The Role of Vascular Smooth Muscle Cells in Arterial Remodeling: Focus on Calcification-Related Processes



Abstract
:
1. Introduction
2. Biology of Vascular Remodeling
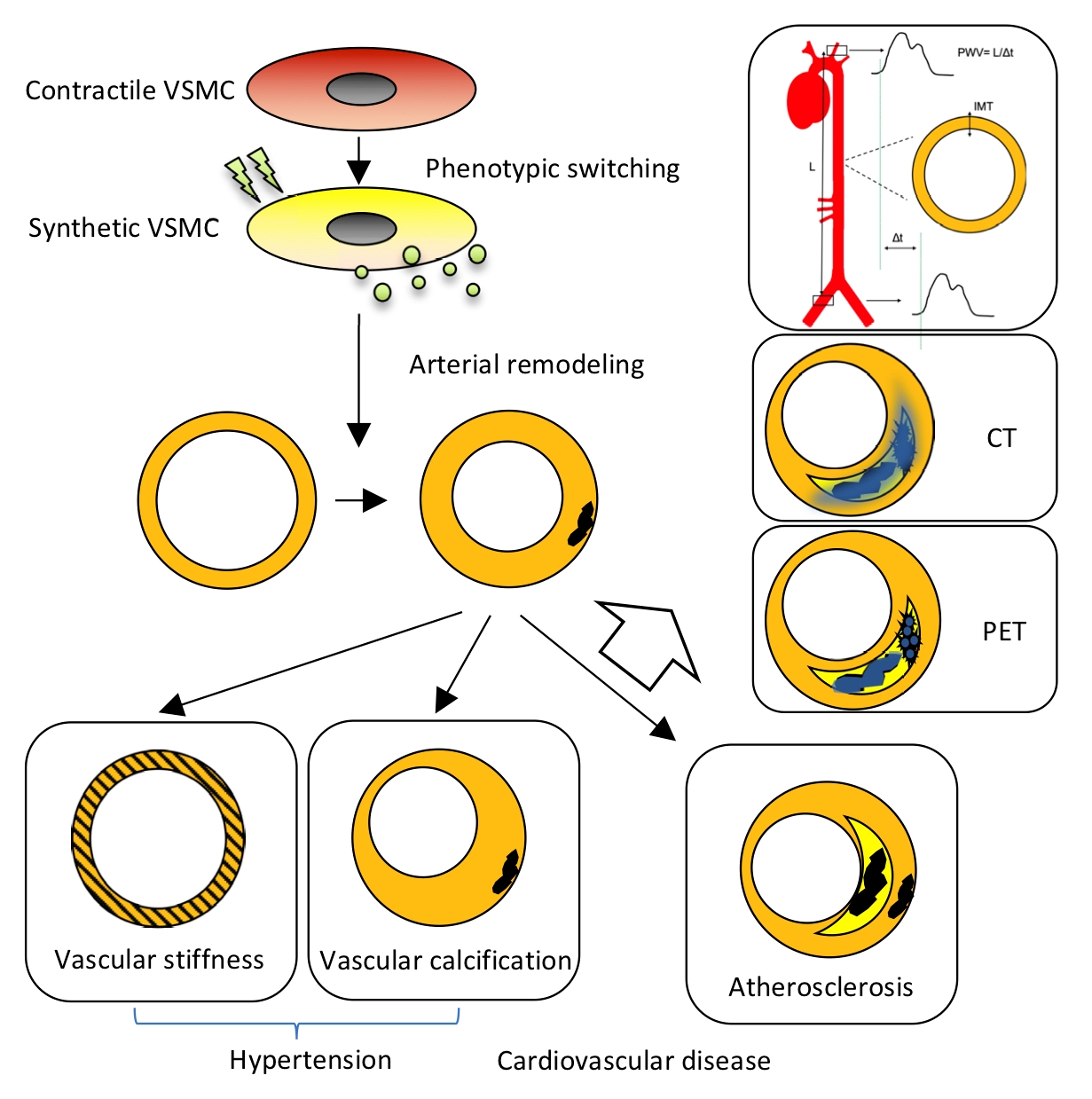
2.1. Arterial Remodeling
2.1.1. Inflammation and Arterial Remodeling
Proliferation
Cell Death
Platelets and Extracellular Vesicles (EVs)
2.2. The Role of VSMC Plasticity in Vascular Remodeling
2.2.1. Biochemical Compounds
2.2.2. Extracellular Components
2.2.3. Biophysical Factors
2.2.4. Transcriptional Regulators
3. Clinical Features of Vascular Remodeling
3.1. Hypertension
3.1.1. Cellular Components and Hypertension
3.1.2. Calcification and Hypertension
3.1.3. Consequences of Vascular Calcification
3.2. Atherosclerosis
3.3. Intimal and Medial Aspects of Vascular Calcification
Cellular Processes of Vascular Calcification
3.4. Fibrosis
4. Assessing Vascular Remodeling and Disease
4.1. Pulse Wave Velocity (PWV), Distensibility Coefficient (DC) and Intima-Media Thickness (IMT)
4.1.1. Pulse Wave Velocity
4.1.2. Distensibility Coefficient
4.1.3. Intima-Media Thickness
4.2. Calcification Imaging by Computed Tomography
4.3. 18F-NaF Positron Emission Tomography
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α-SMA | alpha-smooth muscle actin |
| AGE | advanced glycation end product |
| BMP-2 | bone morphogenetic protein 2 |
| BMP-4 | bone morphogenetic protein 4 |
| CAC | coronary artery calcium |
| CD9 | cluster of differentiation 9 |
| CD34 | cluster of differentiation 34 |
| CD63 | cluster of differentiation 63 |
| CD81 | cluster of differentiation 81 |
| cfPWV | carotid-femoral pulse wave velocity |
| CNN-1 | calponin-1 |
| CT | computed tomography |
| CV | cardiovascular |
| CVD | cardiovascular disease |
| DBP | diastolic blood pressure |
| DC | distensibility coefficient |
| EC | endothelial cell |
| ECM | extracellular matrix |
| EV | extracellular vesicle |
| Gli1 | glioma-associated oncogene |
| IL-1β | interleukin 1 beta |
| IMT | intima-media thickness |
| KLF4 | kruppel-like factor 4 |
| MMP | metalloproteinase |
| MRI | magnetic resonance imaging |
| MSC | mesenchymal stem cell |
| MYC11 | smooth muscle-myosin heavy chain |
| NAD(P)H | nitrate reductase |
| NO | nitric oxide |
| Oct4 | octamer-binding transcription factor 4 |
| p16INK4a | cyclin-dependent kinase inhibitor 2A |
| p21 | cyclin-dependent kinase inhibitor 1 |
| PAR | protease activated receptor |
| PF4 | platelet factor 4 |
| PDGF | platelet derived growth factor |
| PXE | pseudoxanthoma elasicum |
| PWV | pulse wave velocity |
| ROS | reactive oxygen species |
| Runx2 | runt-related transcription factor 2 |
| SBP | systolic blood pressure |
| Sca-1 | Spinocerebellaire ataxie type 1 |
| SM22-α | smooth muscle 22-alpha |
| SMPD3 | sphingomyelin phosphodiesterase 3 |
| smtn | Smoothelin |
| VSMC | vascular smooth muscle cell |
References
- Mazurek, R.; Dave, J.M.; Chandran, R.R.; Misra, A.; Sheikh, A.Q.; Greif, D.M. Vascular Cells in Blood Vessel Wall Development and Disease. Adv. Pharmacol. 2017, 78, 323–350. [Google Scholar] [PubMed]
- Schurgers, L.J.; Akbulut, A.C.; Kaczor, D.M.; Halder, M.; Koenen, R.R.; Kramann, R. Initiation and Propagation of Vascular Calcification Is Regulated by a Concert of Platelet- and Smooth Muscle Cell-Derived Extracellular Vesicles. Front. Cardiovasc. Med. 2018, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Roostalu, U.; Wong, J.K. Arterial smooth muscle dynamics in development and repair. Dev. Biol. 2018, 435, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Gabbiani, G.; Bochaton-Piallat, M.L. Arterial smooth muscle cell heterogeneity: Implications for atherosclerosis and restenosis development. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1510–1520. [Google Scholar] [CrossRef]
- Alexander, M.R.; Owens, G.K. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef]
- van Bussel, F.C.; van Bussel, B.C.; Hoeks, A.P.; Op’t Roodt, J.; Henry, R.M.; Ferreira, I.; Vanmolkot, F.H.; Schalkwijk, C.G.; Stehouwer, C.D.; Reesink, K.D. A control systems approach to quantify wall shear stress normalization by flow-mediated dilation in the brachial artery. PLoS ONE 2015, 10, e0115977. [Google Scholar] [CrossRef]
- van der Heijden, O.W.; Essers, Y.P.; Fazzi, G.; Peeters, L.L.; De Mey, J.G.; van Eys, G.J.J. Uterine artery remodeling and reproductive performance are impaired in endothelial nitric oxide synthase-deficient mice. Biol. Reprod. 2005, 72, 1161–1168. [Google Scholar] [CrossRef]
- Boutouyrie, P.; Laurent, S.; Girerd, X.; Benetos, A.; Lacolley, P.; Abergel, E.; Safar, M. Common carotid artery stiffness and patterns of left ventricular hypertrophy in hypertensive patients. Hypertension 1995, 25, 651–659. [Google Scholar] [CrossRef]
- Rourke, M.F.; Nichols, W.W. Aortic Diameter, Aortic Stiffness, and Wave Reflection Increase With Age and Isolated Systolic Hypertension. Hypertension 2005, 45, 652–658. [Google Scholar]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Henning, R.J.; Bourgeois, M.; Harbison, R.D. Poly(ADP-ribose) Polymerase (PARP) and PARP Inhibitors: Mechanisms of Action and Role in Cardiovascular Disorders. Cardiovasc. Toxicol. 2018, 18, 493–506. [Google Scholar] [CrossRef] [PubMed]
- van Varik, B.J.; Rennenberg, R.J.M.W.; Reutelingsperger, C.P.; Kroon, A.A.; de Leeuw, P.W.; Schurgers, L.J. Mechanisms of arterial remodeling: Lessons from genetic diseases. Front. Genet. 2012, 3, 290. [Google Scholar] [CrossRef] [PubMed]
- Lacolley, P.; Regnault, V.; Segers, P.; Laurent, S. Vascular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease. Physiol. Rev. 2017, 97, 1555–1617. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.I.; Pierre-Paul, D.; Sumpio, B.E.; Gahtan, V. Vascular smooth muscle cell migration: Current research and clinical implications. Vasc. Endovasc. Surg. 2004, 38, 11–23. [Google Scholar] [CrossRef]
- Johnson, J.L. Matrix metalloproteinases: Influence on smooth muscle cells and atherosclerotic plaque stability. Expert Rev. Cardiovasc. Ther. 2014, 5, 265–282. [Google Scholar] [CrossRef]
- Peeters, S.A.; Engelen, L.; Buijs, J.; Chaturvedi, N.; Fuller, J.H.; Schalkwijk, C.G.; Stehouwer, C.D.; Group, E.P.C.S. Plasma levels of matrix metalloproteinase-2, -3, -10, and tissue inhibitor of metalloproteinase-1 are associated with vascular complications in patients with type 1 diabetes: The EURODIAB Prospective Complications Study. Cardiovasc. Diabetol. 2015, 14, 31. [Google Scholar] [CrossRef]
- Reesink, K.D.; Spronck, B. Constitutive interpretation of arterial stiffness in clinical studies: A methodological review. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H693–H709. [Google Scholar] [CrossRef]
- Smulyan, H.; Mookherjee, S.; Safar, M.E. The two faces of hypertension: Role of aortic stiffness. J. Am. Soc. Hypertens. 2016, 10, 175–183. [Google Scholar] [CrossRef]
- Lacolley, P.; Regnault, V.; Nicoletti, A.; Li, Z.; Michel, J.B. The vascular smooth muscle cell in arterial pathology: A cell that can take on multiple roles. Cardiovasc. Res. 2012, 95, 194–204. [Google Scholar] [CrossRef]
- Petsophonsakul, P.; Furmanik, M.; Forsythe, R.; Dweck, M.; Schurink, G.W.; Natour, E.; Reutelingsperger, C.; Jacobs, M.; Mees, B.; Schurgers, L. Role of Vascular Smooth Muscle Cell Phenotypic Switching and Calcification in Aortic Aneurysm Formation. Arterioscler. Thromb. Vasc. Biol. 2019, 27, 1151–1368. [Google Scholar] [CrossRef] [PubMed]
- Pawade, T.A.; Newby, D.E.; Dweck, M.R. Calcification in Aortic Stenosis: The Skeleton Key. J. Am. Coll. Cardiol. 2015, 66, 561–577. [Google Scholar] [CrossRef] [PubMed]
- Neven, E.; Haese, P.C. Vascular calcification in chronic renal failure: What have we learned from animal studies? Circ. Res. 2011, 108, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D. Molecular mechanisms of arterial calcification. Artery Res. 2009, 3, 128–131. [Google Scholar] [CrossRef]
- Tyson, K.L.; Reynolds, J.L.; McNair, R.; Zhang, Q.; Weissberg, P.L.; Shanahan, C.M. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein-Grün, G.; Frey, M.; Thimm, F.; Hofgärtner, W.; Fleckenstein, A. Calcium Overload—An Important Cellular Mechanism in Hypertension and Arteriosclerosis. Drugs 1992, 44, 23–30. [Google Scholar] [CrossRef]
- Shanahan, C.M. Mechanisms of vascular calcification in CKD-evidence for premature ageing? Nat. Rev. Nephrol. 2013, 9, 661–670. [Google Scholar] [CrossRef]
