A Brief History of Mitochondrial Pathologies

Abstract
:1. Introduction
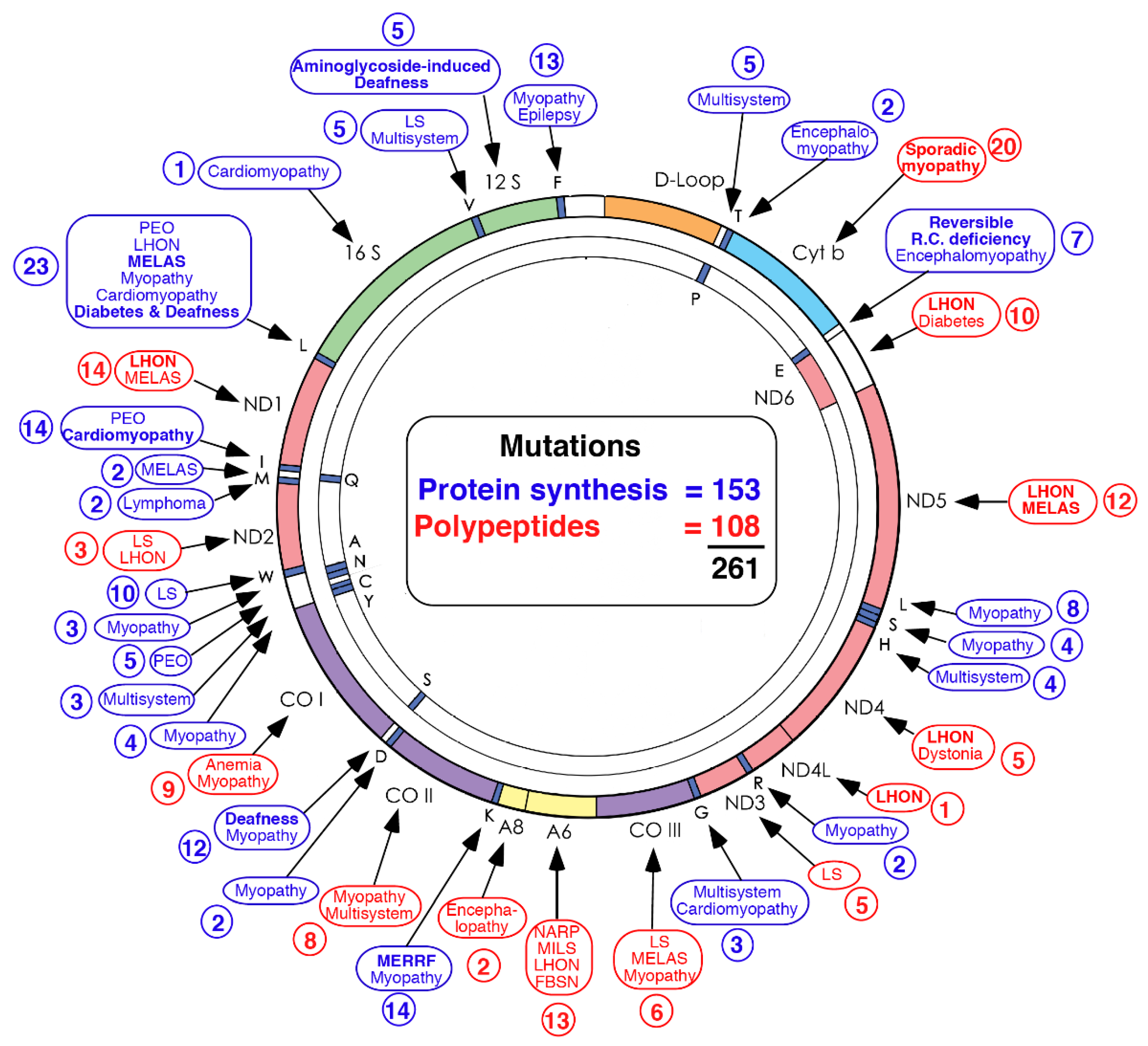
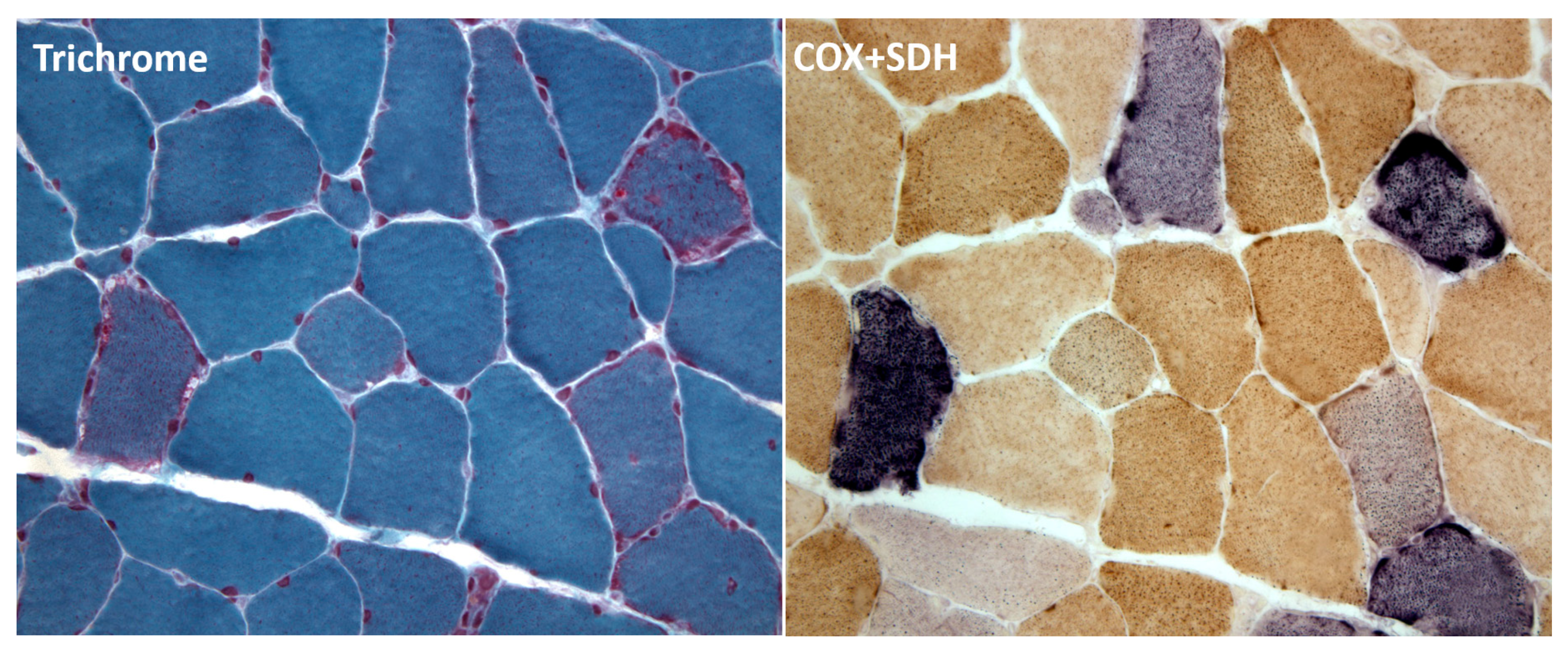
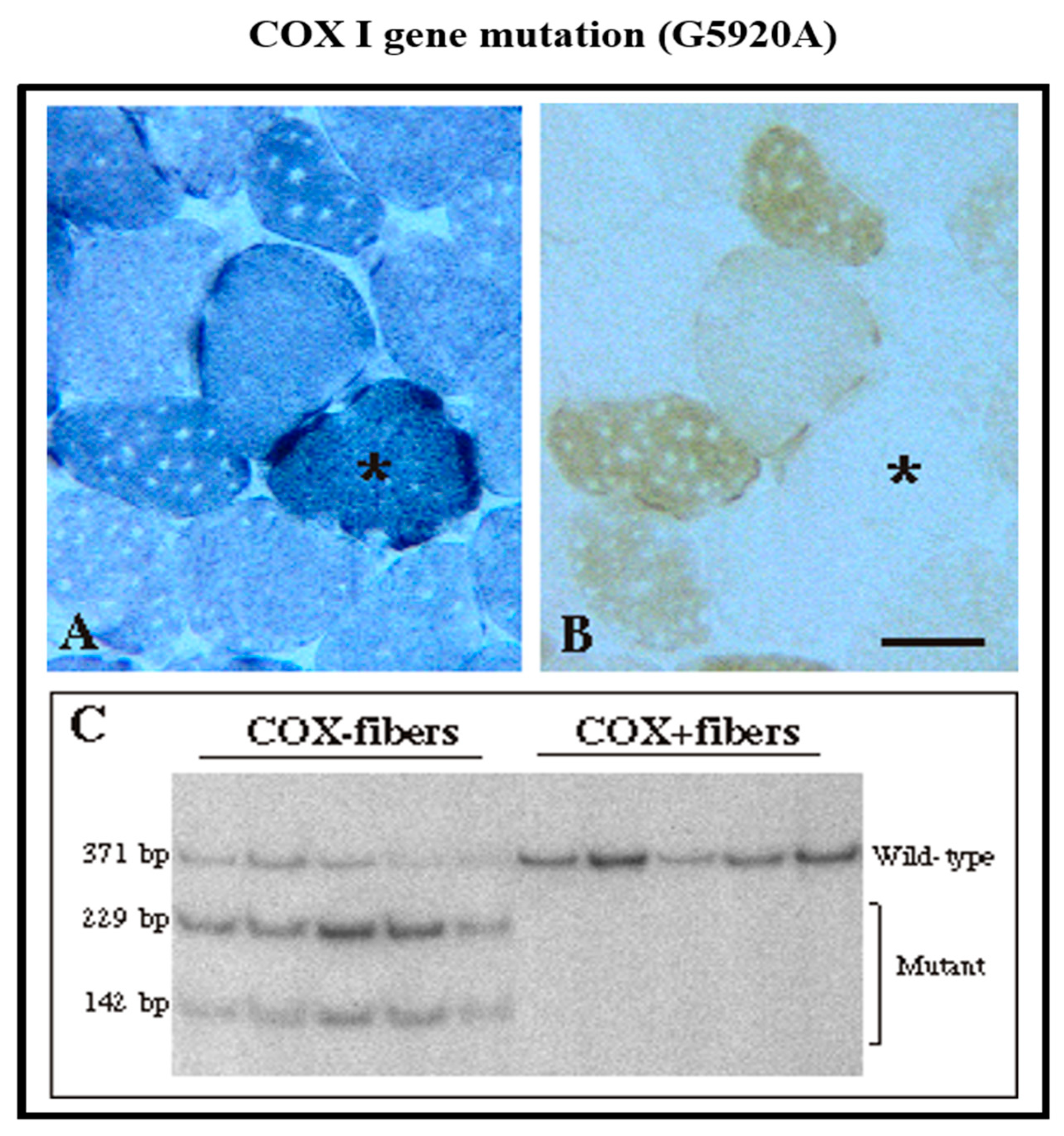
2. Diagnosis of mtDNA-Related Diseases
3. Conclusions
Conflicts of Interest
Abbreviations
| mtDNA | mitochondrial DNA |
| Cytb | Cytochrome b gene |
| COX | cytochrome c oxidase |
| SDH | succinate dehydrogenase |
| IMM | inner mitochondrial membrane |
| PD | Parkinson’s Disease |
References
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J., 2nd; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Moraes, C.T.; DiMauro, S.; Nakase, H.; Bonilla, E.; Schon, E.A.; Rowland, L.P. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology 1988, 38, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases (Mechanisms of Disease). N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A.; Carelli, V.; Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 2013, 9, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Andreu, A.L.; Hanna, M.G.; Reichmann, H.; Bruno, C.; Penn, A.S.; Tanji, K.; Pallotti, F.; Iwata, S.; Bonilla, E.; Lach, B.; et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N. Engl. J. Med. 1999, 341, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.E.; Blanc, H.; Cann, H.M.; Wallace, D.C. Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 6715–6719. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; DiMauro, S.; Shanske, S.; Schon, E.A.; Zeviani, M.; Mariotti, C.; Carrara, F.; Lombes, A.; Laforet, P.; Ogier, H.; et al. Risk of developing a mitochondrial DNA deletion disorder. Lancet 2004, 364, 592–596. [Google Scholar] [CrossRef]
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proc. Natl. Acad. Sci. USA 2018, 115, 13039–13044. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Bonilla, E. Optical imaging techniques (histochemical, immunohistochemical, and in situ hybridization staining methods) to visualize mitochondria. Methods Cell Biol. 2007, 80, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, G.M.; Taylor, R.W.; Walls, T.J.; Tonska, K.; Taylor, G.A.; Shaw, P.J.; Ince, P.G.; Turnbull, D.M. Motor neuron disease in a patient with a mitochondrial tRNAIle mutation. Ann. Neurol. 