B Vitamins and Fatty Acids: What Do They Share with Small Vessel Disease-Related Dementia?


{kind=link}
Abstract
:1. Introduction
2. Vascular Dementia and Small Vessel Disease-Related Dementia
3. State-Of-The-Art: What Do We Know about Nutrients and Brain Degenerative Diseases?
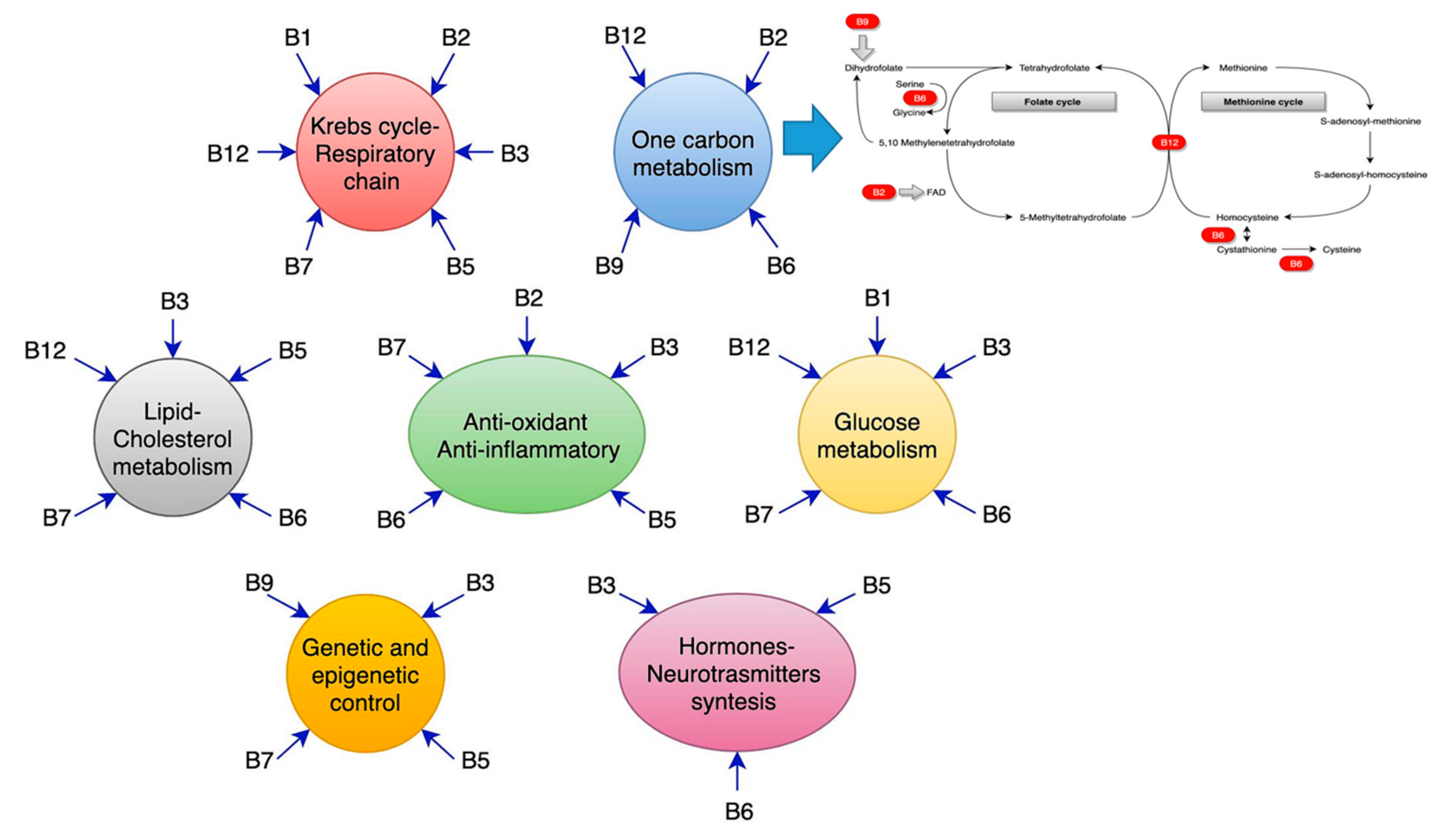
4. Vitamin B1 (Thiamine)
5. Vitamin B2 (Riboflavin)
6. Vitamin B3 (Niacin)
7. Vitamin B5 (Pantothenic Acid)
8. Vitamin B6 (Pyridoxine)
9. Vitamin B7 (Biotin)
10. Vitamin B9 (Folic Acid) and Vitamin B12: Separate or Coexistent Realities?
11. Homocysteine: The Sharing Process of Vitamin B12 and Folate.
12. Conclusions
- Should we apply them to clinical practice?
- When should we employ them? Earlier is better, but when is “early” not intelligent or too wasteful?
- How can we manage clinical trials to be simultaneously efficacious and objective?
- Which are the markers of the evolution from healthy aging towards pathology? In this case, a question that remains open concerns identifying when the small vessel alteration of white matter becomes dementia.