- O’Rourke, M.F.; Hashimoto, J. Mechanical Factors in Arterial Aging: A Clinical Perspective. J. Am. Coll. Cardiol. 2007, 50, 1–13. [Google Scholar] [CrossRef]
- Mulvany, M.J.; Baumbach, G.L.; Aalkjaer, C.; Heagerty, A.M.; Korsgaard, N.; Schiffrin, E.L.; Heistad, D.D. Vascular remodeling. Hypertension 1996, 28, 505–506. [Google Scholar]
- Mulvany, M.J. Small artery remodelling in hypertension: Causes, consequences and therapeutic implications. Med. Biol. Eng. Comp. 2008, 46, 461–467. [Google Scholar] [CrossRef]
- Ward, M.R.; Pasterkamp, G.; Yeung, A.C.; Borst, C. Arterial remodeling. Mechanisms and clinical implications. Circulation 2000, 102, 1186–1191. [Google Scholar] [CrossRef]
- Hansson, G.K.; Libby, P. The immune response in atherosclerosis: A double-edged sword. Nat. Rev. Immunol. 2006, 6, 508–519. [Google Scholar] [CrossRef]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Greenwald, S.E. Ageing of the conduit arteries. J. Pathol. 2007, 211, 157–172. [Google Scholar] [CrossRef]
- Spronck, B.; Heusinkveld, M.H.G.; Donders, W.P.; de Lepper, A.G.W.; O’Roodt, J.; Kroon, A.A.; Delhaas, T.; Reesink, K.D. A constitutive modeling interpretation of the relationship among carotid artery stiffness, blood pressure, and age in hypertensive subjects. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H568–H582. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, J.D.; Harrison, D.G.; Figueroa, C.A.; Lacolley, P.; Laurent, S. Central Artery Stiffness in Hypertension and Aging: A Problem with Cause and Consequence. Circ. Res. 2016, 118, 379–381. [Google Scholar] [CrossRef]
- Sekikawa, A.; Shin, C.; Curb, J.D.; Barinas-Mitchell, E.; Masaki, K.; El-Saed, A.; Seto, T.B.; Mackey, R.H.; Choo, J.; Fujiyoshi, A.; et al. Aortic stiffness and calcification in men in a population-based international study. Atherosclerosis 2012, 222, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Kruger, T.; Oelenberg, S.; Kaesler, N.; Schurgers, L.J.; van de Sandt, A.M.; Boor, P.; Schlieper, G.; Brandenburg, V.M.; Fekete, B.C.; Veulemans, V.; et al. Warfarin Induces Cardiovascular Damage in Mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2618–2624. [Google Scholar] [CrossRef]
- Chi, C.; Li, D.J.; Jiang, Y.J.; Tong, J.; Fu, H.; Wu, Y.H.; Shen, F.M. Vascular smooth muscle cell senescence and age-related diseases: State of the art. Mol. Basis Dis. 2019, 1865, 1810–1821. [Google Scholar] [CrossRef]
- Sans, M.; Moragas, A. Mathematical morphologic analysis of the aortic medial structure. Biomechanical implications. Anal. Quant. Cytol. Histol. 1993, 15, 93–100. [Google Scholar]
- Cliff, W.J. The aortic tunica media in aging rats. Exp. Mol. Pathol. 1970, 13, 172–189. [Google Scholar] [CrossRef]
- Zieman, S.J.; Kass, D.A. Advanced Glycation Endproduct Crosslinking in the Cardiovascular System. Drugs 2004, 64, 459–470. [Google Scholar] [CrossRef]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef] [Green Version]
- Leondes, C.T. Biomechanical Systems: Techniques and Applications, Volume II: Cardiovascular Techniques; CRC Press: Boca Raton, FL, USA, 2000; pp. 6:1–6:61. [Google Scholar]
- Kelly, R.; Hayward, C.; Avolio, A.; O’Rourke, M. Noninvasive determination of age-related changes in the human arterial pulse. Circulation 1989, 80, 1652–1659. [Google Scholar] [CrossRef] [Green Version]
- Laogun, A.A.; Gosling, R.G. In vivo arterial compliance in man. Clin. Phys. Physiol. Meas. 1982, 3, 201–212. [Google Scholar] [CrossRef]
- Zieman, S.J.; Melenovsky, V.; Kass, D.A. Mechanisms, Pathophysiology, and Therapy of Arterial Stiffness. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 932–943. [Google Scholar] [CrossRef] [Green Version]
- Cattell, M.A.; Anderson, J.C.; Hasleton, P.S. Age-related changes in amounts and concentrations of collagen and elastin in normotensive human thoracic aorta. Clin. Chim. Acta 1996, 245, 73–84. [Google Scholar] [CrossRef]
- Sun, Z. Aging, arterial stiffness, and hypertension. Hypertension 2015, 65, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Brandes, R.P.; Fleming, I.; Busse, R. Endothelial aging. Cardiovasc. Res. 2005, 66, 286–294. [Google Scholar] [CrossRef] [Green Version]
- Urschel, K.; Cicha, I.; Daniel, W.G.; Garlichs, C.D. Shear stress patterns affect the secreted chemokine profile in endothelial cells. Clin. Hemorheol. Microcirc. 2012, 50, 143–152. [Google Scholar]
- Qiu, J.; Zheng, Y.; Hu, J.; Liao, D.; Gregersen, H.; Deng, X.; Fan, Y.; Wang, G. Biomechanical regulation of vascular smooth muscle cell functions: From in vitro to in vivo understanding. J. R. Soc. Interface 2014, 11, 20130852. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Clarke, M.C.H.; Figg, N.; Littlewood, T.D.; Bennett, M.R. Smooth muscle cell apoptosis promotes vessel remodeling and repair via activation of cell migration, proliferation, and collagen synthesis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2402–2409. [Google Scholar] [CrossRef] [Green Version]
- Mozos, I.; Luca, C.T. Crosstalk between Oxidative and Nitrosative Stress and Arterial Stiffness. Curr. Vasc. Pharmacol. 2017, 15, 446–456. [Google Scholar] [CrossRef]
- McMaster, W.G.; Kirabo, A.; Madhur, M.S.; Harrison, D.G. Inflammation, immunity, and hypertensive end-organ damage. Circ. Res. 2015, 116, 1022–1033. [Google Scholar] [CrossRef]
- Jain, S.; Khera, R.; Corrales-Medina, V.F.; Townsend, R.R.; Chirinos, J.A. Inflammation and arterial stiffness in humans. Atherosclerosis 2014, 237, 381–390. [Google Scholar] [CrossRef]
- Lutgens, E.; de Muinck, E.D.; Kitslaar, P.J.; Tordoir, J.H.; Wellens, H.J.; Daemen, M.J. Biphasic pattern of cell turnover characterizes the progression from fatty streaks to ruptured human atherosclerotic plaques. Cardiovasc. Res. 1999, 41, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: Effects of telomerase and oxidative stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.K.; Cha, B.Y.; Kim, C.H. In vitro cellular aging is associated with enhanced proliferative capacity, G1 cell cycle modulation, and matrix metalloproteinase-9 regulation in mouse aortic smooth muscle cells. Arch. Biochem. Biophys. 2003, 418, 39–48. [Google Scholar] [CrossRef]
- Clarke, M.C.; Figg, N.; Maguire, J.J.; Davenport, A.P.; Goddard, M.; Littlewood, T.D.; Bennett, M.R. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat. Med. 2006, 12, 1075–1080. [Google Scholar] [CrossRef]
- Clarke, M.C.H.; Littlewood, T.D.; Figg, N.; Maguire, J.J.; Davenport, A.P.; Goddard, M.; Bennett, M.R. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ. Res. 2008, 102, 1529–1538. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.R.; Gibson, D.F.; Schwartz, S.M.; Tait, J.F. Binding and phagocytosis of apoptotic vascular smooth muscle cells is mediated in part by exposure of phosphatidylserine. Circ. Res. 1995, 77, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Morrell, C.N. Platelets as initiators and mediators of inflammation at the vessel wall. Thromb. Res. 2011, 127, 387–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vajen, T.; Benedikter, B.J.; Heinzmann, A.C.A.; Vasina, E.M.; Henskens, Y.; Parsons, M.; Maguire, P.B.; Stassen, F.R.; Heemskerk, J.W.M.; Schurgers, L.J.; et al. Platelet extracellular vesicles induce a pro-inflammatory smooth muscle cell phenotype. J. Extracell. Vesicles 2017, 6, 1322454. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Field, D.J.; Long, X.; Mickelsen, D.; Ko, K.; Ture, S.; Korshunov, V.A.; Miano, J.M.; Morrell, C.N. Platelet factor 4 mediates vascular smooth muscle cell injury responses. Blood 2013, 121, 4417–4427. [Google Scholar] [CrossRef] [Green Version]
- Miano, J.M. Serum response factor: Toggling between disparate programs of gene expression. J. Mol. Cell. Cardiol. 2003, 35, 577–593. [Google Scholar] [CrossRef]
- Rensen, S.S.; Doevendans, P.A.; van Eys, G.J. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth. Heart J. 2007, 15, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Bochaton-Piallat, M.-L.; Ropraz, P.; Gabbiani, F.; Gabbiani, G. Phenotypic Heterogeneity of Rat Arterial Smooth Muscle Cell Clones. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 815–820. [Google Scholar] [CrossRef]