2006, 59, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.T.; Ciacci, F.; Bonilla, E.; Jansen, C.; Hirano, M.; Rao, N.; Lovelace, R.E.; Rowland, L.P.; Schon, E.A.; DiMauro, S. Two novel pathogenic mitochondrial DNA mutations affecting organelle number and protein synthesis. Is the tRNA (Leu(UUR)) gene an etiologic hot spot? J. Clin. Investig. 1993, 92, 2906–2915. [Google Scholar] [CrossRef] [PubMed]
- Karadimas, C.L.; Greenstein, P.; Sue, C.M.; Joseph, J.T.; Tanji, K.; Haller, R.G.; Taivassalo, T.; Davidson, M.M.; Shanske, S.; Bonilla, E.; et al. Recurrent myoglobinuria due to a nonsense mutation in the COX I gene of mitochondrial DNA. Neurology 2000, 55, 644–649. [Google Scholar] [CrossRef] [PubMed]
- King, M.P.; Attardi, G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science 1989, 246, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Servidei, S.; Gellera, C.; Bertini, E.; DiMauro, S.; DiDonato, S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature 1989, 339, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.T.; Shanske, S.; Tritschler, H.J.; Aprille, J.R.; Andreetta, F.; Bonilla, E.; Schon, E.A.; DiMauro, S. mtDNA depletion with variable tissue expression: A novel genetic abnormality in mitochondrial diseases. Am. J. Hum. Genet. 1991, 48, 492–501. [Google Scholar] [PubMed]
- Tritschler, H.J.; Andreetta, F.; Moraes, C.T.; Bonilla, E.; Arnaudo, E.; Danon, M.J.; Glass, S.; Zelaya, B.M.; Vamos, E.; Telerman-Toppet, N.; et al. Mitochondrial myopathy of childhood associated with depletion of mitochondrial DNA. Neurology 1992, 42, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Mandel, H.; Szargel, R.; Labay, V.; Elpeleg, O.; Saada, A.; Shalata, A.; Anbinder, Y.; Berkowitz, D.; Hartman, C.; Barak, M.; et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat. Genet. 2001, 29, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; Taylor, R.W.; Nascimento, A.; Poulton, J.; Fratter, C.; Dominguez-Gonzalez, C.; Evans, J.C.; Loos, M.; Isohanni, P.; Suomalainen, A.; et al. Retrospective natural history of thymidine kinase 2 deficiency. J. Med. Genet. 