Calcium Phosphate Bions Cause Intimal Hyperplasia in Intact Aortas of Normolipidemic Rats through Endothelial Injury

 ,
,
Abstract
:1. Introduction
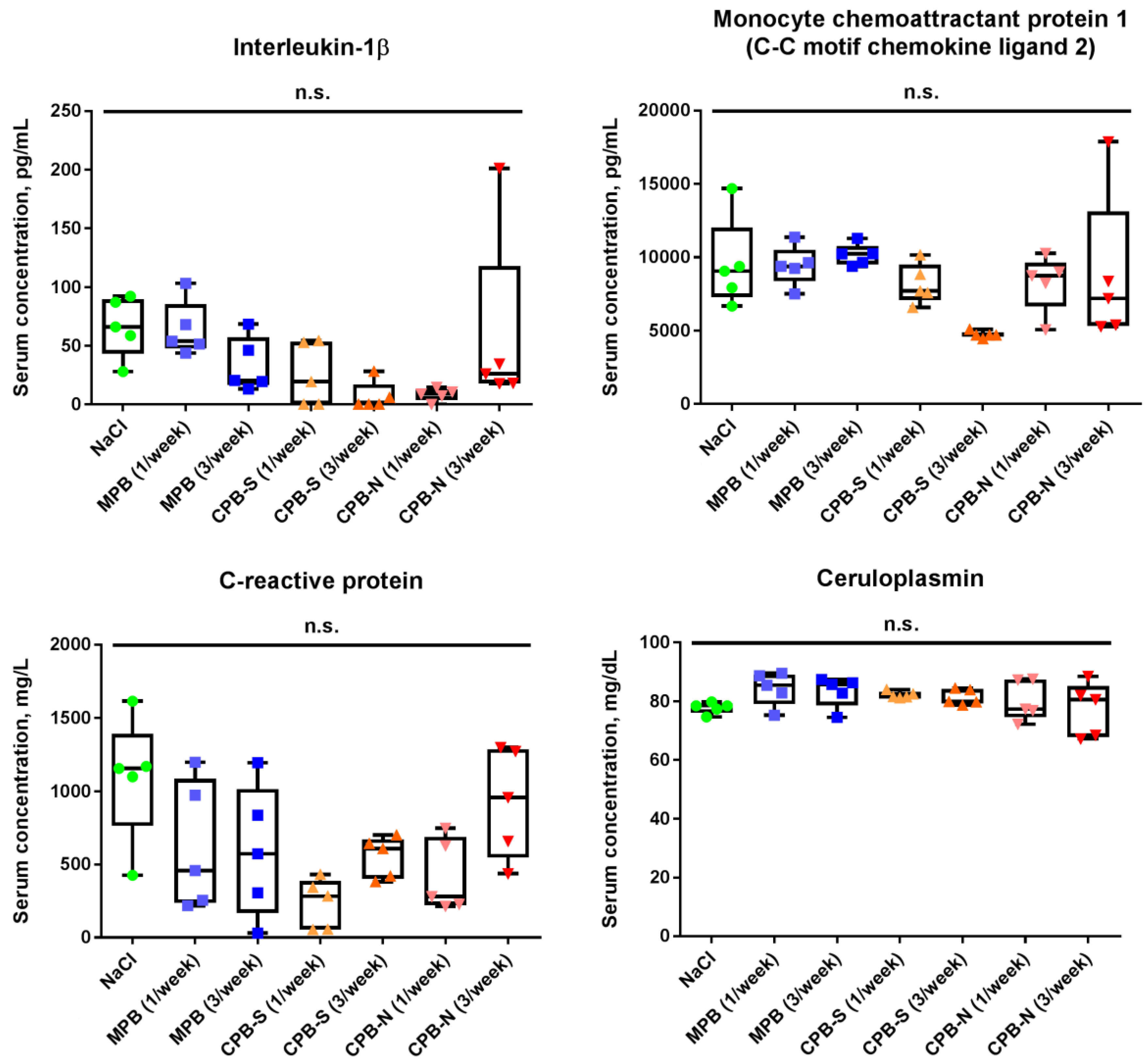
2. Results
3. Discussion
4. Materials and Methods
4.1. Artificial Synthesis of Calcium Phosphate and Magnesium Phosphate Bions (MPBs)
4.2. Animal Model of Bion-Induced Endothelial Injury
4.3. Histological Examination
4.4. Immunofluorescence Staining
4.5. Measurement of Serum Transaminases and Pro-Inflammatory Molecules
4.6. Gene Expression Profiling and Measurement of Cytokines in Cell Culture Supernatant
4.7. Proteomic Profiling
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CPBs | Calcium phosphate bions |
| CKD | Chronic kidney disease |
| CPB-S | Spherical calcium phosphate bions |
| CPB-N | Needle-shaped calcium phosphate bions |
| MPBs | Magnesium phosphate bions |
| ECM | Extracellular matrix |
| CD | Cluster of differentiation |
| αSMA | α-Smooth muscle actin |
| ApoE | Apolipoprotein E |
| LDLR | Low-density lipoprotein receptor |
| DAPI | 4′,6-Diamidino-2-phenylindole |
| AST | Aspartate transaminase |
| ALT | Alanine transaminase |
| IL | Interleukin |
| MCP-1 | Monocyte chemoattractant protein 1 |
| CCL2 | C-C Motif chemokine ligand 2 |
| HCAECs | Human coronary artery endothelial cells |
| HITAECs | Human internal thoracic artery endothelial cells |
| XIAP | X-Linked inhibitor of apoptosis protein |
| ICAM | Intercellular cell adhesion molecule |
| SELP | P-Selectin |
| MMP | Matrix metalloproteinase |
| ACTB | Actin beta |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| B2M | Beta-2 microglobulin |
| SCARF | Scavenger receptor class F member |
| LDLR | Low-density lipoprotein receptor |
| VLDLR | Very low-density lipoprotein receptor |
| VCAM | Vascular cell adhesion molecule |
| PECAM | Platelet endothelial cell adhesion molecule |
| SELE | E-Selectin |
| CDH | Cadherin |
| IL1R | Interleukin-1 receptor |
| TNFRSF | Tumor necrosis factor receptor superfamily |
| NOS | Nitric oxide synthase |
| PXDN | Peroxidasin |
| VWF | von Willebrand factor |
| KDR | Kinase insert domain receptor |
| FAP | Familial adenomatous polyposis |
| ACTA2 | Actin alpha 2 |
| SMTN | Smoothelin |
| VIM | Vimentin |
| COL4A1 | Collagen 4 alpha 1 subunit |
| ZEB | Zinc finger E-box-binding homeobox 1 |
| ESRD | End-stage renal disease |
| EDTA | Ethylenediaminetetraacetic acid |
| TACT | Trial to Assess Chelation Therapy |
| OD | Optical density |
| PBS | Phosphate buffered saline |
References
- Wu, C.Y.; Young, L.; Young, D.; Martel, J.; Young, J.D. Bions: A family of biomimetic mineralo-organic complexes derived from biological fluids. PLoS ONE 2013, 8, e75501. [Google Scholar] [CrossRef] [PubMed]
- Heiss, A.; DuChesne, A.; Denecke, B.; Grötzinger, J.; Yamamoto, K.; Renné, T.; Jahnen-Dechent, W. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J. Biol. Chem. 2003, 278, 13333–13341. [Google Scholar] [CrossRef] [PubMed]
