Genome-Wide Identification and Characterization of FBA Gene Family in Polyploid Crop Brassica napus

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Identification, Properties, and Genomic Distribution of BnaFBA Genes
2.2. Phylogenetic and Structure Analysis of BnaFBAs
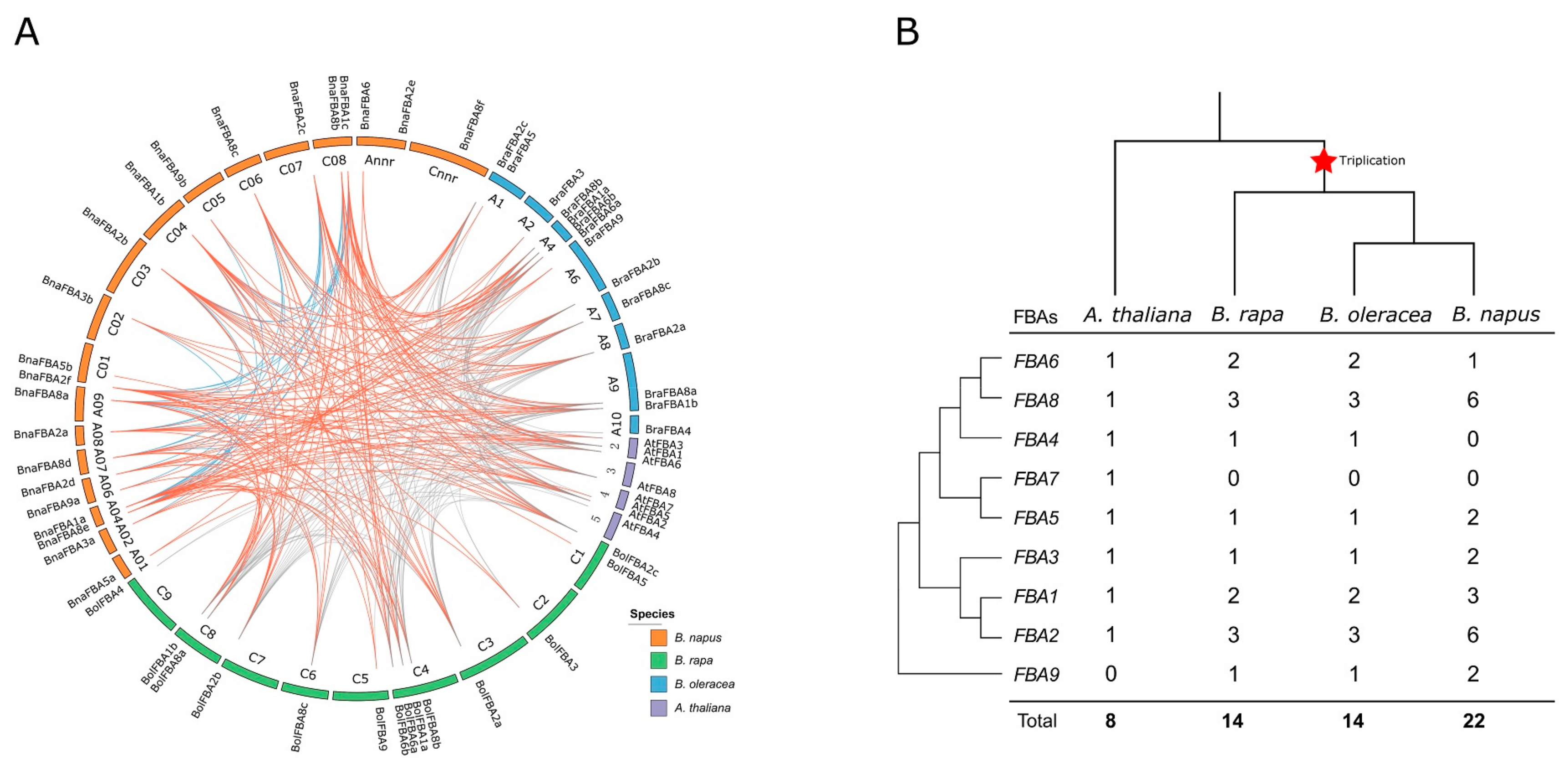
2.3. Synteny and Gene Duplication of BnaFBA Genes
2.4. Cis-Acting Elements in the Putative Promoter Regions of BnaFBA Genes
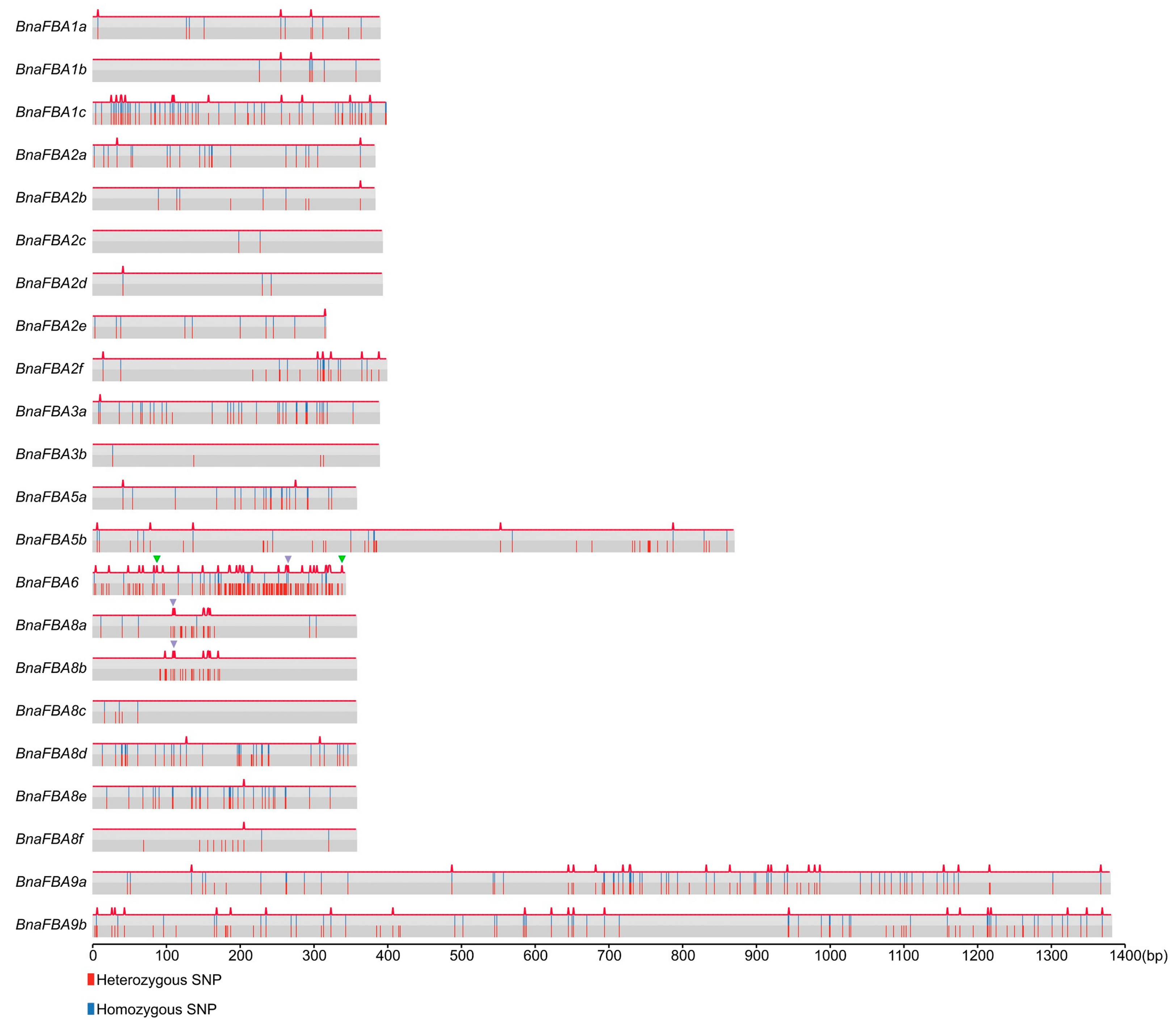
2.5. Natural Variations of BnaFBA Family Genes in B. Napus
2.6. Expression Patterns of BnaFBA Genes
2.7. qRT-PCR Analysis of Selected BnaFBA Genes under Abiotic Stresses
2.8. Co-Regulatory Networks of BnaFBA Genes
3. Discussion
4. Materials and Methods
4.1. Identification and Property Analysis of BnaFBA Genes
4.2. Cis-Acting Regulatory Elements and Subcellular Localization Analysis
4.3. Structure and Chromosomal Localization Analysis
4.4. Multiple Alignments and Phylogenetic Analysis
4.5. Synteny and Duplicate Gene Analysis
4.6. Variations Analysis and Principal Component Analysis
4.7. Expression Analysis of BnaFBA Genes
4.8. Plant Materials and Treatments
4.9. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Analysis
4.10. Pearson Correlation Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABA | Abscisic acid |
| IAA | Indolylacetic acid |
| MeJA | Methyl Jasmonic acid |
| GA | Gibberellin |
| SA | Salicylic acid |
| SLs | Strigolactones |
| EDTA | Ethylene diamine tetraacetic acid |
| CDS | Coding sequence |
| GRAVY | Grand average of hydropathy |
| FPKM | Fragments per kilobase of exon per million fragments mapped |
| PCCs | Pearson correlation coefficients |
| SNP | Single nucleotide polymorphism |
| Indels | Insertions and deletions |
| MW | Molecular weight |
| pI | Isoelectric point |
| aa | Amino acid |
| bp | Base pair |
| kb | Kilobase |
| Mb | Megabase |
| kDa | Kilodalton |
References
- Rutter, W.J. Evolution of aldolase. Fed Proc. 1964, 23, 1248–1257. [Google Scholar] [PubMed]
- Berg, I.A.; Kockelkorn, D.; Ramos-Vera, W.H.; Say, R.F.; Zarzycki, J.; Hügler, M.; Alber, B.E.; Fuchs, G. Autotrophic carbon fixation in archaea. Nat. Rev. Microbiol. 2010, 8, 447. [Google Scholar] [CrossRef] [PubMed]
- Vigeolas, H.; Waldeck, P.; Zank, T.; Geigenberger, P. Increasing seed oil content in oil-seed rape (Brassica napus L.) by over-expression of a yeast glycerol-3-phosphate dehydrogenase under the control of a seed-specific promoter. Plant. Biotechnol. J. 2007, 5, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Shockey, J.M.; Klasson, K.T.; Chapital, D.C.; Mason, C.B.; Scheffler, B.E. Developmental regulation of diacylglycerol acyltransferase family gene expression in tung tree tissues. PloS ONE 2013, 8, e76946. [Google Scholar] [CrossRef]
- Nakahara, K.; Yamamoto, H.; Miyake, C.; Yokota, A. Purification and characterization of class-I and class-II fructose-1, 6-bisphosphate aldolases from the cyanobacterium Synechocystis sp. PCC 6803. Plant Cell physiol. 2003, 44, 326–333. [Google Scholar] [CrossRef]
- Marsh, J.J.; Lebherz, H.G. Fructose-bisphosphate aldolases: An evolutionary history. Trends Biochem. Sci. 1992, 17, 110–113. [Google Scholar] [CrossRef]