Conflicts of Interest
References
- Karakis, I.; Pase, M.P.; Beiser, A.; Booth, S.L.; Jacques, P.F.; Rogers, G.; DeCarli, C.; Vasan, R.S.; Wang, T.J.; Himali, J.J.; et al. Association of serum vitamin D with the risk of incident dementia and subclinical indices of brain aging: The Framingham Heart Study. J. Alzheimer’s Dis. 2016, 51, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Román, G.C.; Tatemichi, T.K.; Erkinjuntti, T.; Cummings, J.L.; Masdeu, J.C.; Garcia, J.H.; Amaducci, L.; Orgogozo, J.M.; Brun, A.; Hofman, A. Vascular dementia: Diagnostic criteria for Research studies. Reports of the NINDS-AIREN International Workshop. Neurology 1993, 43, 250–260. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization for vascular dementia. The ICD-10 Classification of Mental and Behavioral Disorders; Diagnostic Criteria for Research; World Health Organization: Geneva, Switzerland, 1993; pp. 48–50. [Google Scholar]
- Chui, H. Vascular dementia, a new beginning: Shifting focus from clinical phenotype to ischemic brain injury. Neurol. Clin. 2000, 18, 951–978. [Google Scholar] [CrossRef]
- Román, G.C.; Goldstein, M. A population-based study of dementia in 85-year-olds. N. Engl. J. Med. 1993, 329, 63. [Google Scholar]
- Shi, Y.; Wardlaw, J.M. Update on cerebral small vessel disease. A dynamic whole-brain disease. Stroke Vasc. Neurol. 2016, 1, 83–92. [Google Scholar] [CrossRef]
- American Psychiatric Association. Major or Mild Vascular Neurocognitive disorders. In Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Washington, DC, USA, 2013; pp. 612–615. [Google Scholar]
- Sinha, P.; Bharath, S.; Chandra, S.R. DSM-5 in vascular dementia. Comparison with other diagnostic criteria in a retrospective study. EC Neurol. 2015, 2, 135–143. [Google Scholar]
- Wardlaw, J.M.; Smith, E.E.; Biessels, G.J.; Cordonnier, C.; Fazekas, F.; Frayne, R.; Lindley, R.I.; O’Brien, J.T.; Barkhof, F.; Benavente, O.R.; et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013, 12, 822–832. [Google Scholar] [CrossRef]
- De Laat, K.F.; van Norden, A.G.; Gons, R.A.; van Oudheusden, L.J.; van Uden, I.W.; Bloem, B.R.; Zwiers, M.P.; de Leeuw, F.E. Gait in elderly with cerebral small vessel disease. Stroke 2010, 41, 1652–1658. [Google Scholar] [CrossRef]
- Jellinger, K.A. Pathology and pathogenesis of vascular cognitive impairment—Acritical update. Front. Aging Neurosci. 2013, 5, 17. [Google Scholar] [CrossRef]
- Patel, B.; Markus, H.S. Magnetic resonance imaging in cerebral small vessel disease and its use as a surrogate disease marker. Int. J. Stroke 2011, 6, 47–59. [Google Scholar] [CrossRef]
- Erkinjuntti, T.; Inzitari, D.; Pantoni, L.; Wallin, A.; Scheltens, P.; Rockwood, K.; Roman, G.C.; Chui, H.; Desmond, D.W. Resaerch criteria for subcortical vascular dementia in clinical trials. J. Neural Transm. Suppl. 2000, 59, 23–30. [Google Scholar] [PubMed]
- Roman, G.C.; Erkinjunnti, T.; Wallin, A.; Pantoni, L.; Chui, H.C. Subcortical ischemic vascular dementia. Lancet Neurol. 2002, 1, 426–436. [Google Scholar] [CrossRef]
- Jani, B.I.; Rajkumar, C. Ageing and vascular ageing. Postgrad. Med. J. 2006, 82, 357–362. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, J.C. Vascular basis of Alzheimer’s pathogenesis. Ann. N. Y. Acad. Sci. 2002, 977, 196–215. [Google Scholar] [CrossRef] [PubMed]
- Mathias, C.J.; Kimber, J.R. Postural hypotension: Causes, clinical features, investigation, and management. Annu. Rev. Med. 1999, 50, 317–336. [Google Scholar] [CrossRef] [PubMed]
- Roriz-Filho, J.S.; Bernardes-Silva-Filho, S.R.; Rosset, I.; Roriz-Cruz, M. Postural blood pressure dysregulation and dementia: Evidence for a vicious circle and implications for neurocardiovascular rehabilitation. In Cardiac Rehabilitation; Halliday, J.T., Ed.; Novascience Publisher Inc.: New York, NY, USA, 2009; pp. 1–37. ISBN 987-1-60741-918-1. [Google Scholar]
- Pantoni, L.; Garcia, J.H.; Gutierrez, J.A. Cerebral white matter is highly vulnerable to ischemia. Stroke 1996, 27, 1641–1647. [Google Scholar] [CrossRef]
- Schmidt, R.; Schmidt, H.; Haybaeck, J.; Loitfelder, M.; Weis, S.; Cavalieri, M.; Seiler, S.; Enzinger, C.; Ropele, S.; Erkinjuntti, T.; et al. Heterogeneity in age-related white matter changes. Acta Neuropathol. 2011, 122, 171–185. [Google Scholar] [CrossRef]
- Hommet, C.; Mondon, K.; Constans, T.; Beaufils, E.; Desmidt, T.; Camus, V.; Cottier, J.P. Review of cerebral microangiopathy and Alzheimer’s disease: Relation between white matter hyperintensities and microbleeds. Dement. Geriatr. Cogn. Disord. 2011, 32, 367–378. [Google Scholar] [CrossRef]