- Heiss, A.; Eckert, T.; Aretz, A.; Richtering, W.; van Dorp, W.; Schäfer, C.; Jahnen-Dechent, W. Hierarchical role of fetuin-A and acidic serum proteins in the formation and stabilization of calcium phosphate particles. J. Biol. Chem. 2008, 283, 14815–14825. [Google Scholar] [CrossRef] [PubMed]
- Heiss, A.; Jahnen-Dechent, W.; Endo, H.; Schwahn, D. Structural dynamics of a colloidal protein-mineral complex bestowing on calcium phosphate a high solubility in biological fluids. Biointerphases 2007, 2, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Rochette, C.N.; Rosenfeldt, S.; Heiss, A.; Narayanan, T.; Ballauff, M.; Jahnen-Dechent, W. A shielding topology stabilizes the early stage protein-mineral complexes of fetuin-A and calcium phosphate: A time-resolved small-angle X-ray study. Chembiochem 2009, 10, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Kutikhin, A.G.; Velikanova, E.A.; Mukhamadiyarov, R.A.; Glushkova, T.V.; Borisov, V.V.; Matveeva, V.G.; Antonova, L.V.; Filip’ev, D.E.; Golovkin, A.S.; Shishkova, D.K.; et al. Apoptosis-mediated endothelial toxicity but not direct calcification or functional changes in anti-calcification proteins defines pathogenic effects of calcium phosphate bions. Sci. Rep. 2016, 6, 27255. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Shanahan, C.M. Medial Arterial Calcification: An Overlooked Player in Peripheral Arterial Disease. Arter. Thromb. Vasc. Biol. 2016, 36, 1475–1482. [Google Scholar] [CrossRef]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; St Hilaire, C.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Heart. J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef]
- Keyzer, C.A.; de Borst, M.H.; van den Berg, E.; Jahnen-Dechent, W.; Arampatzis, S.; Farese, S.; Bergmann, I.P.; Floege, J.; Navis, G.; Bakker, S.J.; et al. Calcification Propensity and Survival among Renal Transplant Recipients. J. Am. Soc. Nephrol. 2016, 27, 239–248. [Google Scholar] [CrossRef]
- Dahle, D.O.; Åsberg, A.; Hartmann, A.; Holdaas, H.; Bachtler, M.; Jenssen, T.G.; Dionisi, M.; Pasch, A. Serum Calcification Propensity Is a Strong and Independent Determinant of Cardiac and All-Cause Mortality in Kidney Transplant Recipients. Am. J. Transpl. 2016, 16, 204–212. [Google Scholar] [CrossRef]
- Bostom, A.; Pasch, A.; Madsen, T.; Roberts, M.B.; Franceschini, N.; Steubl, D.; Garimella, P.S.; Ix, J.H.; Tuttle, K.R.; Ivanova, A.; et al. Serum Calcification Propensity and Fetuin-A: Biomarkers of Cardiovascular Disease in Kidney Transplant Recipients. Am. J. Nephrol. 2018, 48, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Pasch, A.; Block, G.A.; Bachtler, M.; Smith, E.R.; Jahnen-Dechent, W.; Arampatzis, S.; Chertow, G.M.; Parfrey, P.; Ma, X.; Floege, J. Blood Calcification Propensity, Cardiovascular Events, and Survival in Patients Receiving Hemodialysis in the EVOLVE Trial. Clin. J. Am. Soc. Nephrol. 