2018, 55, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kim, E.; Dai, H.; Stefans, V.; Vogel, H.; Al Jasmi, F.; Schrier Vergano, S.A.; Castro, D.; Bernes, S.; Bhambhani, V.; et al. Clinical and molecular spectrum of thymidine kinase 2-related mtDNA maintenance defect. Mol. Genet. Metab. 2018, 124, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gomez, C.; Levy, R.J.; Sanchez-Quintero, M.J.; Juanola-Falgarona, M.; Barca, E.; Garcia-Diaz, B.; Tadesse, S.; Garone, C.; Hirano, M. Deoxycytidine and Deoxythymidine Treatment for Thymidine Kinase 2 Deficiency. Ann. Neurol. 2017, 81, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Emmanuele, V.; Quinzii, C.M. Emerging therapies for mitochondrial diseases. Essays Biochem. 2018, 62, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Elpeleg, O.; Miller, C.; Hershkovitz, E.; Bitner-Glindzicz, M.; Bondi-Rubinstein, G.; Rahman, S.; Pagnamenta, A.; Eshhar, S.; Saada, A. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am. J. Hum. Genet. 2005, 76, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; Gurgel-Giannetti, J.; Sanna-Cherchi, S.; Krishna, S.; Naini, A.; Quinzii, C.M.; Hirano, M. A Novel SUCLA2 Mutation Presenting as a Complex Childhood Movement Disorder. J. Child. Neurol. 2017, 32, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Van Hove, J.L.; Saenz, M.S.; Thomas, J.A.; Gallagher, R.C.; Lovell, M.A.; Fenton, L.Z.; Shanske, S.; Myers, S.M.; Wanders, R.J.; Ruiter, J.; et al. Succinyl-CoA ligase deficiency: A mitochondrial hepatoencephalomyopathy. Pediatr. Res. 2010, 68, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, B.; Area, E.; Flanigan, K.M.; Ganesh, J.; Jayakar, P.; Swoboda, K.J.; Coku, J.; Naini, A.; Shanske, S.; Tanji, K.; et al. Mitochondrial DNA depletion syndrome due to mutations in the RRM2B gene. Neuromuscul. Disord. 2008, 18, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Takata, A.; Kato, M.; Nakamura, M.; Yoshikawa, T.; Kanba, S.; Sano, A.; Kato, T. Exome sequencing identifies a novel missense variant in RRM2B associated with autosomal recessive progressive external ophthalmoplegia. Genome Biol. 2011, 12, R92. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.; Fassone, E.; Taanman, J.W.; Sadowski, M.; Fratter, C.; Mudanohwo, E.E.; Woodward, C.E.; Sweeney, M.G.; Holton, J.L.; Hanna, M.G.; et al. Kearns-Sayre syndrome caused by defective R1/p53R2 assembly. J. Med. Genet. 2011, 48, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Shaibani, A.; Shchelochkov, O.A.; Zhang, S.; Katsonis, P.; Lichtarge, O.; Wong, L.J.; Shinawi, M. Mitochondrial neurogastrointestinal encephalopathy due to mutations in RRM2B. Arch. Neurol. 2009, 66, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Nishino, I.; Spinazzola, A.; Hirano, M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 1999, 283, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Nishigaki, Y.; Marti, R.; Hirano, M. ND5 is a hot-spot for multiple atypical mitochondrial DNA deletions in mitochondrial neurogastrointestinal encephalomyopathy. Hum. Mol. Genet. 2004, 13, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Halter, J.P.; Michael, W.; Schüpbach, M.; Mandel, H.; Casali, C.; Orchard, K.; Collin, M.; Valcarcel, D.; Rovelli, A.; Filosto, M.; et al. Allogeneic haematopoietic stem cell transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Brain 2015, 138 Pt 10, 2847–2858. [Google Scholar] [CrossRef] [PubMed]
- Spinazzola, A.; Viscomi, C.; Fernandez-Vizarra, E.; Carrara, F.; D’Adamo, P.; Calvo, S.; Marsano, R.M.; Donnini, C.; Weiher, H.; Strisciuglio, P.; et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat. Genet. 2006, 38, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Karadimas, C.L.; Vu, T.H.; Holve, S.A.; Chronopoulou, P.; Quinzii, C.; Johnsen, S.D.; Kurth, J.; Eggers, E.; Palenzuela, L.; Tanji, K.; et al. Navajo neurohepatopathy is caused by a mutation in the MPV17 gene. Am. J. Hum. Genet. 2006, 79, 544–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, K.V.; Ostergaard, E.; Ravn, S.H.; Balslev, T.; Danielsen, E.R.; Vardag, A.; McKiernan, P.J.; Gray, G.; Naviaux, R.K. POLG mutations in Alpers syndrome. Neurology 2005, 65, 1493–1495. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.J.; Naviaux, R.K.; Brunetti-Pierri, N.; Zhang, Q.; Schmitt, E.S.; Truong, C.; Milone, M.; Cohen, B.H.; Wical, B.; Ganesh, J.; et al. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum. Mutat. 2008, 29, E150–E172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suomalainen, A.; Majander, A.; Haltia, M.; Somer, H.; Lonnqvist, J.; Savontaus, M.L.; Peltonen, L. Multiple deletions of mitochondrial DNA in several tissues of a patient with severe retarded depression and familial progressive external ophthalmoplegia. J. Clin. Investig. 1992, 90, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaukonen, J.; Juselius, J.K.; Tiranti, V.; Kyttälä, A.; Zeviani, M.; Comi, G.P.; Keränen, S.; Peltonen, L.; Suomalainen, A. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 2000, 289, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F. Mitochondrial transporters of the SLC25 family and associated diseases: A review. J. Inherit. Metab. Dis. 2014, 37, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Paradas, C.; Camano, P.; Otaegui, D.; Oz, O.; Emmanuele, V.; DiMauro, S.; Hirano, M. Longitudinal clinical follow-up of a large family with the R357P Twinkle mutation. JAMA Neurol. 2013, 70, 1425–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.; Bi, H.; Yao, S.; Wang, Z.; Yuan, Y. Clinical phenotype of autosomal dominant progressive external ophthalmoplegia in a family with a novel mutation in the C10orf2 gene. Muscle Nerve 2010, 41, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Nikali, K.; Suomalainen, A.; Saharinen, J.; Kuokkanen, M.; Spelbrink, J.N.; Lonnqvist, T.; Peltonen, L.L. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum. Mol. Genet. 2005, 14, 2981–2990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakonen, A.H.; Goffart, S.; Marjavaara, S.; Paetau, A.; Cooper, H.; Mattila, K.; Lampinen, M.; Sajantila, A.; Lönnqvist, T.; Spelbrink, J.N.; et al. Infantile-onset spinocerebellar ataxia and mitochondrial recessive ataxia syndrome are associated with neuronal complex I defect and mtDNA depletion. Hum. Mol. Genet. 