- Gross, W.; Lenze, D.; Nowitzki, U.; Weiske, J.; Schnarrenberger, C. Characterization, cloning, and evolutionary history of the chloroplast and cytosolic class I aldolases of the red alga Galdieria sulphuraria. Gene 1999, 230, 7–14. [Google Scholar] [CrossRef]
- Tolan, D.R.; Niclas, J.; Bruce, B.D.; Lebo, R.V. Evolutionary implications of the human aldolase-A,-B,-C, and-pseudogene chromosome locations. Am. J. Hum. Genet. 1987, 41, 907. [Google Scholar]
- Penhoet, E.; Kochman, M.; Valentine, R.; Rutter, W.J. The subunit structure of mammalian fructose diphosphate aldolase. Biochemistry 1967, 6, 2940–2949. [Google Scholar] [CrossRef]
- Henze, K.; Morrison, H.G.; Sogin, M.L.; Müller, M. Sequence and phylogenetic position of a class II aldolase gene in the amitochondriate protist, Giardia lamblia. Gene 1998, 222, 163–168. [Google Scholar] [CrossRef]
- Lv, G.; Guo, X.; Xie, L.; Xie, C.; Zhang, X.; Yang, Y.; Xiao, L.; Tang, Y.; Pan, X.; Guo, A.; et al. Molecular characterization, gene evolution, and expression analysis of the fructose-1, 6-bisphosphate aldolase (FBA) gene family in wheat (Triticum aestivum L.). Front. Plant. Sci. 2017, 8, 1030. [Google Scholar] [CrossRef] [PubMed]
- Stribling, D.; Perham, R.N. Purification and characterization of two fructose diphosphate aldolases from Escherichia coli (Crookes’ strain). Biochem. J. 1973, 131, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, A.M.; Cremona, T.; Preddie, E.C. The aldolases of Chlamydomonas reinhardii. Arch. Biochem. Biophys. 1971, 146, 249–255. [Google Scholar] [CrossRef]
- Lebherz, H.G.; Rutter, W.J. Distribution of fructose diphosphate aldolase variants in biological systems. Biochemistry 1969, 8, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Bukowiecki, A.C.; Anderson, L.E. Multiple forms of aldolase and triose phosphate isomerase in diverse plant species. Plant Sci. Lett. 1974, 3, 381–386. [Google Scholar] [CrossRef]
- Plaxton, W.C. The organization and regulation of plant glycolysis. Annu Rev. Plant. Physiol. Plant. Mol. Biol. 1996, 47, 185–214. [Google Scholar] [CrossRef]
- Lu, W.; Tang, X.; Huo, Y.; Xu, R.; Qi, S.; Huang, J.; Zheng, C.; Wu, C.A. Identification and characterization of fructose 1, 6-bisphosphate aldolase genes in Arabidopsis reveal a gene family with diverse responses to abiotic stresses. Gene 2012, 503, 65–74. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, Y.; Ni, Y.; Meng, Z.; Lu, T.; Li, T. Exogenous calcium alleviates low night temperature stress on the photosynthetic apparatus of tomato leaves. PLoS ONE 2014, 9, e97322. [Google Scholar] [CrossRef]
- Cai, B.; Li, Q.; Xu, Y.; Yang, L.; Bi, H.; Ai, X. Genome-wide analysis of the fructose 1, 6-bisphosphate aldolase (FBA) gene family and functional characterization of FBA7 in tomato. Plant Physiol. Biochem. 2016, 108, 251–265. [Google Scholar] [CrossRef]
- Yamada, S.; Komori, T.; Hashimoto, A.; Kuwata, S.; Imaseki, H.; Kubo, T. Differential expression of plastidic aldolase genes in Nicotiana plants under salt stress. Plant Sci. 2000, 154, 61–69. [Google Scholar] [CrossRef]