- Munoz, D.G.; Hastak, S.M.; Harper, B.; Lee, D.; Hachinski, V.C. Pathologic correlates of increased signals of the centrum ovale on magnetic resonance imaging. Arch. Neurol. 1993, 50, 492–497. [Google Scholar] [CrossRef]
- Salloway, S. Subcortical Vascular Dementia: Binswanger’s and CADASIL; American Academy of Neurology (AAN): Honolulu, HI, USA, 2003; pp. 1–29. [Google Scholar]
- Mirski, M.A. Pharmacology of Blood Pressure Management during Cerebral Ischemia; American Academy of Neurology (AAN): Miami, FL, USA, 2005; pp. 456–469. [Google Scholar]
- Wallin, A.; Blennow, K.; Gottfries, C.G. Neurochemical abnormalities in vascular dementia. Dementia 1989, 1, 120–130. [Google Scholar]
- Moretti, R.; Caruso, P. Small vessel disease to subcortical dementia: A dynamic model, which interfaces aging, cholinergic dysregulation and the neurovascular unit. Vasc. Health Risk Manag. 2019, in press. [Google Scholar]
- Bohnen, N.I.; Muller, M.L.T.M.; Kuwabara, H.; Ocnstantien, G.M.; Studentski, S.A. Age-associated leukoaraiosis and cortical cholinergic deafferentation. Neurology 2009, 72, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Román, G.C. Brain hypoperfusion: A critical factor in vascular dementia. Neurol. Res. 2004, 26, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.S.; Beyreuther, K.; Schmitt, H.P. Synaptophysin immunoreactivity of the cortical neuropil in vascular dementia of Binswanger type compared with the dementia of Alzheimer type and non-demented controls. Dementia 1994, 5, 79–87. [Google Scholar] [CrossRef]
- Ahtiluoto, S.; Polvikoski, T.; Peltonen, M.; Solomon, A.; Tuomilehto, J.; Winblad, B.; Sulkava, R.; Kivipelto, M. Diabetes, Alzheimer disease, and vascular dementia: A population-based neuropathologic study. Neurology 2010, 75, 1195–1202. [Google Scholar] [CrossRef]
- Englund, E.A.; Person, B. Correlations between histopathologic white matter changes and proton MR relaxation times in dementia. Alzheimer Dis. Assoc. Disord. 1987, 1, 156–170. [Google Scholar] [CrossRef]
- Román, G.C. Senile dementia of the Binswanger type: A vascular form of dementia in the elderly. JAMA 1987, 258, 1782–1788. [Google Scholar] [CrossRef]
- Moody, D.M.; Brown, W.R.; Challa, V.R.; Anderson, R.L. Periventricular venous collagenosis: Association with leukoaraiosis. Radiology 1995, 194, 469–476. [Google Scholar] [CrossRef]
- Vinters, H.V.; Ellis, W.G.; Zarow, C.; Zaias, B.W.; Jagust, W.J.; Mack, W.J.; Chui, H.C. Neuropathological substrate of ischemic vascular dementia. J. Neuropathol. Exp. Neurol. 2000, 59, 931–945. [Google Scholar] [CrossRef]
- Garcia, J.H.; Lassen, N.A.; Weiller, C.; Sperling, B.; Nakagawara, J. Ischemic stroke and incomplete infarction. Stroke 1996, 27, 761–765. [Google Scholar] [CrossRef]
- Dalkara, T.; Alarcon-Martinez, L. Cerebral micro-vascular signaling in health and disease. Brain Res. 2015, 1623, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, P.; Gold, G.; Kövari, E.; von Gunten, A.; Imhof, A.; Bouras, C.; Hof, P.R. Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: The Geneva experience. Acta Neuropathol. 2007, 113, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Launer, L.J.; Hughes, T.M.; White, L.R. Microinfarcts, brain atrophy, and cognitive function: The Honolulu Asia Aging Study Autopsy Study. Ann. Neurol. 2011, 70, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Van der Veen, P.H.; Muller, M.; Vincken, K.L.; Hendrikse, J.; Mali, W.P.; van der Graaf, Y.; Geerlings, M.I. Longitudinal relationship between cerebral small vessel disease and cerebral blood flow. The second manifestations of arterial disease-magnetic resonance study. Stroke 2015, 46, 1233–1238. [Google Scholar] [CrossRef]
- Gouw, A.A.; van der Flier, W.M.; Fazekas, F.; van Straaten, E.C.; Pantoni, L.; Poggesi, A.; Inzitari, D.; Erkinjuntti, T.; Wahlund, L.O.; Waldemar, G.; et al. Progression of white matter hyperintensities and incidence of new lacunes over a 3-year period: The leukoaraiosis and disability study. Stroke 2008, 39, 1414–1420. [Google Scholar] [CrossRef]
- Schmidt, R.; Seiler, S.; Loitfelder, M. Longitudinal change of small vessel disease related brain abnormalities. J. Cereb. Blood Flow Metab. 2016, 36, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Muñoz Maniega, S.; Chappell, F.M.; Valdés Hernández, M.C.; Armitage, P.A.; Makin, S.D.; Heye, A.K.; Thrippleton, M.J.; Sakka, E.; Shuler, K.; Dennis, M.S.; et al. Integrity of normal appearing white matter: Influence of age, visible lesion burden and hypertension in patients with small-vessel disease. J. Cereb. Blood Flow Metab. 2016, 37, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Smallwood, A.; Oulhaj, A.; Joachim, C.; Christie, S.; Sloan, C.; Smith, A.D.; Esiri, M. Cerebral subcortical small vessel disease and its relation to cognition in elderly subjects: A pathological study in the Oxford Project to Investigate Memory and Ageing (OPTIMA) cohort. Neuropathol. Appl. Neurobiol. 2012, 38, 337–343. [Google Scholar] [CrossRef]