2017, 12, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Ford, M.L.; Tomlinson, L.A.; Bodenham, E.; McMahon, L.P.; Farese, S.; Rajkumar, C.; Holt, S.G.; Pasch, A. Serum calcification propensity predicts all-cause mortality in predialysis CKD. J. Am. Soc. Nephrol. 2014, 25, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Pruijm, M.; Lu, Y.; Megdiche, F.; Piskunowicz, M.; Milani, B.; Stuber, M.; Bachtler, M.; Vogt, B.; Burnier, M.; Pasch, A. Serum calcification propensity is associated with renal tissue oxygenation and resistive index in patients with arterial hypertension or chronic kidney disease. J. Hypertens. 2017, 35, 2044–2052. [Google Scholar] [CrossRef]
- Nakazato, J.; Hoshide, S.; Wake, M.; Miura, Y.; Kuro-O, M.; Kario, K. Association of calciprotein particles measured by a new method with coronary artery plaque in patients with coronary artery disease: A cross-sectional study. J. Cardiol. 2019, 74, 428–435. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium - Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef]
- Yurdagul, A., Jr.; Finney, A.C.; Woolard, M.D.; Orr, A.W. The arterial microenvironment: The where and why of atherosclerosis. Biochem. J. 2016, 473, 1281–1295. [Google Scholar] [CrossRef]
- Bieghs, V.; Van Gorp, P.J.; Wouters, K.; Hendrikx, T.; Gijbels, M.J.; van Bilsen, M.; Bakker, J.; Binder, C.J.; Lütjohann, D.; Staels, B.; et al. LDL receptor knock-out mice are a physiological model particularly vulnerable to study the onset of inflammation in non-alcoholic fatty liver disease. PLoS ONE 2012, 7, e30668. [Google Scholar] [CrossRef]
- Ampuero, J.; Ranchal, I.; Gallego-Durán, R.; Pareja, M.J.; Del Campo, J.A.; Pastor-Ramírez, H.; Rico, M.C.; Picón, R.; Pastor, L.; García-Monzón, C.; et al. Oxidized low-density lipoprotein antibodies/high-density lipoprotein cholesterol ratio is linked to advanced non-alcoholic fatty liver disease lean patients. J. Gastroenterol. Hepatol. 2016, 31, 1611–1618. [Google Scholar] [CrossRef]
- Ratziu, V.; Munteanu, M.; Charlotte, F.; Bonyhay, L.; Poynard, T. LIDO Study Group. Fibrogenic impact of high serum glucose in chronic hepatitis C. J. Hepatol. 2003, 39, 1049–1055. [Google Scholar] [CrossRef]
- Tada, T.; Toyoda, H.; Sone, Y.; Yasuda, S.; Miyake, N.; Kumada, T.; Tanaka, J. Type 2 diabetes mellitus: A risk factor for progression of liver fibrosis in middle-aged patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Kumar, H.; Bajpai, S.; Singh, R.K.; Tripathi, K. Level of serum IL-12 and its correlation with endothelial dysfunction, insulin resistance, proinflammatory cytokines and lipid profile in newly diagnosed type 2 diabetes. Diabetes Res. Clin. Pr. 2011, 94, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Konukoglu, D.; Hatemi, H.; Bayer, H.; Bağriaçik, N. Relationship between serum concentrations of interleukin-6 and tumor necrosis factor alpha in female Turkish subjects with normal and impaired glucose tolerance. Horm. Metab. Res. 2006, 38, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Y.; Chen, K.; Zhou, Q.; Wei, W.; Wang, Y.; Wang, Y. The role of oxidized low-density lipoprotein in breaking peripheral Th17/Treg balance in patients with acute coronary syndrome. Biochem. Biophys. Res. Commun. 