- Fan, W.; Zhang, Z.; Zhang, Y. Cloning and molecular characterization of fructose-1, 6-bisphosphate aldolase gene regulated by high-salinity and drought in Sesuvium portulacastrum. Plant Cell Rep. 2009, 28, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Functional Analysis of a Fructose-1, 6-diphosphatase Aldolase Gene ALD Y in Rice. Master’s Degree Dissertation, Huazhong Agricultural University, Wuhan, China, June 2014. [Google Scholar]
- Valenti, V.; Pupillo, P.; Scagliarini, S. Compartmentation of aldolase isoforms in maize leaves. J. Exp. Bot. 1987, 38, 1228–1237. [Google Scholar] [CrossRef]
- Zeng, Y.; Tan, X.; Zhang, L.; Jiang, N.; Cao, H. Identification and expression of fructose-1, 6-bisphosphate aldolase genes and their relations to oil content in developing seeds of tea oil tree (Camellia oleifera). PLoS ONE 2014, 9, e107422. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U.; Lerchl, J.; Zrenner, R.; Frommer, W. Manipulation of sink-source relations in transgenic plants. Plant Cell Environ. 1994, 17, 649–658. [Google Scholar] [CrossRef]
- Haake, V.; Zrenner, R.; Sonnewald, U.; Stitt, M. A moderate decrease of plastid aldolase activity inhibits photosynthesis, alters the levels of sugars and starch, and inhibits growth of potato plants. Plant. J. 1998, 14, 147–157. [Google Scholar] [CrossRef]
- Uematsu, K.; Suzuki, N.; Iwamae, T.; Inui, M.; Yukawa, H. Increased fructose 1, 6-bisphosphate aldolase in plastids enhances growth and photosynthesis of tobacco plants. J. Exp. Bot. 2012, 63, 3001–3009. [Google Scholar] [CrossRef]
- Obiadalla-Ali, H.; Fernie, A.R.; Lytovchenko, A.; Kossmann, J.; Lloyd, J.R. Inhibition of chloroplastic fructose 1, 6-bisphosphatase in tomato fruits leads to decreased fruit size, but only small changes in carbohydrate metabolism. Planta 2004, 219, 533–540. [Google Scholar] [CrossRef]
- Konishi, H.; Yamane, H.; Maeshima, M.; Komatsu, S. Characterization of fructose-bisphosphate aldolase regulated by gibberellin in roots of rice seedling. Plant. Mol. Biol. 2004, 56, 839–848. [Google Scholar] [CrossRef]
- Khanna, S.M.; Taxak, P.C.; Jain, P.K.; Saini, R.; Srinivasan, R. Glycolytic enzyme activities and gene expression in Cicer arietinum exposed to water-deficit stress. Appl. Biochem. Biotechnol. 2014, 173, 2241–2253. [Google Scholar] [CrossRef]
- Mutuku, J.M.; Nose, A. Changes in the contents of metabolites and enzyme activities in rice plants responding to Rhizoctonia solani Kuhn infection: Activation of glycolysis and connection to phenylpropanoid pathway. Plant Cell Physiol. 2012, 53, 1017–1032. [Google Scholar] [CrossRef]
- Oelze, M.L.; Muthuramalingam, M.; Vogel, M.O.; Dietz, K.J. The link between transcript regulation and de novo protein synthesis in the retrograde high light acclimation response of Arabidopsis thaliana. BMC Genom. 2014, 15, 320. [Google Scholar] [CrossRef] [PubMed]