- Kramer, J.H.; Reed, B.R.; Mungas, D.; Weiner, M.W.; Chui, H. Executive dysfunction in subcortical ischaemic vascular disease. J. Neurol. Neurosurg. Psychiatry 2002, 72, 217–220. [Google Scholar] [CrossRef] [Green Version]
- Burton, E.; Ballard, C.; Stephens, S.; Kenny, R.A.; Kalaria, R.; Barber, R.; O’Brien, J. Hyperintensities and fronto-subcortical atrophy on MRI are substrates of mild cognitive deficits after stroke. Dement. Geriatr. Cogn. Disord. 2003, 16, 113–118. [Google Scholar] [CrossRef]
- Tullberg, M.; Fletcher, E.; DeCarli, C.; Mungas, D.; Reed, B.R.; Harvey, D.J.; Weiner, M.W.; Chui, H.C.; Jagust, W.J. White matter lesions impair frontal lobe function regard-less of their location. Neurology 2004, 63, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Gold, G.; Kövari, E.; Herrmann, F.R.; Canuto, A.; Hof, P.R.; Michel, J.P.; Bouras, C.; Giannakopoulos, P. Cognitive consequences of thalamic, basal ganglia, and deep white matter lacunes in brain aging and dementia. Stroke 2005, 36, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Golsari, A.; Fiehler, J.; Rosenkranz, M.; Gerloff, C.; Thomalla, G. Dynamics of regional distribution of ischemic lesions in middle cerebral artery trunk occlusion relates to collateral circulation. J. Cereb. Blood Flow Metab. 2010, 31, 36–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijkhuizen, R.M.; Knollema, S.; van der Worp, H.B.; Ter Horst, G.J.; De Wildt, D.J.; Berkelbach van der Sprenkel, J.W.; Tulleken, K.A.; Nicolay, K. Dynamics of cerebral tissue injury and perfusion after temporary hypoxia-ischemia in the rat: Evidence for region-specific sensitivity and delayed damage. Stroke 1998, 29, 695–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.H.; Liu, K.F.; Ye, Z.R.; Gutierrez, J.A. Incomplete infarct and delayed neuronal death after transient middle cerebral artery occlusion in rats. Stroke 1997, 28, 2303–2309. [Google Scholar] [CrossRef]
- Konaka, K.; Miyashita, K.; Naritomi, H. Changes in diffusion-weighted magnetic resonance imaging findings in the acute and subacute phases of anoxic encephalopathy. J. Stroke Cerebrovasc. Dis. 2007, 16, 82–83. [Google Scholar] [CrossRef]
- Ravens, J.R. Vascular changes in the human senile brain. Adv. Neurol. 1978, 20, 487–501. [Google Scholar]
- Klassen, A.C.; Sung, J.H.; Stadlan, E.M. Histological changes in cerebral arteries with increasing age. J. Neuropathol. Exp. Neurol. 1968, 27, 607–623. [Google Scholar] [CrossRef]
- Cummings, J.L. Frontal-subcortical circuits and human behavior. Arch. Neurol. 1993, 50, 873–880. [Google Scholar] [CrossRef]
- Mega, M.S.; Cummings, J.L. Frontal-subcortical circuits and neuropsychiatric disorders. J. Neuropsychiatry Clin. Neurosci. 1994, 6, 358–370. [Google Scholar]
- Yao, H.; Sadoshima, S.; Kuwabara, Y.; Ichiya, Y.; Fujishima, M. Cerebral blood flow and oxygen metabolism in patients with vascular dementia of the Binswanger type. Stroke 1990, 21, 1694–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, A.; Ishii, N.; Nishihara, Y.; Horie, A. Medullary arteries in aging and dementia. Stroke 1991, 22, 442–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tak, S.; Yoon, S.J.; Jang, J.; Yoo, K.; Jeong, Y.; Ye, J.C. Quantitative analysis of hemodynamic and metabolic changes in subcortical vascular dementia using simulataneous near-infrared spectroscopy and FMRI measurements. Neuroimage 2011, 55, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, M.; Cutini, S.; Wahl, M.; Scheid, R.; von Cramon, D.Y. Neurovascular coupling is impaired in cerebral microangiopathy an event related stroop study. Neuroimage 2007, 34, 26–34. [Google Scholar] [CrossRef]
- Bar, K.; Boettger, M.; Seidler, N.; Mentzelh, H.J.; Terborg, C.; Sauer, H. Influence of galantamine on vasomotor reactivity in AD and vascular dementia due to microangiopathy. Stroke 2007, 38, 3186–3192. [Google Scholar] [CrossRef] [Green Version]
- De Reuck, J.; Decoo, D.; Marchau, M.; Santens, P.; Lemahieu, I.; Strijckmans, K. Positron emission tomography in vascular dementia. J. Neurol. Sci. 1998, 154, 55–61. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Murase, K.; Oku, N.; Kitagawa, K.; Imaizumi, M.; Takasawa, M.; Nishikawa, T.; Matsumoto, M.; Hatazawa, J.; Hori, M. Statistical image analysis of cerebral blood flow in vascular dementia with small-vessel disease. J. Nucl. Med. 2003, 44, 505–511. [Google Scholar]