2010, 394, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; Montecucco, F.; Mach, F.; Pontremoli, R.; Viazzi, F. The liver and the kidney: Two critical organs influencing the atherothrombotic risk in metabolic syndrome. Thromb. Haemost. 2013, 110, 940–958. [Google Scholar] [CrossRef]
- Al Rifai, M.; Silverman, M.G.; Nasir, K.; Budoff, M.J.; Blankstein, R.; Szklo, M.; Katz, R.; Blumenthal, R.S.; Blaha, M.J. The association of nonalcoholic fatty liver disease, obesity, and metabolic syndrome, with systemic inflammation and subclinical atherosclerosis: The Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis 2015, 239, 629–633. [Google Scholar] [CrossRef]
- Barrea, L.; Di Somma, C.; Muscogiuri, G.; Tarantino, G.; Tenore, G.C.; Orio, F.; Colao, A.; Savastano, S. Nutrition, inflammation and liver-spleen axis. Crit. Rev. Food Sci. Nutr. 2018, 58, 3141–3158. [Google Scholar] [CrossRef]
- Shishkova, D.K.; Velikanova, E.A.; Krivkina, E.O.; Mironov, A.V.; Kudryavtseva, Y.A.; Kutikhin, A.G. Calcium-phosphate bions do specifically induce hypertrophy of damaged intima in rats. Rus. J. Cardiol. 2018, 23, 33–38. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Beamish, J.A.; He, P.; Kottke-Marchant, K.; Marchant, R.E. Molecular regulation of contractile smooth muscle cell phenotype: Implications for vascular tissue engineering. Tissue Eng. Part B Rev. 2010, 16, 467–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salabei, J.K.; Hill, B.G. Autophagic regulation of smooth muscle cell biology. Redox Biol. 2015, 4, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, M.; Schäfer, C.; Heiss, A.; Gräber, S.; Kinkeldey, A.; Büscher, A.; Schmitt, M.M.; Bornemann, J.; Nimmerjahn, F.; Herrmann, M.; et al. Clearance of fetuin-A--containing calciprotein particles is mediated by scavenger receptor-A. Circ. Res. 2012, 111, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Koppert, S.; Büscher, A.; Babler, A.; Ghallab, A.; Buhl, E.M.; Latz, E.; Hengstler, J.G.; Smith, E.R.; Jahnen-Dechent, W. Cellular Clearance and Biological Activity of Calciprotein Particles Depend on Their Maturation State and Crystallinity. Front. Immunol. 2018, 9, 1991. [Google Scholar] [CrossRef]
- Callegari, A.; Coons, M.L.; Ricks, J.L.; Rosenfeld, M.E.; Scatena, M. Increased calcification in osteoprotegerin-deficient smooth muscle cells: Dependence on receptor activator of NF-κB ligand and interleukin 6. J. Vasc. Res. 2014, 51, 118–131. [Google Scholar] [CrossRef]
- Aghagolzadeh, P.; Bachtler, M.; Bijarnia, R.; Jackson, C.; Smith, E.R.; Odermatt, A.; Radpour, R.; Pasch, A. Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-α. Atherosclerosis 2016, 251, 404–414. [Google Scholar] [CrossRef] [Green Version]