2008, 17, 3822–3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradas, C.; Gutierrez Rios, P.; Rivas, E.; Carbonell, P.; Hirano, M.; DiMauro, S. TK2 mutation presenting as indolent myopathy. Neurology 2013, 80, 504–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pons, R.; Andreetta, F.; Wang, C.H.; Vu, T.H.; Bonilla, E.; DiMauro, S.; De Vivo, D.C. Mitochondrial myopathy simulating spinal muscular atrophy. Pediatr. Neurol. 1996, 15, 153–158. [Google Scholar] [CrossRef]
- Garone, C.; Rubio, J.C.; Calvo, S.E.; Naini, A.; Tanji, K.; Dimauro, S.; Mootha, V.K.; Hirano, M. MPV17 Mutations Causing Adult-Onset Multisystemic Disorder with Multiple Mitochondrial DNA Deletions. Arch. Neurol. 2012, 69, 1648–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronchi, D.; Garone, C.; Bordoni, A.; Gutierrez Rios, P.; Calvo, S.E.; Ripolone, M.; Ranieri, M.; Rizzuti, M.; Villa, L.; Magri, F.; et al. Next-generation sequencing reveals DGUOK mutations in adult patients with mitochondrial DNA multiple deletions. Brain 2012, 135 Pt 11, 3404–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, M.J.; Clark, S.; Man, C.Y.; Hudson, G.; Durham, S.E.; Taylor, R.W.; Nightingale, S.; Turnbull, D.M.; Copeland, W.C.; Chinnery, P.F. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am. J. Hum. Genet. 2006, 78, 1026–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varma, H.; Faust, P.L.; Iglesias, A.D.; Lagana, S.M.; Wou, K.; Hirano, M.; DiMauro, S.; Mansukani, M.M.; Hoff, K.E.; Nagy, P.L.; et al. Whole exome sequencing identifies a homozygous POLG2 missense variant in an infant with fulminant hepatic failure and mitochondrial DNA depletion. Eur. J. Med. Genet. 2016, 59, 540–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, E.P.; Bennett, M.R. The role of mitochondrial DNA damage in the development of atherosclerosis. Free Radic. Biol. Med. 2016, 100, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Uryga, A.; Gray, K.; Bennett, M. DNA Damage and Repair in Vascular Disease. Annu. Rev. Physiol. 2016, 78, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Galitsyna, E.V.; Khasanova, Z.B.; Postnov, A.Y.; Yarygina, E.I.; Orekhov, A.N.; Sobenin, I.A. Role of Mitochondrial Genome Mutations in Pathogenesis of Carotid Atherosclerosis. Oxid. Med. Cell. Longev. 2017, 2017, 6934394. [Google Scholar] [CrossRef] [PubMed]