- Yang, D.; Kim, B.; Park, J.; Kim, S.; Kim, E.; Sohn, H. Analysis of cerebral blood flow of subcortical vascular dementia with single photon emission computed tomography: Adaptation of statistical parametric mapping. J. Neurol. Sci. 2002, 203, 199–205. [Google Scholar] [CrossRef]
- Ramirez-Gomez, C.; Zheng, C.; Reed, B.; Kramer, J.; Mungas, D.; Zarow, C.; Vinters, H.; Ringman, J.M.; Chui, H. Neuropsychological profiles differentiate Alzheimer Disease from Subcortical Ischemic vascular dementia in an autopsy-defined cohort. Dement. Geriatr. Cogn. Disord. 2017, 44, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.L. Vascular subcortical dementias: Clinical aspects. Dementia 1994, 5, 177–180. [Google Scholar] [CrossRef]
- Desmond, D.W.; Erkinjuntti, T.; Sano, M.; Cummings, J.L.; Bowler, J.V.; Pasquier, F.; Moroney, J.T.; Ferris, S.H.; Stern, Y.; Sachdev, P.S.; et al. The cognitive syndrome of vascular dementia: Implications for clinical trials. Alzheimer Dis. Assoc. Disord. 1999, 13, 21–29. [Google Scholar] [CrossRef]
- Sachdev, P.S.; Brodaty, H.; Valenzuela, M.J.; Lorentz, L.; Looi, J.C.; Wen, W.; Zagami, A.S. The neuropsychological profile of vascular cognitive impairment in stroke and TIA patients. Neurology 2004, 62. [Google Scholar] [CrossRef] [PubMed]
- Traykov, L.; Baudic, S.; Thibaudet, M.C.; Rigaud, A.S.; Samgghe, A.; Boller, F. Neuropsychological deficit in early subcortical vascular dementia: Comparison to AD. Dement. Geriatr. Cogn. Disord. 2002, 14, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.; Signori, R. Neural correlates for apathy: Frontal-prefrontal and parietal cortical-subcortical circuits. Front. Aging Neurosci. 2016, 9, 289. [Google Scholar] [CrossRef] [Green Version]
- Ishii, N.; Nashihara, Y.; Imamura, T. Why do frontal lobe symptoms predominate in vascular dementia with lacunes? Neurology 1986, 36, 340–345. [Google Scholar] [CrossRef]
- De Jager, C.A. Critical levels of brain atrophy associated with homocysteine and cognitive decline. Neurobiol. Ageing 2014, 35, S35–S39. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.L. Food for thought: Souvenaid in mild Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 31, 237–238. [Google Scholar] [CrossRef]
- Peng, D. Geriatric Neurology Group; Chinese Society of Geriatrics. Clinical Practice Guideline for Cognitive Impairment of Cerebral Small Vessel Disease Writing Group. Clinical practice guideline for cognitive impairment of cerebral small vessel disease. Aging Med. 2019, 2, 64–73. [Google Scholar] [CrossRef]
- Roe, A.J.; Zhang, S.; Bhadelia, R.A.; Johnson, E.J.; Lichtenstein, A.H.; Rogers, G.T.; Rosenberg, I.H.; Smith, C.E.; Zeisel, S.H.; Scott, T.S. Choline and its metabolites are differently associated with cardiometabolic risk factors, history of cardiovascular disease, and MRI-documented cerebrovascular disease in older adults. Am. J. Clin. Nutr. 2017, 105, 1283–1290. [Google Scholar] [CrossRef]
- Blusztajn, J.K.; Slack, B.E.; Mellott, T.J. Neuroprotective Actions of Dietary Choline. Nutrients 2017, 9, 815. [Google Scholar] [CrossRef] [Green Version]
- Castro, C.A.; Rudy, J.W. Early-life malnutrition selectively retards the development of distal- but not proximal-cue navigation. Dev. Psychobiol. 1987, 20, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Mellott, T.J.; Huleatt, O.M.; Shade, B.N.; Pender, S.M.; Liu, Y.B.; Slack, B.E.; Blusztajn, J.K. Perinatal choline supplementation reduces amyloidosis and increases choline acetyltransferae expression in the hippocampus of the APPswePS1dE9 Alzheimer’s disease model mice. PLoS ONE 2017, 12, e0170450. [Google Scholar]
- Napoli, I.; Blusztajn, J.K.; Mellott, T.J. Prenatal choline supplementation in rats increases the expression of IGF2 and its receptor IGF2R and enhances IGF2-induced acetylcholine release in hippocampus and frontal cortex. Brain Res. 2008, 1237, 124–135. [Google Scholar] [CrossRef]
- Williams, C.L.; Meck, W.H.; Heyer, D.; Loy, R. Hypertrophy of basal forebrain neurons and enhanced visuospatial memory in perinatally choline-supplemented rats. Brain Res. 1998, 794, 225–238. [Google Scholar] [CrossRef]
- Cermak, J.M.; Holler, T.; Jackson, D.A.; Blusztajn, J.K. Prenatal availability of choline modifies development of the hippocampal cholinergic system. FASEB J. 1998, 12, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacheva, V.P.; Mellott, T.J.; Davison, J.M.; Wagner, N.; Lopez-Coviella, I.; Schnitzler, A.C.; Blusztajn, J.K. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J. Biol. Chem. 2007, 282, 31777–31788. [Google Scholar] [CrossRef] [Green Version]