- Rana, D.; van den Boogaart, T.; O’Neill, C.M.; Hynes, L.; Bent, E.; Macpherson, L.; Park, J.Y.; Lim, Y.P.; Bancroft, I. Conservation of the microstructure of genome segments in Brassica napus and its diploid relatives. Plant J. 2004, 40, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Napier, J.A.; Clemente, T.E.; Cahoon, E.B. New frontiers in oilseed biotechnology: Meeting the global demand for vegetable oils for food, feed, biofuel, and industrial applications. Curr. Opin. Biotechnol. 2011, 22, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, F.; Fan, G.; Hu, Q.; Zhou, Y.; Guan, M.; Tong, C.; Li, J.; Du, D.; Qi, C.; Jiang, L.; et al. The high-quality genome of Brassica napus cultivar ‘ZS11′ reveals the introgression history in semi-winter morphotype. Plant J. 2017, 92, 452–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, J.E.; Chapman, B.A.; Rong, J.; Paterson, A.H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 2003, 422, 433. [Google Scholar] [CrossRef]
- Cheng, F.; Wu, J.; Wang, X. Genome triplication drove the diversification of Brassica plants. Hortic Res. 2014, 1, 14024. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Liang, Z.; Yan, T.; Xu, Y.; Xuan, L.; Tang, J.; Zhou, G.; Lohwasser, U.; Hua, S.; Wang, H.; et al. Whole-Genome Resequencing of a Worldwide Collection of Rapeseed Accessions Reveals the Genetic Basis of Ecotype Divergence. Mol. Plant 2019, 12, 30–43. [Google Scholar] [CrossRef] [Green Version]
- Furbank, R.T.; Taylor, W.C. Regulation of photosynthesis in C3 and C4 plants: A molecular approach. Plant Cell 1995, 7, 797. [Google Scholar] [CrossRef] [Green Version]
- Raines, C.A. The Calvin cycle revisited. Photosynth. Res. 2003, 75, 1–10. [Google Scholar] [CrossRef]
- Graciet, E.; Lebreton, S.; Gontero, B. Emergence of new regulatory mechanisms in the Benson-Calvin pathway via protein-protein interactions: A glyceraldehyde-3-phosphate dehydrogenase/CP12/phosphoribulokinase complex. J. Exp. Bot. 2004, 55, 1245–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, J.; Schwender, J. Computational analysis of storage synthesis in developing Brassica napus L. (oilseed rape) embryos: Flux variability analysis in relation to 13C metabolic flux analysis. Plant J. 2011, 67, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.; Sygusch, J. Product binding and role of the C-terminal region in Class I D-fructose 1, 6-bisphosphate aldolase. Nat. Struct. Biol. 1997, 4, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Sauve’, V.; Sygusch, J. Crystallization and preliminary X-ray analysis of native and selenomethionine fructose-1,6-bisphosphate aldolase from Thermus aquaticus. Acta Crystallogr. 2001, 57, 310–313. [Google Scholar]
- Izard, T.; Sygusch, J. Induced fit movements and metal cofactor selectivity of class II aldolases: Structure of Thermus aquaticus fructose-1,6-bisphosphate aldolase. J. Biol. Chem. 2004, 279, 11825–11833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lysak, M.A.; Koch, M.A.; Pecinka, A.; Schubert, I. Chromosome triplication found across the tribe Brassiceae. Genome Res. 2005, 15, 516–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Town, C.D.; Cheung, F.; Maiti, R.; Crabtree, J.; Haas, B.J.; Wortman, J.R.; Hine, E.E.; Althoff, R.; Arbogast, T.S.; Tallon, L.J.; et al. Comparative genomics of Brassica oleracea and Arabidopsis thaliana reveal gene loss, fragmentation, and dispersal after polyploidy. Plant Cell 2006, 18, 1348–1359. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Liu, Y.; Yang, X.; Tong, C.; Edwards, D.; Parkin, I.A.; Zhao, M.; Ma, J.; Yu, J.; Huang, S.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5, 3930. [Google Scholar] [CrossRef]