- Zickler, D.; Luecht, C.; Willy, K.; Chen, L.; Witowski, J.; Girndt, M.; Fiedler, R.; Storr, M.; Kamhieh-Milz, J.; Schoon, J.; et al. Tumour necrosis factor-alpha in uraemic serum promotes osteoblastic transition and calcification of vascular smooth muscle cells via extracellular signal-regulated kinases and activator protein 1/c-FOS-mediated induction of interleukin 6 expression. Nephrol. Dial. Transpl. 2018, 33, 574–585. [Google Scholar] [CrossRef]
- Peng, H.H.; Wu, C.Y.; Young, D.; Martel, J.; Young, A.; Ojcius, D.M.; Lee, Y.H.; Young, J.D. Physicochemical and biological properties of biomimetic mineralo-protein nanoparticles formed spontaneously in biological fluids. Small 2013, 9, 2297–2307. [Google Scholar] [CrossRef]
- Smith, E.R.; Hanssen, E.; McMahon, L.P.; Holt, S.G. Fetuin-A-containing calciprotein particles reduce mineral stress in the macrophage. PLoS ONE 2013, 8, e60904. [Google Scholar] [CrossRef] [Green Version]
- Shannahan, J.H.; Alzate, O.; Winnik, W.M.; Andrews, D.; Schladweiler, M.C.; Ghio, A.J.; Gavett, S.H.; Kodavanti, U.P. Acute phase response, inflammation and metabolic syndrome biomarkers of Libby asbestos exposure. Toxicol. Appl. Pharm. 2012, 260, 105–114. [Google Scholar] [CrossRef]
- Da Cunha, A.A.; Ferreira, A.G.; Wyse, A.T. Increased inflammatory markers in brain and blood of rats subjected to acute homocysteine administration. Metab. Brain Dis. 2010, 25, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Halenova, T.; Raksha, N.; Vovk, T.; Savchuk, O.; Ostapchenko, L.; Prylutskyy, Y.; Kyzyma, O.; Ritter, U.; Scharff, P. Effect of C60 fullerene nanoparticles on the diet-induced obesity in rats. Int. J. Obes. (Lond) 2018, 42, 1987–1998. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Taleb, S.; Mallat, Z.; Tedgui, A. Recent advances on the role of cytokines in atherosclerosis. Arter. Thromb. Vasc. Biol. 2011, 31, 969–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramji, D.P.; Davies, T.S. Cytokines in atherosclerosis: Key players in all stages of disease and promising therapeutic targets. Cytokine Growth Factor. Rev. 2015, 26, 673–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertazzo, S.; Gentleman, E.; Cloyd, K.L.; Chester, A.H.; Yacoub, M.H.; Stevens, M.M. Nano-analytical electron microscopy reveals fundamental insights into human cardiovascular tissue calcification. Nat. Mater 2013, 12, 576–583. [Google Scholar] [CrossRef]
- Schlieper, G.; Grotemeyer, D.; Aretz, A.; Schurgers, L.J.; Krüger, T.; Rehbein, H.; Weirich, T.E.; Westenfeld, R.; Brandenburg, V.M.; Eitner, F.; et al. Analysis of calcifications in patients with coral reef aorta. Ann. Vasc. Surg. 2010, 24, 408–414. [Google Scholar] [CrossRef]
- Schlieper, G.; Aretz, A.; Verberckmoes, S.C.; Krüger, T.; Behets, G.J.; Ghadimi, R.; Weirich, T.E.; Rohrmann, D.; Langer, S.; Tordoir, J.H.; et al. Ultrastructural analysis of vascular calcifications in uremia. J. Am. Soc. Nephrol. 2010, 21, 689–696. [Google Scholar] [CrossRef] [Green Version]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Ocak, G.; van Stralen, K.J.; Rosendaal, F.R.; Verduijn, M.; Ravani, P.; Palsson, R.; Leivestad, T.; Hoitsma, A.J.; Ferrer-Alamar, M.; Finne, P.; et al. Mortality due to pulmonary embolism, myocardial infarction, and stroke among incident dialysis patients. J. Thromb. Haemost. 2012, 10, 2484–2493. [Google Scholar] [CrossRef]