- Lopes, S.; Lewis, A.; Hajkova, P.; Dean, W.; Oswald, J.; Forne, T.; Murrell, A.; Constancia, M.; Bartolomei, M.; Walter, J.; et al. Epigenetic modifications in an imprinting cluster are controlled by a hierarchy of DMRs suggesting long-range chromatin interactions. Hum. Mol. Genet. 2003, 12, 295–305. [Google Scholar] [CrossRef]
- Levenson, J.M.; Roth, T.L.; Lubin, F.D.; Miller, C.A.; Huang, I.C.; Desai, P.; Malone, L.M.; Sweatt, J.D. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 2006, 281, 15763–15773. [Google Scholar] [CrossRef] [Green Version]
- Yossifoff, M.; Kisliouk, T.; Meiri, N. Dynamic changes in DNA methylation during thermal control establishment affect CREB binding to the brain-derived neurotrophic factor promoter. Eur. J. Neurosci. 2008, 28, 2267–2277. [Google Scholar] [CrossRef]
- Lubin, F.D.; Roth, T.L.; Sweatt, J.D. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 2008, 28, 10576–10586. [Google Scholar] [CrossRef]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Webb, W.M.; Sanchez, R.G.; Perez, G.; Butler, A.A.; Hauser, R.M.; Rich, M.C.; O’Bierne, A.L.; Jarome, T.J.; Lubin, F.D. Dynamic association of epigenetic H3K4me3 and DNA 5hmC marks in the dorsal hippocampus and anterior cingulate cortex following reactivation of a fear memory. Neurobiol. Learn. Mem. 2017, 142, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.J.; Rahn, E.J.; Paulukaitis, B.S.; Savell, K.E.; Kordasiewicz, H.B.; Wang, J.; Lewis, J.W.; Posey, J.; Strange, S.K.; Guzman-Karlsson, M.C.; et al. Tcf4 Regulates Synaptic Plasticity, DNA Methylation, and Memory Function. Cell Rep. 2016, 16, 2666–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barany, M.; Chang, Y.C.; Arus, C.; Rustan, T.; Frey, W.H. Increased glycerol-3-phosphorylcholine in post-mortem Alzheimer’s brain. Lancet 1985, 1, 517. [Google Scholar] [CrossRef]
- Blusztajn, J.K.; Gonzalez-Coviella, I.L.; Logue, M.; Growdon, J.H.; Wurtman, R.J. Levels of phospholipid catabolic intermediates, glycerophosphocholine and glycerophosphoethanolamine, are elevated in brains of Alzheimer’s disease but not of Down’s syndrome patients. Brain Res. 1990, 536, 240–244. [Google Scholar] [CrossRef]
- Corbin, K.D.; Zeisel, S.H. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr. Opin. Gastroeneterol. 2012, 28, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Karaca, U.; Schram, M.T.; Houben, A.J.; Muris, D.M.; Stehouwer, C.D. Microvascular dysfunction as a link between obesity, insulin resistance and hypertension. Diabetes Res. Clin. Pract. 2014, 103, 382–387. [Google Scholar] [CrossRef]
- Roberson, L.L.; Aneni, E.C.; Maziak, W.; Agatston, A.; Feldman, T.; Rouseff, M.; Tran, T.; Blaha, M.J.; Santos, R.D.; Sposito, A.; et al. Beyond BMI: The metabolically healthy obese phenotype and its association with clinical/subclinical cardiovascular disease all-cause mortality; a systematic review. BMC Public Health 2014, 14, 14. [Google Scholar] [CrossRef] [Green Version]
- Yuki, D.; Sugiura, Y.; Zaima, N.; Akatsu, H.; Takei, S.; Yao, I.; Maesako, M.; Kinoshita, A.; Yamamoto, T.; Kon, R.; et al. DHA-PC and PSD-95 decrease after loss of synaptophysin and before neuronal loss in patients with Alzheimer’s disease. Sci. Rep. 2014, 4, 7130. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, E.J.; Bongard, V.; Beiser, A.S.; Lamon-Fava, S.; Robins, S.J.; Au, R.; Tucker, K.L.; Kyle, D.J.; Wilson, P.W.; Wolf, P.A. Plasma phosphatidylcholine docosahexaenoic acid content and risk of dementia and Alzheimer disease: The Framingham Heart Study. Arch. Neurol. 2006, 63, 1545–1550. [Google Scholar] [CrossRef]
- Trushina, E.; Dutta, T.; Persson, X.M.; Mielke, M.M.; Petersen, R.C. Identification of altered metabolic pathways in plasma and CSF in mild cognitive impairment and Alzheimer’s disease using metabolomics. PLoS ONE 2013, 8, e63644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Dominguez, R.; Garcia-Barrera, T.; Gomez-Ariza, J.L. Combination of metabolomic and phospholipid-profiling approaches for the study of Alzheimer’s disease. J. Proteom. 2014, 104, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Zhong, X.; Cheema, A.K.; Orquiza, M.H.; Chidambaram, S.; Tan, M.T.; Gresenz, C.R.; Fitzgerald, K.T.; Nalls, M.A.; Singleton, A.B.; et al. Plasma 24-metabolite Panel Predicts Preclinical Transition to Clinical Stages of Alzheimer’s Disease. Front. Neurol. 2015, 6, 237. [Google Scholar] [CrossRef] [PubMed]