- Paterson, A.H.; Freeling, M.; Tang, H.; Wang, X. Insights from the comparison of plant genome sequences. Annu. Rev. Plant Biol. 2010, 61, 349–372. [Google Scholar] [CrossRef]
- Xu, X.; Liu, X.; Ge, S.; Jensen, J.D.; Hu, F.; Li, X.; Dong, Y.; Gutenkunst, R.N.; Fang, L.; Li, J.; et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 2011, 30, 105. [Google Scholar] [CrossRef]
- Lin, T.; Zhu, G.; Zhang, J.; Xu, X.; Yu, Q.; Zheng, Z.; Zhang, Z.; Lun, Y.; Li, S.; Wang, X.; et al. Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 2014, 46, 1220. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y.; et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015, 33, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagawa, Y.; Tamoi, M.; Shigeoka, S. Overexpression of a cyanobacterial fructose-1,6-/sedoheptulose-1,7-bisphosphatase in tobacco enhances photosynthesis and growth. Nat. Biotechnol. 2001, 19, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Wei, L.; Long, Z.; Chen, L.; Wang, Q. Increased activity of only an individual non-regulated enzyme fructose-1, 6-bisphosphate aldolase in Anabaena sp. strain PCC 7120 stimulates photosynthetic yield. Acta Physiol. Plant 2008, 30, 897. [Google Scholar] [CrossRef]
- Wan, H.; Cui, Y.; Ding, Y.; Mei, J.; Dong, H.; Zhang, W.; Wu, S.; Liang, Y.; Zhang, C.; Li, J.; et al. Time-Series Analyses of Transcriptomes and Proteomes Reveal Molecular Networks Underlying Oil Accumulation in Canola. Front. Plant Sci. 2017, 7, 2007. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Rombauts, S.; Déhais, P.; Van Montagu, M.; Rouzé, P. PlantCARE, a plant cis-acting regulatory element database. Nucleic Acids Res. 1999, 27, 295–296. [Google Scholar] [CrossRef] [Green Version]
- Chou, K.C.; Shen, H.B. Cell-PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 2008, 3, 153. [Google Scholar] [CrossRef]
- Kayum, M.A.; Park, J.I.; Nath, U.K.; Saha, G.; Biswas, M.K.; Kim, H.T.; Nou, I.S. Genome-wide characterization and expression profiling of PDI family gene reveals function as abiotic and biotic stress tolerance in Chinese cabbage (Brassica rapa ssp. pekinensis). BMC Genom. 2017, 18, 885. [Google Scholar] [CrossRef] [Green Version]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL Workspace: A web-based environment for protein structure homology modeling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Hao, Z.; Lv, D.; Ge, Y.; Shi, J.; Weijers, D.; Yu, G.; Chen, J. RIdeogram: Drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Prepr. 2019, 7, e27928v1. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Wang, X.; Bowers, J.E.; Ming, R.; Alam, M.; Paterson, A.H. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 2008, 18, 1944–1954. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Bio. 2019, 20, 38. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Genome Project Data Processing, Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Picard Toolkit. Broad Institute, GitHub Repository. Available online: http://broadinstitute.github.io/picard/ (accessed on 5 August 2019).