- Hao, H.; Ropraz, P.; Verin, V.; Camenzind, E.; Geinoz, A.; Pepper, M.S.; Gabbiani, G.; Bochaton-Piallat, M.-L. Heterogeneity of Smooth Muscle Cell Populations Cultured From Pig Coronary Artery. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1093–1099. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Fan, Y.S.; Chow, L.H.; Van Den Diepstraten, C.; van Der Veer, E.; Sims, S.M.; Pickering, J.G. Innate diversity of adult human arterial smooth muscle cells: Cloning of distinct subtypes from the internal thoracic artery. Circ. Res. 2001, 89, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Wanjare, M.; Kuo, F.; Gerecht, S. Derivation and maturation of synthetic and contractile vascular smooth muscle cells from human pluripotent stem cells. Cardiovasc. Res. 2013, 97, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Gerthoffer, W.T. Mechanisms of vascular smooth muscle cell migration. Circ. Res. 2007, 100, 607–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frismantiene, A.; Philippova, M.; Erne, P.; Resink, T.J. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell. Signal. 2018, 52, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chaabane, C.; Boukais, K.; Francis, G.A.; Bochaton-Piallat, M.L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 2018, 114, 540–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schattemann, G.C.; Loushin, C.; Li, T.; Hart, C.E. PDGF-A is required for normal murine cardiovascular development. Dev. Biol. 1996, 176, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; van Putten, V.; Zarinetchi, F.; Nicks, E.M.; Thaler, S.; Heasley, E.L.; Nemenoff, A.R. Suppression of smooth-muscle α-actin expression by platelet-derived growth factor in vascular smooth-muscle cells involves Ras and cytosolic phospholipase A2. Biochem. J. 1997, 327, 709–716. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Y.; Liu, C.P.; Shiao, W.C.; Jayakumar, T.; Li, Y.S.; Chang, N.C.; Huang, S.Y.; Hsieh, C.Y. Inhibitory effect of PDGF-BB and serum-stimulated responses in vascular smooth muscle cell proliferation by hinokitiol via up-regulation of p21 and p53. Arch. Med. Sci. 2018, 14, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Ishigami, M.; Swertfeger, D.K.; Granholm, N.A.; Hui, D.Y. Apolipoprotein E inhibits platelet-derived growth factor-induced vascular smooth muscle cell migration and proliferation by suppressing signal transduction and preventing cell entry to G1 phase. J. Biol. Chem. 1998, 273, 20156–20161. [Google Scholar] [CrossRef] [Green Version]
- Kotani, M.; Fukuda, N.; Ando, H.; Hu, W.Y.; Kunimoto, S.; Saito, S.; Kanmatsuse, K. Chimeric DNA–RNA hammerhead ribozyme targeting PDGF A-chain mRNA specifically inhibits neointima formation in rat carotid artery after balloon injury. Cardiovasc. Res. 2003, 57, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Deguchi, J.; Namba, T.; Hamada, H.; Nakaoka, T.; Abe, J.; Sato, O.; Miyata, T.; Makuuchi, M.; Kurokawa, K.; Takuwa, Y. Targeting endogenous platelet-derived growth factor B-chain by adenovirus-mediated gene transfer potently inhibits in vivo smooth muscle proliferation after arterial injury. Gene Ther. 1999, 6, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar]
- Hautmann, M.B.; Madsen, C.S.; Owens, G.K. A transforming growth factor beta (TGFbeta) control element drives TGFbeta-induced stimulation of smooth muscle alpha-actin gene expression in concert with two CArG elements. J. Biol. Chem. 1997, 272, 10948–10956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapustin, A.N.; Chatrou, M.L.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; de Rosales, R.T.M.; Alvarez-Hernandez, D.; et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borissoff, J.I.; Spronk, H.M.H.; ten Cate, H. The hemostatic system as a modulator of atherosclerosis. N. Engl. J. Med. 2011, 364, 1746–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, F.-F.; Shang, X.-K.; Du, X.-L.; Chen, S. Rapamycin Treatment Attenuates Angiotensin II -induced Abdominal Aortic Aneurysm Formation via VSMC Phenotypic Modulation and Down-regulation of ERK1/2 Activity. Curr. Med. Sci. 2018, 38, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Yang, J.; Zhang, Y.; Dong, M.; Wang, S.; Zhang, Q.; Liu, F.F.; Zhang, K.; Zhang, C. Angiotensin-converting enzyme 2 and angiotensin 1–7: Novel therapeutic targets. Nat. Rev. Cardiol. 2014, 11, 413–426. [Google Scholar] [CrossRef]
- Moiseeva, E.P. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc. Res. 2001, 52, 372–386. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Wei, Y.; Gao, C.; Chen, X.; Kong, W.; Kong, D.; Zhao, Q. ECM-mimetic heparin glycosamioglycan-functionalized surface favors constructing functional vascular smooth muscle tissue in vitro. Colloids Surf. B Biointerfaces 2016, 146, 280–288. [Google Scholar] [CrossRef]
- Sazonova, O.v.; Isenberg, B.C.; Herrmann, J.; Lee, K.L.; Purwada, A.; Valentine, A.D.; Buczek-Thomas, J.A.; Wong, J.Y.; Nugent, M.A. Extracellular matrix presentation modulates vascular smooth muscle cell mechanotransduction. Matrix Biol. 2015, 41, 36–43. [Google Scholar] [CrossRef]
- Finney, A.C.; Stokes, K.Y.; Pattillo, C.B.; Orr, A.W. Integrin signaling in atherosclerosis. Cell. Mol. Life Sci. 2017, 74, 2263–2282. [Google Scholar] [CrossRef]
- Ichii, T.; Koyama, H.; Tanaka, S.; Kim, S.; Shioi, A.; Okuno, Y.; Raines, E.W.; Iwao, H.; Otani, S.; Nishizawa, Y. Fibrillar collagen specifically regulates human vascular smooth muscle cell genes involved in cellular responses and the pericellular matrix environment. Circ. Res. 2001, 88, 460–467. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.H.; Lin, K.T.; Chang, C.H.; Peng, H.C.; Huang, T.F. The integrin α2β1 agonist, aggretin, promotes proliferation and migration of VSMC through NF-kB translocation and PDGF production. Br. J. Pharmacol. 2009, 156, 846–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thyberg, J.; Hultgårdh-Nilsson, A. Fibronectin and the basement membrane components laminin and collagen type IV influence the phenotypic properties of subcultured rat aortic smooth muscle cells differently. Cell Tissue Res. 1994, 276, 263–271. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, W.N.; Davis, J.B.; Geissler, R.; Cottrill, C.M.; Noonan, J.A.; Todd, E.P. Supravalvular aortic stenosis. Clinical and pathologic observations in six patients. Arch. Pathol. Lab. Med. 1985, 109, 179–185. [Google Scholar]
- Bunton, T.E.; Biery, N.J.; Myers, L.; Gayraud, B.; Ramirez, F.; Dietz, H.C. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ. Res. 2001, 88, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Bouvet, C.; Moreau, S.; Blanchette, J.; de Blois, D.; Moreau, P. Sequential activation of matrix metalloproteinase 9 and transforming growth factor beta in arterial elastocalcinosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 856–862. [Google Scholar] [CrossRef]
- Pai, A.; Leaf, E.M.; El-Abbadi, M.; Giachelli, C.M. Elastin degradation and vascular smooth muscle cell phenotype change precede cell loss and arterial medial calcification in a uremic mouse model of chronic kidney disease. Am. J. Pathol. 2011, 178, 764–773. [Google Scholar] [CrossRef]
- Li, S.; Lao, J.; Benjamin, C.; Li, Y.S.; Zhao, Y.; Chu, J.; Chen, K.-D.; Tsou, T.C.; Peck, K.; Chien, S. Genomic analysis of smooth muscle cells in 3-dimensional collagen matrix1. FASEB J. 2002, 17, 97–99. [Google Scholar] [CrossRef]
- Tijore, A.; Behr, J.M.; Irvine, S.A.; Baisane, V.; Venkatraman, S. Bioprinted gelatin hydrogel platform promotes smooth muscle cell contractile phenotype maintenance. Biomed. Microdevics. 2018, 20, 32. [Google Scholar] [CrossRef]
- Sarkar, S.; Dadhania, M.; Rourke, P.; Desai, T.A.; Wong, J.Y. Vascular tissue engineering: Microtextured scaffold templates to control organization of vascular smooth muscle cells and extracellular matrix. Acta Biomater. 2005, 1, 93–100. [Google Scholar] [CrossRef]
- van Engeland, N.C.A.; Pollet, A.M.A.O.; den Toonder, J.M.J.; Bouten, C.V.C.; Stassen, O.M.J.A.; Sahlgren, C.M. A biomimetic microfluidic model to study signalling between endothelial and vascular smooth muscle cells under hemodynamic conditions. Lab Chip 2018, 18, 1607–1620. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, C.J.; Williams, B. Mechanical Strain–Induced Extracellular Matrix Production by Human Vascular Smooth Muscle Cells. Hypertension 2000, 36, 319–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Kaestner, K.H.; Owens, G.K. Conditional Deletion of Krüppel-Like Factor 4 Delays Downregulation of Smooth Muscle Cell Differentiation Markers but Accelerates Neointimal Formation Following Vascular Injury. Circ. Res. 2008, 102, 1548–1557. [Google Scholar] [CrossRef] [PubMed]