- Proitsi, P.; Kim, M.; Whiley, L.; Simmons, A.; Sattlecker, M.; Velayudhan, L.; Lupton, M.K.; Soininen, H.; Kloszewska, I.; Mecocci, P.; et al. Association of blood lipids with Alzheimer’s disease: A comprehensive lipidomics analysis. Alzheimers Dement. 2016, 13, 140–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heude, B.; Ducimetiere, P.; Berr, C. Cognitive decline and fatty acid composition of erythrocyte membranes—The EVA Study. Am. J. Clin. Nutr. 2003, 77, 803–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.N.; Ma, D.; Shui, G.; Wong, P.; Cazenave-Gassiot, A.; Zhang, X.; Wenk, M.R.; Goh, E.L.; Silver, D.L. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 2014, 509, 503–506. [Google Scholar] [CrossRef]
- Guemez-Gamboa, A.; Nguyen, L.N.; Yang, H.; Zaki, M.S.; Kara, M.; Ben-Omran, T.; Akizu, N.; Rosti, R.O.; Rosti, B.; Scott, E.; et al. Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome. Nat. Genet. 2015, 47, 809–813. [Google Scholar] [CrossRef] [Green Version]
- Goedecke, L.; Fernadnez-Hernando, C. MicroRNAs: A connection between cholesterol metabolism and neurodegeneration. Neurobiol. Dis. 2014, 72, 48–53. [Google Scholar] [CrossRef] [Green Version]
- Scheltens, P.; Kamphuis, P.J.; Verhey, F.R.; Olde Rikkert, M.G.M.; Wurtman, R.J.; Wilkinson, D.; Twisk, J.W.R.; Kurz, A. Efficacy of a medical food in mild Alzheimer’s Disease: A randomized, controlled trial. Alzheimer’s Dement. 2010, 6, 1–10. [Google Scholar] [CrossRef]
- Scheltens, P.; Twisk, J.W.R.; Blesa, R.; Scarpini, E.; von Arnim, C.A.F.; Bongers, A.; Harrison, J.; Swinkels, S.H.N.; Stam, C.J.; de Waal, H.; et al. Efficacy of Souvenaid in mild Alzheimer’s disease: Results from a randomized, controlled trial. J. Alzheimer’s Dis. 2014, 31, 225–226. [Google Scholar] [CrossRef] [Green Version]
- Rijpma, A.; Meulenbroek, O.; van Hees, A.M.J.; Sijben, J.W.C.; Vellas, B.; Shah, R.C.; Bennett, D.A.; Scheltens, P.; Olde Rikkert, M.G.M. Effects of Souvenaid on plasma micronutiren levels and fatty acid profiles in mild and mild-to-mderate AD. Alzheimer’s Res. Ther. 2015, 7, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olde Rikkert, M.G.; Verhey, F.R.; Blesa, R.; von Arnim, C.A.; Bongers, A.; Harrison, J.; Sijben, J.; Scarpini, E.; Vandewoude, M.F.; Vellas, B.; et al. Tolerability and safety of Souvenaid in Patients with Mild Alzheimer’s Disease: Results of multi center, 24-week, open label extension study. J. Alzheimer’s Dis. 2015, 44, 471–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchetti, A.; Perotta, D.; Cravello, L.; Ranieri, P.; Trabucchi, M. Effectiveness of a specific nutritional supplement on cognitive, behavioral and functional sympotms in mild cognitive impairment and AD: caregivers’ judgements. Results of an observational survey. JGG 2018, 66, 68–74. [Google Scholar]
- Yanai, H. Effects of N-3 Polyunsaturated Fatty Acids on Dementia. J. Clin. Med. Res. 2017, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.C.; Schneider, J.A.; Tangney, C.C. Thoughts on B-vitamins and dementia. J. Alzheimer’s Dis. 2006, 9, 429–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suwa, M.; Yamaguchi, S.; Komori, T.; Kajimoto, S.; Kino, M. The Association between Cerebral White Matter Lesions and Plasma Omega-3 to Omega-6 Polyunsaturated Fatty Acids Ratio to Cognitive Impairment Development. Biomed Res. Int. 2015, 2015, 153437. [Google Scholar] [CrossRef] [Green Version]
- Bowman, G.; Silbert, L.C.; Dodge, H.H.; Lahna, D.; Hagen, K.; Murchison, C.F.; Howieson, D.; Kaye, J.; Quinn, J.F.; Shinto, L. Randomized Trial of Marine n-3 Polyunsaturated Fatty Acids for the Prevention of Cerebral Small Vessel Disease and Inflammation in Aging (PUFA Trial): Rationale, Design and Baseline Results. Nutrients 2019, 11, 735. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Ren, H.; Yao, X.; Shi, Z.; Liang, F.; Kang, J.X.; Wan, J.B.; Pei, Z.; Su, K.P.; Su, H. Enriched Brain Omega-3 Polyunsaturated Fatty Acids Confer Neuroprotection against Microinfarction. EBioMedicine 2018, 32, 50–61. [Google Scholar] [CrossRef]
- Victor, M.; Adam, R.D.; Collins, G.H. The Wernicke-Korsakoff Syndrome and Related Neurologic Disorders Due to Alcoholism and Malnutrition, 2nd ed.; Davis Publications: Philadelphia, PA, USA, 1989. [Google Scholar]
- Victor, M.; Adams, R.D.; Collins, G.H. The Wernicke-Korsakoff syndrome. A clinical and pathological study of 245 patients, 82 with post-mortem examinations. Contemp. Neurol. Ser. 1971, 7, 201–206. [Google Scholar]
- Alzheimer’s Society. What is alcohol-related brain damage. Factsheet 438LP. 2015. Available online: https://www.alzheimers.org.uk (accessed on 15 November 2019).
- UK Chief Medical Officers’ Alcohol Guidelines Review. Available online: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/489795/summary.pdf (accessed on 15 September 2016).