- Bundy, J.D.; Cai, X.; Scialla, J.J.; Dobre, M.A.; Chen, J.; Hsu, C.Y.; Leonard, M.B.; Go, A.S.; Rao, P.S.; Lash, J.P.; et al. Serum Calcification Propensity and Coronary Artery Calcification Among Patients With CKD: The CRIC (Chronic Renal Insufficiency Cohort) Study. Am. J. Kidney Dis. 2019, 73, 806–814. [Google Scholar] [CrossRef]
- Cai, M.M.X.; Smith, E.R.; Kent, A.; Huang, L.; Hewitson, T.D.; McMahon, L.P.; Holt, S.G. Calciprotein Particle Formation in Peritoneal Dialysis Effluent Is Dependent on Dialysate Calcium Concentration. Perit. Dial. Int. 2018, 38, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Iwazu, Y.; Shiizaki, K.; Akimoto, T.; Kotani, K.; Kurabayashi, M.; Kurosu, H.; Kuro-O, M. Identification and quantification of plasma calciprotein particles with distinct physical properties in patients with chronic kidney disease. Sci. Rep. 2018, 8, 1256. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Hewitson, T.D.; Cai, M.M.X.; Aghagolzadeh, P.; Bachtler, M.; Pasch, A.; Holt, S.G. A novel fluorescent probe-based flow cytometric assay for mineral-containing nanoparticles in serum. Sci. Rep. 2017, 7, 5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Anokhina, V.; Dieudonne, G.; Abramowitz, M.K.; Kashyap, R.; Yan, C.; Wu, T.T.; de Mesy Bentley, K.L.; Miller, B.L.; Bushinsky, D.A. Patients with advanced chronic kidney disease and vascular calcification have a large hydrodynamic radius of secondary calciprotein particles. Nephrol. Dial. Transpl. 2019, 34, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Viegas, C.S.B.; Santos, L.; Macedo, A.L.; Matos, A.A.; Silva, A.P.; Neves, P.L.; Staes, A.; Gevaert, K.; Morais, R.; Vermeer, C.; et al. Chronic Kidney Disease Circulating Calciprotein Particles and Extracellular Vesicles Promote Vascular Calcification: A Role for GRP (Gla-Rich Protein). Arter. Thromb. Vasc. Biol. 2018, 38, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.R.; Hewitson, T.D.; Hanssen, E.; Holt, S.G. Biochemical transformation of calciprotein particles in uraemia. Bone 2018, 110, 355–367. [Google Scholar] [CrossRef]
- Lamas, G.A.; Goertz, C.; Boineau, R.; Mark, D.B.; Rozema, T.; Nahin, R.L.; Lindblad, L.; Lewis, E.F.; Drisko, J.; Lee, K.L. TACT Investigators. Effect of disodium EDTA chelation regimen on cardiovascular events in patients with previous myocardial infarction: The TACT randomized trial. JAMA 2013, 309, 1241–1250. [Google Scholar] [CrossRef] [Green Version]