- Cherepanova, O.A.; Pidkovka, N.A.; Sarmento, O.F.; Yoshida, T.; Gan, Q.; Adiguzel, E.; Bendeck, M.P.; Berliner, J.; Leitinger, N.; Owens, G.K. Oxidized Phospholipids Induce Type VIII Collagen Expression and Vascular Smooth Muscle Cell Migration. Circ. Res. 2009, 104, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.C.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Kotchen, T.A. Historical trends and milestones in hypertension research: A model of the process of translational research. Hypertension 2011, 58, 522–538. [Google Scholar] [CrossRef] [Green Version]
- Franklin, S.S.; Wong, N.D. Hypertension and cardiovascular disease: Contributions of the framingham heart study. Glob. Heart 2013, 8, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Dawber, T.R.; Kannel, W.B.; Revotskie, N.; Stokes, J.; Kagan, A.; Gordon, T. Some factors associated with the development of coronary heart disease: Six years’ follow-up experience in the Framingham study. Am. J. Public Health Nations Health 1959, 49, 1349–1356. [Google Scholar] [CrossRef]
- Lewington, S.; Clarke, R.; Qizilbash, N.; Peto, R.; Collins, R.; Collaboration, P.S. Age-specific relevance of usual blood pressure to vascular mortality: A meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002, 360, 1903–1913. [Google Scholar]
- Rapsomaniki, E.; Timmis, A.; George, J.; Pujades-Rodriguez, M.; Shah, A.D.; Denaxas, S.; White, I.R.; Caulfield, M.J.; Deanfield, J.E.; Smeeth, L.; et al. Blood pressure and incidence of twelve cardiovascular diseases: Lifetime risks, healthy life-years lost, and age-specific associations in 1·25 million people. Lancet 2014, 383, 1899–1911. [Google Scholar] [CrossRef] [Green Version]
- Rahimi, K.; Emdin, C.A.; MacMahon, S. The epidemiology of blood pressure and its worldwide management. Circ. Res. 2015, 116, 925–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najjar, S.S.; Scuteri, A.; Shetty, V.; Wright, J.G.; Muller, D.C.; Fleg, J.L.; Spurgeon, H.P.; Ferrucci, L.; Lakatta, E.G. Pulse wave velocity is an independent predictor of the longitudinal increase in systolic blood pressure and of incident hypertension in the Baltimore Longitudinal Study of Aging. J. Am. Coll. Cardiol. 2008, 51, 1377–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, S.S.; Levy, D. Aging, blood pressure, and heart failure: What are the connections? Hypertension 2011, 58, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Verwoert, G.C.; Franco, O.H.; Hoeks, A.P.G.; Reneman, R.S.; Hofman, A.; V Duijn, C.M.; Sijbrands, E.J.G.; Witteman, J.C.M.; Mattace-Raso, F.U.S. Arterial stiffness and hypertension in a large population of untreated individuals: The Rotterdam Study. J. Hypertens. 2014, 32, 1606–1612; discussion 1612. [Google Scholar] [CrossRef]
- Harvey, A.; Montezano, A.C.; Touyz, R.M. Vascular biology of ageing-Implications in hypertension. J. Mol. Cell. Cardiol. 2015, 83, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Gimbrone, M.A.; García-Cardeña, G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc. Pathol. 2013, 22, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, G.F. Increased Aortic Stiffness: An Unfavorable Cardiorenal Connection. Hypertension 2004, 43, 151–153. [Google Scholar] [CrossRef] [Green Version]
- Dai, G.; Kaazempur-Mofrad, M.R.; Natarajan, S.; Zhang, Y.; Vaughn, S.; Blackman, B.R.; Kamm, R.D.; García-Cardeña, G.; Gimbrone, M.A. Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc. Natl. Acad. Sci. USA 2004, 101, 14871–14876. [Google Scholar] [CrossRef] [Green Version]
- Passerini, A.G.; Polacek, D.C.; Shi, C.; Francesco, N.M.; Manduchi, E.; Grant, G.R.; Pritchard, W.F.; Powell, S.; Chang, G.Y.; Stoeckert, C.J.; et al. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc. Natl. Acad. Sci. USA 2004, 101, 2482–2487. [Google Scholar] [CrossRef] [Green Version]
- Blackman, B.R.; García-Cardeña, G.; Gimbrone, M.A. A New In Vitro Model to Evaluate Differential Responses of Endothelial Cells to Simulated Arterial Shear Stress Waveforms. J. Biomech. Eng. 2002, 124, 397–407. [Google Scholar] [CrossRef]
- Wang, Y.X.; Xiang, C.; Liu, B.; Zhu, Y.; Luan, Y.; Liu, S.T.; Qin, K.R. A multi-component parallel-plate flow chamber system for studying the effect of exercise-induced wall shear stress on endothelial cells. Biomed. Eng. Online 2016, 15, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, B. Mechanical influences on vascular smooth muscle cell function. J. Hypertens. 1998, 16, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Kapustin, A.N.; Schoppet, M.; Schurgers, L.J.; Reynolds, J.L.; McNair, R.; Heiss, A.; Jahnen-Dechent, W.; Hackeng, T.M.; Schlieper, G.; Harrison, P.; et al. Prothrombin Loading of Vascular Smooth Muscle Cell–Derived Exosomes Regulates Coagulation and CalcificationHighlights. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e22–e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutcheson, J.D.; Goettsch, C.; Pham, T.; Iwashita, M.; Aikawa, M.; Singh, S.A.; Aikawa, E. Enrichment of calcifying extracellular vesicles using density-based ultracentrifugation protocol. J. Extracell. Vesicles 2014, 3, 25129. [Google Scholar] [CrossRef]
- Tanimura, A.; McGregor, D.H.; Anderson, H.C. Matrix Vesicles in Atherosclerotic Calcification. Proc. Soc. Exp. Biol. Med. 2016, 172, 173–177. [Google Scholar] [CrossRef]
- Chen, N.X.; O’Neill, K.D.; Moe, S.M. Matrix vesicles induce calcification of recipient vascular smooth muscle cells through multiple signaling pathways. Kidney Int. 2018, 93, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Carlo, A.S.; Nykjaer, A.; Willnow, T.E. Sorting receptor sortilin-a culprit in cardiovascular and neurological diseases. J. Mol. Med. (Berl.) 2014, 92, 905–911. [Google Scholar] [CrossRef]
- Goettsch, C.; Hutcheson, J.D.; Aikawa, M.; Iwata, H.; Pham, T.; Nykjaer, A.; Kjolby, M.; Rogers, M.; Michel, T.; Shibasaki, M.; et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J. Clin. Invest. 2016, 126, 1323–1336. [Google Scholar] [CrossRef]
- Boström, K.I.; Rajamannan, N.M.; Towler, D.A. The regulation of valvular and vascular sclerosis by osteogenic morphogens. Circ. Res. 2011, 109, 564–577. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Proudfoot, D.; Tyson, K.L.; Cary, N.R.; Edmonds, M.; Weissberg, P.L. Expression of mineralisation-regulating proteins in association with human vascular calcification. Z. Kardiol. 2000, 89, 63–68. [Google Scholar] [CrossRef]
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yan, S.M.; Zheng, H.L.; Hu, D.H.; Zhang, Y.T.; Guan, Q.H.; Ding, Q.L. A Mechanism Underlying Hypertensive Occurrence in the Metabolic Syndrome: Cooperative Effect of Oxidative Stress and Calcium Accumulation in Vascular Smooth Muscle Cells. Horm. Metab. Res. 2013, 46, 126–132. [Google Scholar] [PubMed]
- Fleckenstein, A.; Fleckenstein-Grün, G.; Frey, M.; Thimm, F. Experimental antiarteriosclerotic effects of calcium antagonists. J. Clin. Pharmacol. 1990, 30, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; He, L.; Qu, J.; Zhou, Y. Regulation of Vascular Smooth Muscle Cell Stiffness and Adhesion by [Ca2+]i: An Atomic Force Microscopy-Based Study. Microsc. Microanal. 2018, 24, 708–712. [Google Scholar] [CrossRef]
- Godfraind, T. Discovery and Development of Calcium Channel Blockers. Front. Pharmacol. 2017, 8, 286. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.X.; Kircelli, F.; O’Neill, K.D.; Chen, X.; Moe, S.M. Verapamil inhibits calcification and matrix vesicle activity of bovine vascular smooth muscle cells. Kidney Int. 2010, 77, 436–442. [Google Scholar] [CrossRef] [Green Version]
- Caballero-Gonzalez, F.J. Calcium Channel Blockers in the Management of Hypertension in the Elderly. Cardiovasc. Hematol. Agents Med. Chem. 2014, 12, 160–165. [Google Scholar] [CrossRef]
- Niederhoffer, N.; Marque, V.; Lartaud-Idjouadiene, I.; Duvivier, C.; Peslin, R.; Atkinson, J. Vasodilators, aortic elasticity, and ventricular end-systolic stress in nonanesthetized unrestrained rats. Hypertension 1997, 30, 1169–1174. [Google Scholar] [CrossRef]