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Q.; Zhang, X.K.; Zhang, K.; An, H.; Hu, K.N.; Wen, J.; Shen, J.S.; Ma, C.Z.; Yi, B.; Tu, J.X.; et al. Comparative Analysis of the Brassica napus Root and Leaf Transcript Profiling in Response to Drought Stress. Int. J. Mol. Sci. 2015, 16, 18752–18777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, I.J.; Tong, C.; Becker, M.G.; Mao, X.; Huang, J.; de Kievit, T.; Fernando, W.G.D.; Liu, S.; Belmonte, M.F. RNA sequencing of Brassica napus reveals cellular redox control of Sclerotinia infection. J. Exp. Bot. 2017, 68, 5079–5091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, N.; Hu, C.; Wan, L.; Hu, Q.; Xiong, J.L.; Zhang, C.L. Strigolactones Improve Plant Growth, Photosynthesis, and Alleviate Oxidative Stress under Salinity in Rapeseed (Brassica napus L.) by Regulating Gene Expression. Front. Plant Sci. 2017, 8, 1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.Y.; Huang, C.Q.; Zhang, L.; Liu, H.F.; Yu, J.H.; Hu, Z.Y.; Hua, W. Systematic Analysis of Hsf Family Genes in the Brassica napus Genome Reveals Novel Responses to Heat, Drought and High CO2 Stresses. Front. Plant Sci. 2017, 8, 1174. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Locus Name | Gene Location 1 | Transcript Length (bp) | Protein Length (aa) | MW (kDa) | pI | GRAVY | Domain 2 (Start-End aa) | Homolog of Arabidopsis | Classifi-cation | Subcellular Localization |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BnaFBA1a | BnaA04g12130D | chrA04:10353159–10354905:− | 1173 | 390 | 42.2 | 6.92 | −0.194 | PF00274 (55–390) | AT2G21330 (AtFBA1) | I | Chloroplast |
| BnaFBA1b | BnaC04g33570D | chrC04:35146154–35147860:+ | 1173 | 390 | 42.16 | 6.92 | −0.19 | PF00274 (55–390) | AT2G21330 (AtFBA1) | I | Chloroplast |
| BnaFBA1c | BnaC08g35820D | chrC08:33457869–33459923:+ | 1197 | 398 | 42.81 | 6.78 | −0.155 | PF00274 (54–398) | AT2G21330 (AtFBA1) | I | Chloroplast |
| BnaFBA2a | BnaA08g16820D | chrA08:13562506–13564430:+ | 1152 | 383 | 41.3 | 5.7 | −0.179 | PF00274 (44–383) | AT4G38970 (AtFBA2) | I | Chloroplast |
| BnaFBA2b | BnaC03g60280D | chrC03:49437707–49439536:− | 1152 | 383 | 41.33 | 5.7 | −0.185 | PF00274 (44–383) | AT4G38970 (AtFBA2) | I | Chloroplast |
| BnaFBA2c | BnaC07g47470D | chrC07:44670819–44672806:+ | 1182 | 393 | 42.58 | 6.87 | −0.196 | PF00274 (54–393) | AT4G38970 (AtFBA2) | I | Chloroplast |
| BnaFBA2d | BnaA06g37230D | chrA06:24314148–24316076:+ | 1182 | 393 | 42.64 | 7.61 | −0.208 | PF00274 (54–393) | AT4G38970 (AtFBA2) | I | Chloroplast |
| BnaFBA2e | BnaAnng40850D | chrAnn_random:46872917–46874555:+ | 951 | 317 | 34.18 | 6.77 | −0.036 | PF00274 (54–306) | AT4G38970 (AtFBA2) | I | Chloroplast |
| BnaFBA2f | BnaC01g00070D | chrC01:20706–22753:− | 1200 | 399 | 42.95 | 6.07 | −0.137 | PF00274 (44–399) | AT4G38970 (AtFBA2) | I | Chloroplast |
| BnaFBA3a | BnaA02g27140D | chrA02:20044496–20046087:+ | 1170 | 389 | 42.3 | 8.47 | −0.263 | PF00274 (45–389) | AT2G01140 (AtFBA3) | I | Chloroplast |
| BnaFBA3b | BnaC02g33660D | chrC02:36012928–36014598:− | 1170 | 389 | 42.28 | 8.47 | −0.257 | PF00274 (45–389) | AT2G01140 (AtFBA3) | I | Chloroplast |