- Volobueva, A.; Grechko, A.; Yet, S.F.; Sobenin, I.; Orekhov, A. Changes in Mitochondrial Genome Associated with Predisposition to Atherosclerosis and Related Disease. Biomolecules 2019, 9, 377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Sazonova, M.D.; Khasanova, Z.B.; Shkurat, T.P.; Karagodin, V.P.; Orekhov, A.N.; Sobenin, I.A. Creation of Cybrid Cultures Containing mtDNA Mutations m.12315G>A and m.1555G>A, Associated with Atherosclerosis. Biomolecules 2019, 9, 499. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Gene | mtDNA Depletion | mtDNA Multiple Deletions |
|---|---|---|
| DGUOK | Hepato-cerebral syndrome | Adult-onset PEO |
| TK2 | Lethal infantile myopathy | Later-onset myopathies |
| SUCLA2 | Infantile encephalomyopathy | Later-onset movement disorder |
| SUCLG1 | Hepato-cerebral syndrome | |
| RRM2B | Infantile encephalomyopathy | Adult-onset PEO; KSS; MNGIE |
| TYMP | MNGIE | |
| MPV17 | Hepato-cerebral syndrome; NNH | Adult-onset PEO |
| POLG | Alpers–Huttenlocher syndrome | Adult-onset PEO; SANDO; MIRAS; PD |
| POLG2 | Fatal infantile liver failure | Adult-onset PEO |
| TWNK | - | AD PEO; IOSCA |
| ANT1 | - | Adult-onset PEO, dementia |
| OPA1 | DOA | Adult PEO-plus |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
DiMauro, S. A Brief History of Mitochondrial Pathologies. Int. J. Mol. Sci. 2019, 20, 5643. https://doi.org/10.3390/ijms20225643
DiMauro S. A Brief History of Mitochondrial Pathologies. International Journal of Molecular Sciences. 2019; 20(22):5643. https://doi.org/10.3390/ijms20225643
Chicago/Turabian StyleDiMauro, Salvatore. 2019. "A Brief History of Mitochondrial Pathologies" International Journal of Molecular Sciences 20, no. 22: 5643. https://doi.org/10.3390/ijms20225643
APA StyleDiMauro, S. (2019). A Brief History of Mitochondrial Pathologies. International Journal of Molecular Sciences, 20(22), 5643. https://doi.org/10.3390/ijms20225643