- London, G.M.; Guérin, A.P.; Marchais, S.J.; Métivier, F.; Pannier, B.; Adda, H. Arterial media calcification in end-stage renal disease: Impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transpl. 2003, 18, 1731–1740. [Google Scholar] [CrossRef]
- Odink, A.E.; Mattace-Raso, F.U.; van der Lugt, A.; Hofman, A.; Hunink, M.G.M.; Breteler, M.M.; Krestin, G.P.; Witteman, J.C. The association of arterial stiffness and arterial calcification: The Rotterdam study. J. Hum. Hypertens. 2008, 22, 205–207. [Google Scholar] [CrossRef] [Green Version]
- Guérin, A.P.; London, G.M.; Marchais, S.J.; Metivier, F. Arterial stiffening and vascular calcifications in end-stage renal disease. Nephrol. Dial. Transplant 2000, 15, 1014–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenseil, J.E.; Mecham, R.P. Elastin in large artery stiffness and hypertension. J. Cardiovasc. Transl. Res. 2012, 5, 264–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaess, B.M.; Rong, J.; Larson, M.G.; Hamburg, N.M.; Vita, J.A.; Levy, D.; Benjamin, E.J.; Vasan, R.S.; Mitchell, G.F. Aortic stiffness, blood pressure progression, and incident hypertension. JAMA 2012, 308, 875–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehgel, N.L.; Zhu, Y.; Sun, Z.; Trzeciakowski, J.P.; Hong, Z.; Hunter, W.C.; Vatner, D.E.; Meininger, G.A.; Vatner, S.F. Increased vascular smooth muscle cell stiffness: A novel mechanism for aortic stiffness in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1281–H1287. [Google Scholar] [CrossRef]
- Csiszar, A.; Lehoux, S.; Ungvari, Z. Hemodynamic Forces, Vascular Oxidative Stress, and Regulation of BMP-2/4 Expression. Antioxid. Redox. Signal. 2009, 11, 1683–1697. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.D. Biomechanics of plaque rupture: Progress, problems, and new frontiers. Ann. Biomed. Eng. 2002, 30, 524–536. [Google Scholar] [CrossRef]
- Tang, D.; Yang, C.; Mondal, S.; Liu, F.; Canton, G.; Hatsukami, T.S.; Yuan, C. A negative correlation between human carotid atherosclerotic plaque progression and plaque wall stress: In vivo MRI-based 2D/3D FSI models. J. Biomech. 2008, 41, 727–736. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, P.N. Molecular Biology of Atherosclerosis. Physiol. Rev. 2013, 93, 1317–1542. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis--an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Chatrou, M.L.L.; Cleutjens, J.P.; van der Vusse, G.J.; Roijers, R.B.; Mutsaers, P.H.A.; Schurgers, L.J. Intra-Section Analysis of Human Coronary Arteries Reveals a Potential Role for Micro-Calcifications in Macrophage Recruitment in the Early Stage of Atherosclerosis. PLoS ONE 2015, 10, e0142335. [Google Scholar] [CrossRef]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, D.; Szklo, M.; Cushman, M.; Holvoet, P.; Polak, J.; Bahrami, H.; Jenny, N.S.; Ouyang, P. Association of endothelial and oxidative stress with metabolic syndrome and subclinical atherosclerosis: Multi-ethnic study of atherosclerosis. Eur. J. Clin. Nutr. 2011, 65, 818–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, R.; Glomset, J.A. Atherosclerosis and the arterial smooth muscle cell: Proliferation of smooth muscle is a key event in the genesis of the lesions of atherosclerosis. Science 1973, 180, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, H. Calcification of the media of the human aorta and its relation to intimal arteriosclerosis, ageing and disease. Am. J. Pathol. 2007, 1–23. [Google Scholar]
- Sangiorgi, G.; Rumberger, J.A.; Severson, A.; Edwards, W.D.; Gregoire, J.; Fitzpatrick, L.A.; Schwartz, R.S. Arterial Calcification and Not Lumen Stenosis Is Highly Correlated With Atherosclerotic Plaque Burden in Humans: A Histologic Study of 723 Coronary Artery Segments Using Nondecalcifying Methodology. J. Am. Coll. Cardiol. 1998, 31, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Pundziute, G.; Schuijf, J.D.; Jukema, J.W.; Decramer, I.; Sarno, G.; Vanhoenacker, P.K.; Reiber, J.H.C.; Schalij, M.J.; Wijns, W.; Bax, J.J. Head-to-head comparison of coronary plaque evaluation between multislice computed tomography and intravascular ultrasound radiofrequency data analysis. JACC. Cardiovasc. Interv. 2008, 1, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Granillo, G.A.; Carrascosa, P.; Bruining, N.; Waksman, R.; Garcia-Garcia, H.M. Defining the non-vulnerable and vulnerable patients with computed tomography coronary angiography: Evaluation of atherosclerotic plaque burden and composition. Eur. Heart J. Cardiovasc. Imaging 2016, 17, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Nissen, S.E. Effect of intensive lipid lowering on progression of coronary atherosclerosis: Evidence for an early benefit from the Reversal of Atherosclerosis with Aggressive Lipid Lowering (REVERSAL) trial. Am. J. Cardiol. 2005, 96, 61F–68F. [Google Scholar] [CrossRef]
- Ridker, P.M.; Pradhan, A.; MacFadyen, J.G.; Libby, P.; Glynn, R.J. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: An analysis from the JUPITER trial. Lancet 2012, 380, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Puri, R.; Libby, P.; Nissen, S.E.; Wolski, K.; Ballantyne, C.M.; Barter, P.J.; Chapman, M.J.; Erbel, R.; Raichlen, J.S.; Uno, K.; et al. Long-term effects of maximally intensive statin therapy on changes in coronary atheroma composition: Insights from SATURN. Eur. Heart J. Cardiovasc. Imaging 2014, 15, 380–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Räber, L.; Taniwaki, M.; Zaugg, S.; Kelbæk, H.; Roffi, M.; Holmvang, L.; Noble, S.; Pedrazzini, G.; Moschovitis, A.; Lüscher, T.F.; et al. Effect of high-intensity statin therapy on atherosclerosis in non-infarct-related coronary arteries (IBIS-4): A serial intravascular ultrasonography study. Eur. Heart J. 2015, 36, 490–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rennenberg, R.J.M.W.; Kessels, A.G.H.; Schurgers, L.J.; van Engelshoven, J.M.A.; de Leeuw, P.W.; Kroon, A.A. Vascular calcifications as a marker of increased cardiovascular risk: A meta-analysis. Vasc. Health Risk Manag. 2009, 5, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essalihi, R.; Dao, H.H.; Yamaguchi, N.; Moreau, P. A new model of isolated systolic hypertension induced by chronic warfarin and vitamin K1 treatment. Am. J. Hypertens. 2003, 16, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Essalihi, R.; Ouellette, V.; Hao Dao, H.; McKee, M.D.; Moreau, P. Phenotypic Modulation of Vascular Smooth Muscle Cells During Medial Arterial Calcification: A Role for Endothelin? J. Cardiovasc. Pharmacol. 2004, 44, S147–S150. [Google Scholar] [CrossRef]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; St Hilaire, C.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Heart J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef]
- Ehara, S.; Kobayashi, Y.; Yoshiyama, M.; Shimada, K.; Shimada, Y.; Fukuda, D.; Nakamura, Y.; Yamashita, H.; Yamagishi, H.; Takeuchi, K.; et al. Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: An intravascular ultrasound study. Circulation 2004, 110, 3424–3429. [Google Scholar] [CrossRef] [Green Version]
- Vengrenyuk, Y.; Carlier, S.; Xanthos, S.; Cardoso, L.; Ganatos, P.; Virmani, R.; Einav, S.; Gilchrist, L.; Weinbaum, S. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proc. Natl. Acad. Sci. USA 2006, 103, 14678–14683. [Google Scholar] [CrossRef] [Green Version]
- Schurgers, L.J.; Joosen, I.A.; Laufer, E.M.; Chatrou, M.L.L.; Herfs, M.; Winkens, M.H.M.; Westenfeld, R.; Veulemans, V.; Krueger, T.; Shanahan, C.M.; et al. Vitamin K-antagonists accelerate atherosclerotic calcification and induce a vulnerable plaque phenotype. PLoS ONE 2012, 7, e43229. [Google Scholar] [CrossRef] [Green Version]
- Iyemere, V.P.; Proudfoot, D.; Weissberg, P.L.; Shanahan, C.M. Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J. Int. Med. 2006, 260, 192–210. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L.; Sheppard, D.; Duffield, J.S.; Violette, S. Therapy for fibrotic diseases: Nearing the starting line. Sci. Trans. Med. 2013, 5, sr1–sr167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmoulière, A.; Varga, J.; de Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef] [PubMed]