| BnaFBA5a | BnaA01g15640D | chrA01:8017642–8018970:+ | 1077 | 358 | 38.23 | 6.38 | −0.072 | PF00274 (11–358) | AT4G26530 (AtFBA5) | I | Cytoplasm |
| BnaFBA5b | BnaC01g18640D | chrC01:12962252–12967551:+ | 2613 | 870 | 95.97 | 6.82 | −0.107 | PF00485 (52–237); PF14681 (266–458); PF00274 (523–870) | AT4G26530 (AtFBA5) | I | Cytoplasm |
| BnaFBA6 | BnaAnng07310D | chrAnn_random:7183202–7186036:+ | 1032 | 343 | 36.91 | 5.8 | −0.189 | PF00274 (11–338) | AT2G36460 (AtFBA6) | I | Cytoplasm |
| BnaFBA8a | BnaA09g33290D | chrA09:24581809–24583398:− | 1077 | 358 | 38.45 | 6.28 | −0.197 | PF00274 (11–358) | AT3G52930 (AtFBA8) | I | Cytoplasm |
| BnaFBA8b | BnaC08g24100D | chrC08:26197819–26199427:- | 1077 | 358 | 38.45 | 6.28 | −0.197 | PF00274 (11–358) | AT3G52930 (AtFBA8) | I | Cytoplasm |
| BnaFBA8c | BnaC06g14270D | chrC06:17065486–17066935:− | 1077 | 358 | 38.42 | 6.22 | −0.177 | PF00274 (11–358) | AT3G52930 (AtFBA8) | I | Cytoplasm |
| BnaFBA8d | BnaA07g15900D | chrA07:13618484–13619930:− | 1077 | 358 | 38.42 | 6.22 | −0.177 | PF00274 (11–358) | AT3G52930 (AtFBA8) | I | Cytoplasm |
| BnaFBA8e | BnaA04g05120D | chrA04:3768229–3769737:+ | 1077 | 358 | 38.49 | 6.28 | −0.218 | PF00274 (11–358) | AT3G52930 (AtFBA8) | I | Cytoplasm |
| BnaFBA8f | BnaCnng50220D | chrCnn_random:49773855–49775405:+ | 1077 | 358 | 38.49 | 6.28 | −0.218 | PF00274 (11–358) | AT3G52930 (AtFBA8) | I | Cytoplasm |
| BnaFBA9a | BnaA06g12420D | chrA06:6440041–6449829:− | 4143 | 1380 | 147.66 | 5.82 | 0.067 | PF01116 (1104–1379) | AT1G18270 (ketose-bisphosphate aldolase class-II family protein) | II | Chloroplast |
| BnaFBA9b | BnaC05g14020D | chrC05:8111222–8120981:− | 4149 | 1382 | 148 | 5.75 | 0.076 | PF01116 (1106–1381) | AT1G18270 (ketose-bisphosphate aldolase class-II family protein) | II | Cytoplasm |
| Gene | Cis-Acting Elements 1 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ABRE | ARE | AuxRE | Circadian | DIRE | DRE | DSRE | ERE | GARE | HSRE | LRE | LTRE | MeJARE | SARE | WRE | |
| BnaFBA1a | 13 | 3 | 1 | 1 | 26 | 1 | 2 | ||||||||
| BnaFBA1b | 11 | 7 | 2 | 1 | 2 | 16 | 2 | 8 | 2 | ||||||
| BnaFBA1c | 5 | 3 | 1 | 1 | 1 | 21 | 4 | 1 | |||||||
| BnaFBA2a | 4 | 1 | 2 | 1 | 3 | 1 | 16 | 1 | 10 | ||||||
| BnaFBA2b | 4 | 2 | 1 | 3 | 2 | 18 | 12 | ||||||||
| BnaFBA2c | 4 | 1 | 2 | 1 | 1 | 2 | 14 | 4 | |||||||
| BnaFBA2d | 4 | 3 | 3 | 1 | 2 | 14 | 4 | ||||||||
| BnaFBA2e | 4 | 6 | 1 | 1 | 2 | 13 | 1 | 1 | |||||||
| BnaFBA2f | 12 | 4 | 1 | 1 | 19 | 2 | 4 | ||||||||
| BnaFBA3a | 2 | 2 | 1 | 1 | 1 | 1 | 10 | 1 | 2 | ||||||
| BnaFBA3b | 3 | 1 | 2 | 1 | 2 | 1 | 9 | 2 | |||||||
| BnaFBA5a | 5 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 13 | 2 | |||||
| BnaFBA5b | 1 | 3 | 1 | 2 | 1 | 10 | 10 | 1 | |||||||
| BnaFBA6 | 1 | 1 | 3 | 1 | 2 | 1 | 13 | 4 | 1 | ||||||
| BnaFBA8a | 7 | 5 | 2 | 1 | 2 | 1 | 16 | 1 | 4 | 1 | |||||
| BnaFBA8b | 1 | 2 | 1 | 1 | 2 | 1 | 1 | 10 | 2 | ||||||
| BnaFBA8c | 2 | 9 | 1 | 4 | 2 | 8 | 1 | 1 | |||||||
| BnaFBA8d | 2 | 4 | 1 | 1 | 1 | 5 | 9 | 1 | 2 | ||||||