- Escolar, E.; Lamas, G.A.; Mark, D.B.; Boineau, R.; Goertz, C.; Rosenberg, Y.; Nahin, R.L.; Ouyang, P.; Rozema, T.; Magaziner, A.; et al. The effect of an EDTA-based chelation regimen on patients with diabetes mellitus and prior myocardial infarction in the Trial to Assess Chelation Therapy (TACT). Circ. Cardiovasc. Qual. Outcomes 2014, 7, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Ujueta, F.; Arenas, I.A.; Escolar, E.; Diaz, D.; Boineau, R.; Mark, D.B.; Golden, P.; Lindblad, L.; Kim, H.; Lee, K.L.; et al. The effect of EDTA-based chelation on patients with diabetes and peripheral artery disease in the Trial to Assess Chelation Therapy (TACT). J. Diabetes Complicat. 2019, 33, 490–494. [Google Scholar] [CrossRef]
- Yamada, H.; Kuro-O, M.; Ishikawa, S.E.; Funazaki, S.; Kusaka, I.; Kakei, M.; Hara, K. Daily variability in serum levels of calciprotein particles and their association with mineral metabolism parameters: A cross-sectional pilot study. Nephrology (Carlton) 2018, 23, 226–230. [Google Scholar] [CrossRef]
- Avila, M.D.; Escolar, E.; Lamas, G.A. Chelation therapy after the trial to assess chelation therapy: Results of a unique trial. Curr. Opin. Cardiol. 2014, 29, 481–488. [Google Scholar] [CrossRef]
- Foreman, H.; Trujillo, T.T. The metabolism of C14 labeled ethylenediaminetetraacetic acid in human beings. J. Lab. Clin. Med. 1954, 43, 566–571. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primers | R2 | Efficiency, % |
|---|---|---|---|
| ACTB | F: 5′-CATCGAGCACGGCATCGTCA-3′ R: 5′-TAGCACAGCCTGGACAGCAAC-3′ | 0.996 | 92.834 |
| GAPDH | F: 5′-AGCCACATCGCTCAGACAC-3′ R: 5′-GCCCAATACGACCAAATCC-3′ | 0.994 | 105.854 |
| B2M | F: 5′-TCCATCCGACATTGAAGTTG-3′ R: 5′-CGGCAGGCATACTCATCTT-3′ | 0.990 | 76.601 |
| SCARF1 | F: 5′-CCGATCAGACCTCAAGGACAG-3′ R: 5′-CCCAGGGTAGCTTGTGGGA-3′ | 0.997 | 97.848 |
| CD36 | F: 5′-GGCTGTGACCGGAACTGTG-3′ R: 5′-AGGTCTCCAACTGGCATTAGAA-3′ | 0.992 | 89.779 |
| LDLR | F: 5′-ACGGCGTCTCTTCCTATGACA-3′ R: 5′-CCCTTGGTATCCGCAACAGA-3′ | 0.991 | 97.402 |
| VLDLR | F: 5′-AGAAAAGCCAAATGTGAACCCT-3′ R: 5′-CACTGCCGTCAACACAGTCT-3′ | 0.990 | 90.926 |
| VCAM1 | F: 5′-CGTCTTGGTCAGCCCTTCCT-3′ R: 5′-ACATTCATATACTCCCGCATCCTTC-3′ | 0.987 | 86.482 |
| ICAM1 | F: 5′-TTGGGCATAGAGACCCCGTT-3′ R: 5′-GCACATTGCTCAGTTCATACACC-3′ | 0.993 | 107.375 |
| PECAM1 | F: 5′-TGGCGCATGCCTGTAGTA-3′ R: 5′-TCCGTTTCCTGGGTTCAA-3′ | 0.987 | 85.560 |
| SELE1 | F: 5′-GCACAGCCTTGTCCAACC-3′ R: 5′-ACCTCACCAAACCCTTCG-3′ | 0.990 | 95.994 |
| SELP1 | F: 5′-ATGGGTGGGAACCAAAAAGG-3′ R: 5′-GGCTGACGGACTCTTGATGTAT-3′ | 0.989 | 94.008 |
| CDH5 | F: 5′-AAGCGTGAGTCGCAAGAATG-3′ R: 5′-TCTCCAGGTTTTCGCCAGTG-3′ | 0.996 | 91.622 |
| IL1R1 | F: 5′-GGCTGAAAAGCATAGAGGGAAC-3′ R: 5′-CTGGGCTCACAATCACAGG-3′ | 0.988 | 98.004 |
| IL1R2 | F: 5′-TGGCACCTACGTCTGCACTACT-3′ R: 5′-TTGCGGGTATGAGATGAACG-3′ | 0.988 | 89.115 |
| TNFRSF1A | F: 5′-CCAGGAGAAACAGAACACCGT-3′ R: 5′-AAACCAATGAAGAGGAGGGATAA-3′ | 0.995 | 87.900 |
| TNFRSF1B | F: 5′-GTCCACACGATCCCAACAC-3′ R: 5′-CACACCCACAATCAGTCCAA-3′ | 0.981 | 96.901 |
| NOS3 | F: 5′-GTGATGGCGAAGCGAGTGAAG-3′ R: 5′-CCGAGCCCGAACACACAGAAC-3′ | 0.992 | 90.406 |
| PXDN | F: 5′-AGCCAGCCATCACCTGGAAC-3′ R: 5′-TTCCGGGCCACACACTCATA-3′ | 0.987 | 91.478 |