- Liberman, M.; Pesaro, A.E.P.; Carmo, L.S.; Serrano Jr, C.V. Vascular calcification: Pathophysiology and clinical implications. Einstein 2013, 11, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Byon, C.H.; Yuan, K.; Chen, J.; Mao, X.; Heath, J.M.; Javed, A.; Zhang, K.; Anderson, P.G.; Chen, Y. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ. Res. 2012, 111, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Block, G.A.; Port, F.K. Re-evaluation of risks associated with hyperphosphatemia and hyperparathyroidism in dialysis patients: Recommendations for a change in management. Am. J. Kidney Dis. 2000, 35, 1226–1237. [Google Scholar] [CrossRef]
- Zhao, M.M.; Xu, M.J.; Cai, Y.; Zhao, G.; Guan, Y.; Kong, W.; Tang, C.; Wang, X. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 2011, 79, 1071–1079. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, B.K.; Pandey, K.B.; Abidi, A.B.; Rizvi, S.I. Markers of Oxidative Stress during Diabetes Mellitus. J. Biomark. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Ren, X.; Jiang, Y.; Jin, H.; Liu, N.; Li, J. Advanced glycation end products accelerate rat vascular calcification through RAGE/oxidative stress. BMC Cardiovasc. Disord. 2013, 13, 13. [Google Scholar] [CrossRef] [Green Version]
- Tada, Y.; Yano, S.; Yamaguchi, T.; Okazaki, K.; Ogawa, N.; Morita, M.; Sugimoto, T. Advanced glycation end products-induced vascular calcification is mediated by oxidative stress: Functional roles of NAD(P)H-oxidase. Horm. Metab. Res. 2013, 45, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Girerd, X.; Mourad, J.J.; Copie, X.; Moulin, C.; Acar, C.; Safar, M.; Laurent, S. Noninvasive detection of an increased vascular mass in untreated hypertensive patients. Am. J. Hypertens. 1994, 7, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Fridez, P.; Zulliger, M.; Bobard, F.; Montorzi, G.; Miyazaki, H.; Hayashi, K.; Stergiopulos, N. Geometrical, functional, and histomorphometric adaptation of rat carotid artery in induced hypertension. J. Biomech. 2003, 36, 671–680. [Google Scholar] [CrossRef]
- Safar, M.; Chamiot-Clerc, P.; Dagher, G.; Renaud, J.F. Pulse pressure, endothelium function, and arterial stiffness in spontaneously hypertensive rats. Hypertension 2001, 38, 1416–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arribas, S.M.; Hinek, A.; González, M.C. Elastic fibres and vascular structure in hypertension. Pharmacol. Ther. 2006, 111, 771–791. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem. Cell 2015, 16, 51–66. [Google Scholar] [CrossRef] [Green Version]
- Kramann, R.; Goettsch, C.; Wongboonsin, J.; Iwata, H.; Schneider, R.K.; Kuppe, C.; Kaesler, N.; Chang-Panesso, M.; Machado, F.G.; Gratwohl, S.; et al. Adventitial MSC-like Cells Are Progenitors of Vascular Smooth Muscle Cells and Drive Vascular Calcification in Chronic Kidney Disease. Cell Stem. Cell 2016, 19, 628–642. [Google Scholar] [CrossRef] [Green Version]
- Bersi, M.R.; Bellini, C.; Wu, J.; Montaniel, K.R.C.; Harrison, D.G.; Humphrey, J.D. Excessive Adventitial Remodeling Leads to Early Aortic Maladaptation in Angiotensin-Induced Hypertension. Hypertension 2016, 67, 890–896. [Google Scholar] [CrossRef] [Green Version]
- Majesky, M.W.; Horita, H.; Ostriker, A.; Lu, S.; Regan, J.N.; Bagchi, A.; Dong, X.R.; Poczobutt, J.; Nemenoff, R.A.; Weiser-Evans, M.C.M. Differentiated Smooth Muscle Cells Generate a Subpopulation of Resident Vascular Progenitor Cells in the Adventitia Regulated by Klf4Novelty and Significance. Circ. Res. 2017, 120, 296–311. [Google Scholar] [CrossRef]
- Wu, J.; Montaniel, K.R.C.; Saleh, M.A.; Xiao, L.; Chen, W.; Owens, G.K.; Humphrey, J.D.; Majesky, M.W.; Paik, D.T.; Hatzopoulos, A.K.; et al. Origin of Matrix-Producing Cells That Contribute to Aortic Fibrosis in Hypertension. Hypertension 2016, 67, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Cecelja, M.; Chowienczyk, P. Role of arterial stiffness in cardiovascular disease. JRSM Cardiovasc. Dis. 2012, 1, 11. [Google Scholar] [CrossRef]
- Lee, H.Y.; Oh, B.H. Aging and arterial stiffness. Circ. J. 2010, 74, 2257–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancia, G.; Fagard, R.; Narkiewicz, K.; Redon, J.; Zanchetti, A.; Bohm, M.; Christiaens, T.; Cifkova, R.; de Backer, G.; Dominiczak, A.; et al. The task force for the management ofarterial hypertension of the european society ofhypertension (esh) and of the european society of cardiology (esc). J. Hypertens. 2013, 31, 1281–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nürnberger, J.; Keflioglu-Scheiber, A.; Opazo Saez, A.M.; Wenzel, R.R.; Philipp, T.; Schäfers, R.F. Augmentation index is associated with cardiovascular risk. J. Hypertens. 2002, 20, 2407–2414. [Google Scholar] [CrossRef] [PubMed]
- Vlachopoulos, C.; Ioakeimidis, N.; Aznaouridis, K.; Terentes-Printzios, D.; Rokkas, K.; Aggelis, A.; Panagiotakos, D.; Stefanadis, C. Prediction of Cardiovascular Events With Aortic Stiffness in Patients With Erectile Dysfunction. Hypertension 2014, 64, 672–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, B.; Rabkin, S.W. Brachial-ankle pulse wave velocity is the only index of arterial stiffness that correlates with a mitral valve indices of diastolic dysfunction, but no index correlates with left atrial size. Cardiol. Res. Pract. 2013, 2013, 986847. [Google Scholar] [CrossRef] [PubMed]
- Spronck, B.; Heusinkveld, M.H.G.; Vanmolkot, F.H.; Roodt, J.O.; Hermeling, E.; Delhaas, T.; Kroon, A.A.; Reesink, K.D. Pressure-dependence of arterial stiffness: Potential clinical implications. J. Hypertens. 2015, 33, 330–338. [Google Scholar] [CrossRef]
- Spronck, B.; Avolio, A.P.; Tan, I.; Butlin, M.; Reesink, K.D.; Delhaas, T. Arterial stiffness index beta and cardio-ankle vascular index inherently depend on blood pressure but can be readily corrected. J. Hypertens. 2017, 35, 98–104. [Google Scholar] [CrossRef]
- Spronck, B.; Delhaas, T.; de Lepper, A.G.; Giroux, J.; Goldwasser, F.; Boutouyrie, P.; Alivon, M.; Reesink, K.D. Patient-specific blood pressure correction technique for arterial stiffness: Evaluation in a cohort on anti-angiogenic medication. Hypertens. Res. 2017, 40, 752–757. [Google Scholar] [CrossRef]
- Dao, H.H.; Essalihi, R.; Bouvet, C.; Moreau, P. Evolution and modulation of age-related medial elastocalcinosis: Impact on large artery stiffness and isolated systolic hypertension. Cardiovasc. Res. 2005, 66, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Laucyte-Cibulskiene, A.; Petraviciute, M.; Gudynaite, M.; Gumbys, L.; Valanciene, D.; Galiauskiene, K.; Ryliskyte, L.; Rimsevicius, L.; Miglinas, M.; Strupas, K. Mismatch between stiffness in elastic and muscular arteries as a predictor of vascular calcification in dialysis patients. Aging Clin. Exp. Res. 2018, 30, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Sehgel, N.L.; Sun, Z.; Hong, Z.; Hunter, W.C.; Hill, M.A.; Vatner, D.E.; Vatner, S.F.; Meininger, G.A. Augmented Vascular Smooth Muscle Cell Stiffness and Adhesion When Hypertension Is Superimposed on Aging. Hypertension 2014, 65, 370–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imanishi, M.; Tomita, S.; Ishizawa, K.; Kihira, Y.; Ueno, M.; Izawa-Ishizawa, Y.; Ikeda, Y.; Yamano, N.; Tsuchiya, K.; Tamaki, T. Smooth muscle cell-specific Hif-1α deficiency suppresses angiotensin II-induced vascular remodelling in mice. Cardiovasc. Res. 2014, 102, 460–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, W.; Fontaine, V.; Pueyo, M.E.; Laquay, N.; Messika-Zeitoun, D.; Philippe, M.; Arnal, J.F.; Jacob, M.P.; Michel, J.B. Molecular plasticity of vascular wall during N(G)-nitro-L-arginine methyl ester-induced hypertension: Modulation of proinflammatory signals. Hypertension 2000, 36, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, S.; Boutouyrie, P. Arterial stiffness and stroke in hypertension: Therapeutic implications for stroke prevention. CNS Drug. 2005, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Segers, P.; Swillens, A.; Taelman, L.; Vierendeels, J. Wave reflection leads to over- and underestimation of local wave speed by the PU- and QA-loop methods: Theoretical basis and solution to the problem. Physiol. Meas. 2014, 35, 847–861. [Google Scholar] [CrossRef]
- Hermeling, E.; Reesink, K.D.; Kornmann, L.M.; Reneman, R.S.; Hoeks, A.P. The dicrotic notch as alternative time-reference point to measure local pulse wave velocity in the carotid artery by means of ultrasonography. J. Hypertens. 2009, 27, 2028–2035. [Google Scholar] [CrossRef]
