubin, E.M.; Kent, W.J.; Haussler, D. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature 2006, 441, 87–90. [Google Scholar] [CrossRef]
- Santangelo, A.M.; de Souza, F.S.; Franchini, L.F.; Bumaschny, V.F.; Low, M.J.; Rubinstein, M. Ancient exaptation of a CORE-SINE retroposon into a highly conserved mammalian neuronal enhancer of the proopiomelanocortin gene. PLoS Genet. 2007, 3, 1813–1826. [Google Scholar] [CrossRef]
- Sasaki, T.; Nishihara, H.; Hirakawa, M.; Fujimura, K.; Tanaka, M.; Kokubo, N.; Kimura-Yoshida, C.; Matsuo, I.; Sumiyama, K.; Saitou, N.; et al. Possible involvement of SINEs in mammalian brain formation. Proc. Natl. Acad. Sci. USA 2008, 105, 4220–4225. [Google Scholar] [CrossRef]
- Nakanishi, A.; Kobayashi, N.; Suzuki-Hirano, A.; Nishihara, H.; Sasaki, T.; Hirakawa, M.; Sumiyama, K.; Shimogori, T.; Okada, N. A SINE-derived element constitutes a unique modular enhancer for mammalian diencephalic Fgf8. PLoS ONE 2012, 7, e43785. [Google Scholar] [CrossRef] [PubMed]
- Scarpato, M.; Angelini, C.; Cocca, E.; Pallotta, M.M.; Morescalchi, M.A.; Capriglione, T. Short interspersed DNA elements and miRNAs: A novel hidden gene regulation layer in zebrafish? Chromosome Res. 2015, 23, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, A.; Plazzi, F.; Mantovani, B. Evolution of two short interspersed elements in Callorhinchus milii (Chondrichthyes, Holocephali) and related elements in sharks and the coelacanth. Genome Biol. Evol. 2017, 9, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Riggs, A.D. Tiggers and DNA transposon fossils in the human genome. Proc. Natl. Acad. Sci. USA 1996, 93, 1443–1448. [Google Scholar] [CrossRef]
- Feschotte, C.; Zhang, X.; Wessler, S.R. Miniature inverted-repeat transposable elements (MITEs) and their relationship with established DNA transposons. In Mobile DNA II; Craig, N., Craigie, R., Gellert, M., Lambowitz, A., Eds.; A.S.M. Press: Washington, DC, USA, 2002; pp. 1147–1158. [Google Scholar]
- Pace, J.K., II; Feschotte, C. The evolutionary history of human DNA transposons: Evidence for intense activity in the primate lineage. Genome Res. 2007, 17, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Subramanian, S. Mutation rates in mammalian genomes. Proc. Natl. Acad. Sci. USA 2002, 99, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Ray, D.A.; Feschotte, C.; Pagan, H.J.T.; Smith, J.D.; Pritham, E.J.; Arensburger, P.; Atkinson, P.W.; Craig, N.L. Multiple waves of recent DNA transposon activity in the bat, Myotis lucifugus. Genome Res. 2008, 18, 717–728. [Google Scholar] [CrossRef]
- Platt, R.N., II; Vandewege, M.W.; Ray, D.A. Mammalian transposable elements and their impacts on genome evolution. Chromosome Res. 2018, 26, 25–43. [Google Scholar] [CrossRef] [Green Version]
- Pritham, E.J.; Feschotte, C. Massive amplification of rolling-circle transposons in the lineage of the bat Myotis lucifugus. Proc. Natl. Acad. Sci. USA 2007, 104, 1895–1900. [Google Scholar] [CrossRef]
- Ray, D.A.; Pagan, H.J.T.; Thompson, M.L.; Stevens, R.D. Bats with hats: Evidence for recent DNA transposon activity in genus Myotis. Mol. Biol. Evol. 2007, 24, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Pagan, H.J.; Smith, J.D.; Hubley, R.M.; Ray, D.A. PiggyBac-ing on a primate genome: Novel elements, recent activity and horizontal transfer. Genome Biol. Evol. 2010, 2, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Malik, H.S.; Eickbush, T.H. Phylogenetic analysis of ribonuclease H domains suggests a late, chimeric origin of LTR retrotransposable elements and retroviruses. Genome Res. 2001, 11, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Ostertag, E.M.; Goodier, J.L.; Zhang, Y.; Kazazian, H.H., Jr. SVA elements are nonautonomous retrotransposons that cause disease in humans. Am. J. Hum. Genet. 2003, 73, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Brown, N.P.; Leroy, C.; Sander, C. MView: A Web compatible database search or multiple alignment viewer. Bioinformatics 1998, 14, 380–381. [Google Scholar] [CrossRef]
- Charif, D.; Lobry, J. SeqinR 1.0-2: A contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis. In Structural Approaches to Sequence Evolution: Molecules, Networks, Populations; Bastolla, U., Porto, M., Roman, H., Vendruscolo, M., Eds.; Springer: New York, NY, USA, 2007; pp. 207–232. [Google Scholar]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013–2015. Available online: http://www.repeatmasker.org (accessed on 9 November 2019).