| IL1B | F: 5′-TGGCTTATTACAGTGGCAATG-3′ R: 5′-GTGGTGGTCGGAGATTCG-3′ | 0.997 | 107.422 |
| IL6 | F: 5′-GGCACTGGCAGAAAACAACC-3′ R: 5′-GCAAGTCTCCTCATTGAATCC-3′ | 0.992 | 97.406 |
| IL8 | F: 5′-CAGAGACAGCAGAGCACAC-3′ R: 5′-AGTTCTTTAGCACTCCTTGGC-3′ | 0.992 | 105.816 |
| IL12A | F: 5′-GCCTTCACCACTCCCAAAAC-3′ R: 5′-TGTCTGGCCTTCTGGAGCAT-3′ | 0.989 | 89.617 |
| IL23 | F: 5′-CTCAGGGACAACAGTCAGTTC-3′ R: 5′-ACAGGGCTATCAGGGAGCA-3′ | 0.981 | 92.297 |
| VWF | F: 5′-CCTTGACCTCGGACCCTTATG-3′ R: 5′-GATGCCCGTTCACACCACT-3′ | 0.996 | 107.880 |
| KDR | F: 5′-TGCCTACCTCACCTGTTTC-3′ R: 5′-GGCTCTTTCGCTTACTGTTC-3′ | 0.980 | 90.308 |
| FAP | F: 5′-TCAACTGTGATGGCAAGAGCA-3′ R: 5′-TAGGAAGTGGGTCATGTGGGT-3′ | 0.980 | 107.368 |
| ACTA2 | F: 5′-GTGTTGCCCCTGAAGAGCAT-3′ R: 5′-GCTGGGACATTGAAAGTCTCA-3′ | 0.982 | 107.327 |
| SMTN | F: 5′-GGGATCGTGTCCACAAGTTCA-3′ R: 5′-GCTACTCCTCGTTGCTCCTT-3′ | 0.999 | 96.724 |
| CDH2 | F: 5′-GCTTCTGGTGAAATCGCATTA-3′ R: 5′-AGTCTCTCTTCTGCCTTTGTAG-3′ | 0.994 | 93.226 |
| VIM | F: 5′-CGCCAGATGCGTGAAATGG-3′ R: 5′-ACCAGAGGGAGTGAATCCAGA-3′ | 0.989 | 90.249 |
| COL4A1 | F: 5′-GGACTACCTGGAACAAAAGGG-3′ R: 5′-GCCAAGTATCTCACCTGGATCA-3′ | 0.992 | 91.030 |
| MMP2 | F: 5′-CCGTGTTTGCCATCTGTTTTAG-3′ R: 5′-AGGTTCTCTTGCTGTTTACTTTGGA-3′ | 0.981 | 94.465 |
| SNAI1 | F: 5′-CAGACCCACTCAGATGTCAAGAA-3′ R: 5′-GGGCAGGTATGGAGAGGAAGA-3′ | 0.993 | 94.588 |
| SNAI2 | F: 5′-ACTCCGAAGCCAAATGACAA-3′ R: 5′-CTCTCTCTGTGGGTGTGTGT-3′ | 0.986 | 87.981 |
| TWIST1 | F: 5′-GTCCGCAGTCTTACGAGGAG-3′ R: 5′-GCTTGAGGGTCTGAATCTTGCT-3′ | 0.996 | 96.188 |
| ZEB1 | F: 5′-GATGATGAATGCGAGTCAGATGC-3′ R: 5′-ACAGCAGTGTCTTGTTGTTGT-3′ | 0.983 | 90.857 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shishkova, D.; Velikanova, E.; Sinitsky, M.; Tsepokina, A.; Gruzdeva, O.; Bogdanov, L.; Kutikhin, A. Calcium Phosphate Bions Cause Intimal Hyperplasia in Intact Aortas of Normolipidemic Rats through Endothelial Injury. Int. J. Mol. Sci. 2019, 20, 5728. https://doi.org/10.3390/ijms20225728
Shishkova D, Velikanova E, Sinitsky M, Tsepokina A, Gruzdeva O, Bogdanov L, Kutikhin A. Calcium Phosphate Bions Cause Intimal Hyperplasia in Intact Aortas of Normolipidemic Rats through Endothelial Injury. International Journal of Molecular Sciences. 2019; 20(22):5728. https://doi.org/10.3390/ijms20225728
Chicago/Turabian StyleShishkova, Daria, Elena Velikanova, Maxim Sinitsky, Anna Tsepokina, Olga Gruzdeva, Leo Bogdanov, and Anton Kutikhin. 2019. "Calcium Phosphate Bions Cause Intimal Hyperplasia in Intact Aortas of Normolipidemic Rats through Endothelial Injury" International Journal of Molecular Sciences 20, no. 22: 5728. https://doi.org/10.3390/ijms20225728
APA StyleShishkova, D., Velikanova, E., Sinitsky, M., Tsepokina, A., Gruzdeva, O., Bogdanov, L., & Kutikhin, A. (2019). Calcium Phosphate Bions Cause Intimal Hyperplasia in Intact Aortas of Normolipidemic Rats through Endothelial Injury. International Journal of Molecular Sciences, 20(22), 5728. https://doi.org/10.3390/ijms20225728