- Mirault, T.; Pernot, M.; Frank, M.; Couade, M.; Niarra, R.; Azizi, M.; Emmerich, J.; Jeunemaître, X.; Fink, M.; Tanter, M.; et al. Carotid stiffness change over the cardiac cycle by ultrafast ultrasound imaging in healthy volunteers and vascular Ehlers-Danlos syndrome. J. Hypertens. 2015, 33, 1890–1896. [Google Scholar] [CrossRef]
- de Korte, C.L.; Fekkes, S.; Nederveen, A.J.; Manniesing, R.; Hansen, H.R.H.G. Review: Mechanical Characterization of Carotid Arteries and Atherosclerotic Plaques. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 2016, 63, 1613–1623. [Google Scholar] [CrossRef]
- Couade, M.; Pernot, M.; Prada, C.; Messas, E.; Emmerich, J.; Bruneval, P.; Criton, A.; Fink, M.; Tanter, M. Quantitative assessment of arterial wall biomechanical properties using shear wave imaging. Ultrasound Med. Biol. 2010, 36, 1662–1676. [Google Scholar] [CrossRef]
- Grotenhuis, H.B.; Westenberg, J.J.M.; Steendijk, P.; van der Geest, R.J.; Ottenkamp, J.; Bax, J.J.; Jukema, J.W.; de Roos, A. Validation and reproducibility of aortic pulse wave velocity as assessed with velocity-encoded MRI. J. Magn. Reson. Imaging 2009, 30, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Polak, J.F.; Szklo, M.; Kronmal, R.A.; Burke, G.L.; Shea, S.; Zavodni, A.E.H.; O’Leary, D.H. The Value of Carotid Artery Plaque and Intima-Media Thickness for Incident Cardiovascular Disease: The Multi-Ethnic Study of Atherosclerosis. J. Am. Heart Assoc. 2013, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakiri, T.; Yano, Y.; Sato, Y.; Hatakeyama, K.; Marutsuka, K.; Fujimoto, S.; Kitamura, K.; Kario, K.; Asada, Y. Usefulness of carotid intima-media thickness measurement as an indicator of generalized atherosclerosis: Findings from autopsy analysis. Atherosclerosis 2012, 225, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.R.; Raichlen, J.S.; Riley, W.A.; Evans, G.W.; Palmer, M.K.; O’Leary, D.H.; Grobbee, D.E.; Bots, M.L.; METEOR Study Group. Effect of Rosuvastatin on Progression of Carotid Intima-Media Thickness in Low-Risk Individuals With Subclinical Atherosclerosis. JAMA 2007, 297, 1344–1353. [Google Scholar] [CrossRef] [Green Version]
- de Groot, E.; Jukema, J.W.; Montauban van Swijndregt, A.D.; Zwinderman, A.H.; Ackerstaff, R.G.A.; van der Steen, A.F.W.; Bom, N.; Lie, K.I.; Bruschke, A.V.G. B-Mode Ultrasound Assessment of Pravastatin Treatment Effect on Carotid and Femoral Artery Walls and Its Correlations With Coronary Arteriographic Findings: A Report of the Regression Growth Evaluation Statin Study (REGRESS). J. Am. Coll. Cardiol. 1998, 31, 1561–1567. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, M.W.; Polak, J.F.; Kavousi, M.; Mathiesen, E.B.; Völzke, H.; Tuomainen, T.-P.; Sander, D.; Plichart, M.; Catapano, A.L.; Robertson, C.M.; et al. Carotid intima-media thickness progression to predict cardiovascular events in the general population (the PROG-IMT collaborative project): A meta-analysis of individual participant data. Lancet 2012, 379, 2053–2062. [Google Scholar] [CrossRef] [Green Version]
- Ravani, A.; Werba, J.P.; Frigerio, B.; Sansaro, D.; Amato, M.; Tremoli, E.; Baldassarre, D. Assessment and Relevance of Carotid Intima-Media Thickness (C-IMT) in Primary and Secondary Cardiovascular Prevention. Curr. Pharm. Des. 2015, 21, 1164–1171. [Google Scholar] [CrossRef] [Green Version]
- Zaid, M.; Fujiyoshi, A.; Kadota, A.; Abbott, R.D.; Miura, K. Coronary Artery Calcium and Carotid Artery Intima Media Thickness and Plaque: Clinical Use in Need of Clarification. J. Atheroscler. Thromb. 2017, 24, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Iglesias del Sol, A.; Bots, M.L.; Grobbee, D.E.; Hofman, A.; Witteman, J.C.M. Carotid intima-media thickness at different sites: Relation to incident myocardial infarction. The Rotterdam Study. Eur. Heart J. 2002, 23, 934–940. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, D.H.; Bots, M.L. Imaging of atherosclerosis: Carotid intima-media thickness. Eur. Heart J. 2010, 31, 1682–1689. [Google Scholar] [CrossRef] [Green Version]
- Skilton, M.R.; Sérusclat, A.; Sethu, A.H.A.U.; Brun, S.; Bernard, S.; Balkau, B.; Moulin, P.; Bonnet, F. Noninvasive measurement of carotid extra-media thickness: Associations with cardiovascular risk factors and intima-media thickness. JACC Cardiovasc. Imaging 2009, 2, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, S.; Budoff, M.J.; Katz, R.; Blumenthal, R.S.; Bertoni, A.G.; Nasir, K.; Szklo, M.; Barr, R.G.; Wong, N.D. Impact of subclinical atherosclerosis on cardiovascular disease events in individuals with metabolic syndrome and diabetes: The multi-ethnic study of atherosclerosis. Diabetes Care 2011, 34, 2285–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulos, N.M.; Gardin, J.M.; Malik, S.; Postley, J.; Wong, N.D. Carotid Plaque Characterization, Stenosis, and Intima-Media Thickness According to Age and Gender in a Large Registry Cohort. Am. J. Cardiol. 2016, 117, 1185–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agatston, A.S.; Janowitz, W.R.; Hildner, F.J.; Zusmer, N.R.; Viamonte, M.; Detrano, R. Quantification of coronary artery calcium using ultrafast computed tomography. J. Am. Coll. Cardiol. 1990, 15, 827–832. [Google Scholar] [CrossRef] [Green Version]
- Popovic, W.J.; Brown, J.E.; Adamson, J.W. The influence of thyroid hormones on in vitro erythropoiesis. Mediation by a receptor with beta adrenergic properties. J. Clin. Invest. 1977, 60, 907–913. [Google Scholar] [CrossRef]
- Greenland, P.; Bonow, R.O.; Brundage, B.H.; Budoff, M.J.; Eisenberg, M.J.; Grundy, S.M.; Lauer, M.S.; Post, W.S.; Raggi, P.; Redberg, R.F.; et al. ACCF/AHA 2007 clinical expert consensus document on coronary artery calcium scoring by computed tomography in global cardiovascular risk assessment and in evaluation of patients with chest pain: A report of the American College of Cardiology Foundation Clinical Expert Consensus Task Force (ACCF/AHA Writing Committee to Update the 2000 Expert Consensus Document on Electron Beam Computed Tomography). Circulation 2007, 115, 402–426. [Google Scholar]
- Fujiyoshi, A.; Miura, K.; Ohkubo, T.; Kadowaki, T.; Kadowaki, S.; Zaid, M.; Hisamatsu, T.; Sekikawa, A.; Budoff, M.J.; Liu, K.; et al. Cross-Sectional Comparison of Coronary Artery Calcium Scores Between Caucasian Men in the United States and Japanese Men in JapanThe Multi-Ethnic Study of Atherosclerosis and the Shiga Epidemiological Study of Subclinical Atherosclerosis. Am. J. Epidemiol. 2014, 180, 590–598. [Google Scholar] [CrossRef] [Green Version]
- Raggi, P.; Callister, T.Q.; Shaw, L.J. Progression of coronary artery calcium and risk of first myocardial infarction in patients receiving cholesterol-lowering therapy. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1272–1277. [Google Scholar] [CrossRef]
- Raggi, P. Cardiovascular disease: Coronary artery calcification predicts risk of CVD in patients with CKD. Nat. Rev. Nephrol. 2017, 13, 324–326. [Google Scholar] [CrossRef]
- Yamamoto, H.; Kitagawa, T.; Kihara, Y. Clinical Implications of the Coronary Artery Calcium Score in Japanese Patients. J. Atheroscler. Thromb. 2014, 21, 1101–1108. [Google Scholar] [CrossRef] [Green Version]
- Krueger, T.; Schlieper, G.; Schurgers, L.; Cornelis, T.; Cozzolino, M.; Jacobi, J.; Jadoul, M.; Ketteler, M.; Rump, L.C.; Stenvinkel, P.; et al. Vitamin K1 to slow vascular calcification in haemodialysis patients (VitaVasK trial): A rationale and study protocol. Nephrol. Dial. Trans. 2014, 29, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Vossen, L.M.; Schurgers, L.J.; van Varik, B.J.; Kietselaer, B.L.J.H.; Vermeer, C.; Meeder, J.G.; Rahel, B.M.; van Cauteren, Y.J.M.; Hoffland, G.A.; Rennenberg, R.J.M.W.; et al. Menaquinone-7 Supplementation to Reduce Vascular Calcification in Patients with Coronary Artery Disease: Rationale and Study Protocol (VitaK-CAC Trial). Nutrients 2015, 7, 8905–8915. [Google Scholar] [CrossRef] [Green Version]