- Bailly-Bechet, M.; Haudry, A.; Lerat, E. “One code to find them all”: A Perl tool to conveniently parse RepeatMasker output files. Mob. DNA 2014, 5, 13. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Huang, C.R.L.; Burns, K.H.; Boeke, J.D. Active transposition in genomes. Annu. Rev. Genet. 2012, 46, 651–675. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Hedegs, S.B.; Marin, J.; Suleski, M.; Paymer, M.; Kumar, S. Tree of life reveals clock-like speciation and diversification. Mol. Biol. Evol. 2015, 32, 835–845. [Google Scholar]
- Kapitonov, V.V.; Jurka, J. Molecular paleontology of transposable elements in the Drosophila melanogaster genome. Proc. Natl. Acad. Sci. USA 2003, 100, 6569–6574. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | MER6 | MER6A | ||||
|---|---|---|---|---|---|---|
| No. of Insertions | Genome Coverage (%) | Divergence from the Consensus (%) | No. of Insertions | Genome Coverage (%) | Divergence from the Consensus (%) | |
| Homo sapiens | 428 | 0.01 | 19.2 | 545 | 0.01 | 19.0 |
| Pan troglodytes | 411 | 0.01 | 19.2 | 538 | 0.01 | 19.1 |
| Callithrix jacchus | 364 | 0.009 | 21.9 | 482 | 0.009 | 21.6 |
| Macaca mulatta | 422 | 0.01 | 20.2 | 544 | 0.01 | 19.8 |
| Carlito syrichta | 428 | 0.009 | 22.9 | 648 | 0.01 | 22.1 |
| Microcebus murinus | 385 | 0.011 | 19.8 | 544 | 0.012 | 19.3 |
| Pteropus vampyrus | 873 | 0.023 | 18.6 | |||
| Myotis lucifugus | 544 | 0.015 | 19.2 | |||
| Condylura cristata | 425 | 0.012 | 30.2 | |||
| Erinaceus europaeus | 136 | 0.002 | 34.9 | |||
| Solenodon paradoxus | 871 | 0.022 | 27.6 | |||
| Species | mRNA | lncRNA | Miscellaneous RNA | |||
|---|---|---|---|---|---|---|
| Total | 5′ UTR | CDS | 3′ UTR | |||
| Homo sapiens | 39 | 15 | -- | 24 | 40 | 4 |
| Pan troglodytes | 33 | 6 | 1 | 26 | 11 | 4 |
| Callithrix jacchus | 9 | 2 | 1 | 6 | 5 | 2 |
| Macaca mulatta | 12 | 3 | -- | 9 | 7 | -- |
| Carlito syrichta | 1 | -- | -- | 1 | 1 | -- |
| Microcebus murinus | 23 | 18 | -- | 5 | 17 | 2 |
| Pteropus vampyrus | 34 | 10 | -- | 24 | 11 | 9 |
| Myotis lucifugus | 16 | 4 | -- | 12 | -- | -- |
| Condylura cristata | 3 | -- | -- | 3 | -- | -- |
| Erinaceus europaeus | -- | -- | -- | -- | -- | -- |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luchetti, A.; Lomiento, M.; Mantovani, B. Riding the Wave: The SINE-Specific V Highly-Conserved Domain Spread into Mammalian Genomes Exploiting the Replication Burst of the MER6 DNA Transposon. Int. J. Mol. Sci. 2019, 20, 5607. https://doi.org/10.3390/ijms20225607
Luchetti A, Lomiento M, Mantovani B. Riding the Wave: The SINE-Specific V Highly-Conserved Domain Spread into Mammalian Genomes Exploiting the Replication Burst of the MER6 DNA Transposon. International Journal of Molecular Sciences. 2019; 20(22):5607. https://doi.org/10.3390/ijms20225607
Chicago/Turabian StyleLuchetti, Andrea, Mariana Lomiento, and Barbara Mantovani. 2019. "Riding the Wave: The SINE-Specific V Highly-Conserved Domain Spread into Mammalian Genomes Exploiting the Replication Burst of the MER6 DNA Transposon" International Journal of Molecular Sciences 20, no. 22: 5607. https://doi.org/10.3390/ijms20225607
APA StyleLuchetti, A., Lomiento, M., & Mantovani, B. (2019). Riding the Wave: The SINE-Specific V Highly-Conserved Domain Spread into Mammalian Genomes Exploiting the Replication Burst of the MER6 DNA Transposon. International Journal of Molecular Sciences, 20(22), 5607. https://doi.org/10.3390/ijms20225607