| BnaFBA8e | 4 | 2 | 1 | 1 | 1 | 3 | 1 | 15 | 4 | 1 | |||||
| BnaFBA8f | 3 | 3 | 1 | 1 | 1 | 11 | 8 | ||||||||
| BnaFBA9a | 15 | 2 | 1 | 1 | 1 | 1 | 1 | 23 | 10 | 1 | 1 | ||||
| BnaFBA9b | 3 | 3 | 1 | 1 | 1 | 13 | 1 | 2 | |||||||
| Gene | CDS Length (bp) | Sample Size | No. of Polymorphic Sites 1 | Sequence Variation Ratio (%) | Nucleotide Diversity | Tajima’s D 4 | |
|---|---|---|---|---|---|---|---|
| π/bp 2 | θw/bp 3 | ||||||
| BnaFBA1a | 1173 | 401 | 11 | 0.94 | 0.00376 | 0.00187 | 2.28214 * |
| BnaFBA1b | 1173 | 469 | 7 | 0.60 | 0.00286 | 0.00155 | 2.56152 ** |
| BnaFBA1c | 1197 | 198 | 60 | 5.01 | 0.01299 | 0.00762 | 2.14201 * |
| BnaFBA2a | 1152 | 163 | 23 | 2.00 | 0.00657 | 0.00394 | 1.45126 |
| BnaFBA2b | 1152 | 505 | 10 | 0.87 | 0.00279 | 0.00211 | 0.98657 |
| BnaFBA2c | 1182 | 780 | 2 | 0.17 | 0.00113 | 0.00092 | 1.89306 |
| BnaFBA2d | 1182 | 727 | 3 | 0.25 | 0.00145 | 0.00098 | 1.9818 * |
| BnaFBA2e | 951 | 289 | 10 | 1.05 | 0.0044 | 0.00239 | 2.43882 * |
| BnaFBA2f | 1200 | 552 | 21 | 1.75 | 0.00509 | 0.00298 | 1.90478 |
| BnaFBA3a | 1170 | 249 | 34 | 2.91 | 0.00894 | 0.00665 | 0.81257 |
| BnaFBA3b | 1170 | 210 | 5 | 0.43 | 0.00057 | 0.00054 | 0.17874 |
| BnaFBA5a | 1077 | 307 | 21 | 1.95 | 0.00783 | 0.00535 | 1.49221 |
| BnaFBA5b | 2613 | 228 | 42 | 1.61 | 0.0053 | 0.00349 | 1.63186 |
| BnaFBA6 | 1032 | 132 | 130 | 12.60 | 0.04158 | 0.02255 | 1.99668 |
| BnaFBA8a | 1077 | 379 | 29 | 2.69 | 0.00959 | 0.00706 | 1.32819 |
| BnaFBA8b | 1077 | 529 | 29 | 2.69 | 0.01251 | 0.0056 | 3.68226 *** |
| BnaFBA8c | 1077 | 726 | 5 | 0.46 | 0.00241 | 0.00143 | 1.43186 |
| BnaFBA8d | 1077 | 251 | 38 | 3.53 | 0.01206 | 0.00807 | 1.33435 |
| BnaFBA8e | 1077 | 234 | 31 | 2.88 | 0.0108 | 0.00598 | 1.93228 |
| BnaFBA8f | 1077 | 429 | 12 | 1.11 | 0.0033 | 0.00208 | 1.81155 |
| BnaFBA9a | 4143 | 100 | 79 | 1.91 | 0.00467 | 0.00355 | 0.97966 |
| BnaFBA9b | 4149 | 43 | 86 | 2.07 | 0.00673 | 0.00511 | 1.19193 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, W.; Liu, H.; Zhang, L.; Hu, Z.; Liu, J.; Hua, W.; Xu, S.; Liu, J. Genome-Wide Identification and Characterization of FBA Gene Family in Polyploid Crop Brassica napus. Int. J. Mol. Sci. 2019, 20, 5749. https://doi.org/10.3390/ijms20225749
Zhao W, Liu H, Zhang L, Hu Z, Liu J, Hua W, Xu S, Liu J. Genome-Wide Identification and Characterization of FBA Gene Family in Polyploid Crop Brassica napus. International Journal of Molecular Sciences. 2019; 20(22):5749. https://doi.org/10.3390/ijms20225749
Chicago/Turabian StyleZhao, Wei, Hongfang Liu, Liang Zhang, Zhiyong Hu, Jun Liu, Wei Hua, Shouming Xu, and Jing Liu. 2019. "Genome-Wide Identification and Characterization of FBA Gene Family in Polyploid Crop Brassica napus" International Journal of Molecular Sciences 20, no. 22: 5749. https://doi.org/10.3390/ijms20225749
APA StyleZhao, W., Liu, H., Zhang, L., Hu, Z., Liu, J., Hua, W., Xu, S., & Liu, J. (2019). Genome-Wide Identification and Characterization of FBA Gene Family in Polyploid Crop Brassica napus. International Journal of Molecular Sciences, 20(22), 5749. https://doi.org/10.3390/ijms20225749