- Peeters, F.E.C.M.; van Mourik, M.J.W.; Meex, S.J.R.; Bucerius, J.; Schalla, S.M.; Gerretsen, S.C.; Mihl, C.; Dweck, M.R.; Schurgers, L.J.; Wildberger, J.E.; et al. Bicuspid Aortic Valve Stenosis and the Effect of Vitamin K2 on Calcification Using 18F-Sodium Fluoride Positron Emission Tomography/Magnetic Resonance: The BASIK2 Rationale and Trial Design. Nutrients 2018, 10, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geleijnse, J.M.; Vermeer, C.; Grobbee, D.E.; Schurgers, L.J.; Knapen, M.H.J.; van der Meer, I.M.; Hofman, A.; Witteman, J.C.M. Dietary Intake of Menaquinone Is Associated with a Reduced Risk of Coronary Heart Disease: The Rotterdam Study. J. Nutr. 2004, 134, 3100–3105. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, V.M.; Reinartz, S.; Kaesler, N.; Krüger, T.; Dirrichs, T.; Kramann, R.; Peeters, F.; Floege, J.; Keszei, A.; Marx, N.; et al. Slower Progress of Aortic Valve Calcification With Vitamin K Supplementation: Results From a Prospective Interventional Proof-of-Concept Study. Circulation 2017, 135, 2081–2083. [Google Scholar] [CrossRef]
- Houslay, E.S.; Cowell, S.J.; Prescott, R.J.; Reid, J.; Burton, J.; Northridge, D.B.; Boon, N.A.; Newby, D.E.; Scottish Aortic Stenosis and Lipid Lowering Therapy, Impact on Regression trial Investigators. Progressive coronary calcification despite intensive lipid-lowering treatment: A randomised controlled trial. Heart 2006, 92, 1207–1212. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Qureshi, A.R.; Parini, P.; Hurt-Camejo, E.; Ripsweden, J.; Brismar, T.B.; Barany, P.; Jaminon, A.M.; Schurgers, L.J.; Heimbürger, O.; et al. Does statins promote vascular calcification in chronic kidney disease? Eur. J. Clin. Investig. 2017, 47, 137–148. [Google Scholar] [CrossRef]
- Terry, J.G.; Carr, J.J.; Kouba, E.O.; Davis, D.H.; Menon, L.; Bender, K.; Chandler, E.T.; Morgan, T.; Crouse, J.R. Effect of simvastatin (80 mg) on coronary and abdominal aortic arterial calcium (from the coronary artery calcification treatment with zocor [CATZ] study). Am. J. Cardiol. 2007, 99, 1714–1717. [Google Scholar] [CrossRef]
- Criqui, M.H.; Denenberg, J.O.; Ix, J.H.; McClelland, R.L.; Wassel, C.L.; Rifkin, D.E.; Carr, J.J.; Budoff, M.J.; Allison, M.A. Calcium density of coronary artery plaque and risk of incident cardiovascular events. JAMA 2014, 311, 271–278. [Google Scholar] [CrossRef]
- Rumberger, J.A.; Simons, D.B.; Fitzpatrick, L.A.; Sheedy, P.F.; Schwartz, R.S. Coronary Artery Calcium Area by Electron-Beam Computed Tomography and Coronary Atherosclerotic Plaque Area. Circulation 1995, 92, 2157–2162. [Google Scholar] [CrossRef]
- Cho, I.; Ó Hartaigh, B.; Gransar, H.; Valenti, V.; Lin, F.Y.; Achenbach, S.; Berman, D.S.; Budoff, M.J.; Callister, T.Q.; Al-Mallah, M.H.; et al. Prognostic implications of coronary artery calcium in the absence of coronary artery luminal narrowing. Atherosclerosis 2017, 262, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amann, K. Media calcification and intima calcification are distinct entities in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1599–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vos, A.; Kockelkoren, R.; de Vis, J.B.; van der Schouw, Y.T.; van der Schaaf, I.C.; Velthuis, B.K.; Mali, W.P.T.M.; de Jong, P.A.; DUST study group. Risk factors for atherosclerotic and medial arterial calcification of the intracranial internal carotid artery. Atherosclerosis 2018, 276, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alluri, K.; Joshi, P.H.; Henry, T.S.; Blumenthal, R.S.; Nasir, K.; Blaha, M.J. Scoring of coronary artery calcium scans: History, assumptions, current limitations, and future directions. Atherosclerosis 2015, 239, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Yoshioka, K.; Takagi, H.; Schuijf, J.D.; Arakita, K. Novel developments in non-invasive imaging of peripheral arterial disease with CT: Experience with state-of-the-art, ultra-high-resolution CT and subtraction imaging. Clin. Radiol. 2018, 74, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberger, Z.D.; Valle, J.A.; Dandekar, V.K.; Chan, P.S.; Ko, D.T.; Nallamothu, B.K. Are changes in carotid intima-media thickness related to risk of nonfatal myocardial infarction? A critical review and meta-regression analysis. Am. Heart J. 2010, 160, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Criqui, M.H.; Knox, J.B.; Denenberg, J.O.; Forbang, N.I.; McClelland, R.L.; Novotny, T.E.; Sandfort, V.; Waalen, J.; Blaha, M.J.; Allison, M.A. Coronary Artery Calcium Volume and Density: Potential Interactions and Overall Predictive Value: The Multi-Ethnic Study of Atherosclerosis. JACC Cardiovasc. Imaging 2017, 10, 845–854. [Google Scholar] [CrossRef]
- Kim, K.P.; Einstein, A.J.; Berrington de González, A. Coronary artery calcification screening: Estimated radiation dose and cancer risk. Arch. Int. Med. 2009, 169, 1188–1194. [Google Scholar] [CrossRef] [Green Version]
- Bucerius, J.; Dijkgraaf, I.; Mottaghy, F.M.; Schurgers, L.J. Target identification for the diagnosis and intervention of vulnerable atherosclerotic plaques beyond 18F-fluorodeoxyglucose positron emission tomography imaging: Promising tracers on the horizon. Eur. J. Nucl. Med. Mol Imaging 2019, 46, 251–265. [Google Scholar] [CrossRef] [Green Version]
- Blau, M.; Ganatra, R.; Bender, M.A. 18F-fluoride for bone imaging. Semin. Nucl. Med. 1972, 2, 31–37. [Google Scholar] [CrossRef]
- Dweck, M.R.; Chow, M.W.L.; Joshi, N.V.; Williams, M.C.; Jones, C.; Fletcher, A.M.; Richardson, H.; White, A.; McKillop, G.; van Beek, E.J.R.; et al. Coronary arterial 18F-sodium fluoride uptake: A novel marker of plaque biology. J. Am. Coll. Cardiol. 2012, 59, 1539–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, N.V.; Vesey, A.T.; Williams, M.C.; Shah, A.S.V.; Calvert, P.A.; Craighead, F.H.M.; Yeoh, S.E.; Wallace, W.; Salter, D.; Fletcher, A.M.; et al. 18F-fluoride positron emission tomography for identification of ruptured and high-risk coronary atherosclerotic plaques: A prospective clinical trial. Lancet 2014, 383, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Irkle, A.; Vesey, A.T.; Lewis, D.Y.; Skepper, J.N.; Bird, J.L.E.; Dweck, M.R.; Joshi, F.R.; Gallagher, F.A.; Warburton, E.A.; Bennett, M.R.; et al. Identifying active vascular microcalcification by 18F-sodium fluoride positron emission tomography. Nat. Commun. 2015, 6, 7495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutcheson, J.D.; Maldonado, N.; Aikawa, E. Small entities with large impact: Microcalcifications and atherosclerotic plaque vulnerability. Curr. Opin. Lipidol. 2014, 25, 327–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Gene Name | Abbreviation | Expression | |
|---|---|---|---|---|
| Contractile | Synthetic | |||
| Alpha smooth muscle actin | ACTA-2 | ⍺-SMA | + | + |
| Smooth muscle myosin heavy chain | MYH11 | SMMHC | + | − |
| Smooth muscle 22 alpha | TAGLN | SM22-⍺ | + | − |
| Smoothelin | SMTN | Smtn | + | − |
| Calponin | CNN-1 | CNN-1 | + | − |
| Tumor necrosis factor alpha | TNFA | TNF-⍺ | + | |
| S100 calcium binding protein A4 | S100A4 | S100A4 | − | + |
| Monocyte chemoattractant protein 1 | CCL2 | MCP-1 | − | + |
| Factors Involved in VSMC Phenotype Switching | Phenotype | |
|---|---|---|
| Biochemical compounds | Contractile | Synthetic |
| PDGF | − | + |
| TGF-β | + | − |
| PARs | − | + |
| TNF-⍺ | + | + |
| Angiotensin II | + | + |
| Extracellular matrix components | ||
| Integrin: ⍺1β1, ⍺7β1, ⍺8β1 | + | − |
| Integrin: ⍺2β1, ⍺5β1, ⍺vβ3 | − | + |
| Collagen type I | − | + |
| Collagen type IV | + | - |
| Elastin | + | - |
| Heparin | + | - |
| Fibronectin | − | + |
| Laminin | + | − |
| Physical factors | ||
| Tensile stress | − | + |
| Shear stress | − | + |
| Transcription | ||
| KLF4 | − | + |
| Oct4 | − | + |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaminon, A.; Reesink, K.; Kroon, A.; Schurgers, L. The Role of Vascular Smooth Muscle Cells in Arterial Remodeling: Focus on Calcification-Related Processes. Int. J. Mol. Sci. 2019, 20, 5694. https://doi.org/10.3390/ijms20225694
Jaminon A, Reesink K, Kroon A, Schurgers L. The Role of Vascular Smooth Muscle Cells in Arterial Remodeling: Focus on Calcification-Related Processes. International Journal of Molecular Sciences. 2019; 20(22):5694. https://doi.org/10.3390/ijms20225694
Chicago/Turabian StyleJaminon, Armand, Koen Reesink, Abraham Kroon, and Leon Schurgers. 2019. "The Role of Vascular Smooth Muscle Cells in Arterial Remodeling: Focus on Calcification-Related Processes" International Journal of Molecular Sciences 20, no. 22: 5694. https://doi.org/10.3390/ijms20225694
APA StyleJaminon, A., Reesink, K., Kroon, A., & Schurgers, L. (2019). The Role of Vascular Smooth Muscle Cells in Arterial Remodeling: Focus on Calcification-Related Processes. International Journal of Molecular Sciences, 20(22), 5694. https://doi.org/10.3390/ijms20225694