- Gupta, S.; Warner, J. Alcohol-related dementia: A 21st-century silent epidemic? Br. J. Psychiatry 2008, 193, 351–353. [Google Scholar] [CrossRef] [Green Version]
- Sechi, G.; Serra, A. Wernicke’s encephalopathy: New clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007, 6, 442–455. [Google Scholar] [CrossRef]
- Pech, N.; Meyer, F.; Lippert, H.; Manger, T.; Stroh, C. Complications, reoperations, and nutrient deficiencies two years after sleeve gastrectomy. J. Obes. 2012, 2012, 828737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, C.G.; Sheedy, D.L.; Lara, A.I.; Garrick, T.M.; Hilton, J.M.; Raisanen, J. Prevalence of Wernicke-Korsakoff syndrome in Australia: Has thiamine fortification made a difference? Med. J. Aust. 1998, 168, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.D. Mechanisms of vitamin deficiency in chronic alcohol misusers and the development of the Wernicke-Korsakoff syndrome. Alcohol Alcohol. Oxf. Suppl. 2000, 35, 2–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majchrzak, D.; Singer, I.; Männer, M.; Rust, P.; Genser, D.; Wagner, K.H.; Elmadfa, I. B-vitamin status and concentrations of homocysteine in Austrian omnivores, vegetarians and vegans. Ann. Nutr. Metab. 2006, 50, 485–491. [Google Scholar] [CrossRef]
- Moretti, R.; Caruso, P.; Dal Ben, M.; Gazzin, S.; Tiribelli, C. Thiamine and alcohol for brain pathology: Super-imposing or different causative factors for brain damage? Curr. Drug Abuse Rev. 2017, 10, 44–51. [Google Scholar] [CrossRef]
- Butterworth, R.F.; Giguère, J.F.; Besnard, A.M. Activities of thiamine-dependent enzymes in two experimental models of thiamine-deficiency encephalopathy 2 alpha-Ketoglutarate dehydrogenase. Neurochem. Res. 1986, 11, 567–577. [Google Scholar] [CrossRef]
- Dreyfus, P.M. Clinical application of blood transketolase determinations. N. Engl. J. Med. 1962, 267, 596–598. [Google Scholar] [CrossRef]
- Hazell, A.S.; Todd, K.G.; Butterworth, R.F. Mechanisms of neuronal cell death in Wernicke’s encephalopathy. Metab. Brain Dis. 1998, 13, 97–122. [Google Scholar] [CrossRef]
- Butterworth, R.F.; Kril, J.J.; Harper, C.G. Thiamine-dependent enzyme changes in the brains of alcoholics: Relationship to the Wernicke-Korsakoff syndrome. Alcohol. Clin. Exp. Res. 1993, 17, 1084–1088. [Google Scholar] [CrossRef]
- Peterson, C.; Héroux, M.; Lavoie, J.; Butterworth, R.F. Loss of (3H) kainate and of NMDA-displaceable (3H)glutamate binding sites in brain in thiamine deficiency: Results of a quantitative autoradiographic study. Neurochem. Res. 1995, 20, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, D.D.; Rao, V.L.; Butterworth, R.F. Alterations in serotonin parameters in brain of thiamine-deficient rats are evident prior to the appearance of neurological symptoms. J. Neurochem. 1996, 67, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Todd, K.G.; Butterworth, R.F. Evaluation of the role of NMDA-mediated excitotoxicity in the selective neuronal loss in experimental Wernicke encephalopathy. Exp. Neurol. 1998, 149, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Jhala, S.S.; Wang, D.; Hazell, A.S. Thiamine deficiency results in release of soluble factors that disrupt mitochondrial membrane potential and downregulate the glutamate transporter splice-variant GLT-1b in cultured astrocytes. Biochem. Biophys. Res. Commun. 2014, 6, 335–341. [Google Scholar] [CrossRef]
- Sheng, M.; McFadden, G.; Greenberg, M.E. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron 1990, 4, 571–582. [Google Scholar] [CrossRef]
- Estus, S.; Zaks, W.J.; Freeman, R.S.; Gruda, M.; Bravo, R.; Johnson, E.M. Altered gene expression in neurons during programmed cell death: Identification of c-jun as necessary for neuronal apoptosis. J. Cell. Biol. 1994, 127, 1717–1727. [Google Scholar] [CrossRef] [Green Version]
- Guerrini, I.; Thomson, A.D.; Cook, C.C.; McQuillin, A.; Sharma, V.; Kopelman, M.; Reynolds, G.; Jauhar, P.; Harper, C.; Gurling, H.M. Direct genomic PCR sequencing of the high affinity thiamine transporter (SLC19A2) gene identifies three genetic variants in Wernicke Korsakoff syndrome (WKS). Am. J. Med. Genet B Neuropsychiatr. Genet. 2005, 137, 17. [Google Scholar] [CrossRef]
- Guerrini, I.; Cook, C.C.; Kest, W.; Devitgh, A.; McQuillin, A.; Curtis, D.; Gurling, H.M. Genetic linkage analysis supports the presence of two susceptibility loci for alcoholism and heavy drinking on chromosome 1p22.1-11.2 and 1q21.3-24.2. BMC Genet. 2005, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Guerrini, I.; Thomson, A.D.; Gurling, H.M. Molecular genetics of alcohol related brain damage. Alcohol Alcohol. 2009, 44, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Witt, E.D.; Goldman-Rakic, P.S. Intermittent thiamine deficiency in the rhesus monkey. I. Progression of neurological signs and neuroanatomical lesions. Ann. Neurol. 1983, 13, 376–395. [Google Scholar] [CrossRef]
- Summers, J.A.; Pullan, P.T.; Kril, J.J.; Harper, C.G. Increased central immunoreactive beta-endorphin content in patients with Wernicke-Korsakoff syndrome and in alcoholics. J. Clin. Pathol. 1991, 44, 126–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G.E.; Hirsch, J.H.; Fonzetti, P.; Jordon, B.D.; Cirio, R.T.; Elder, J. Vitamin B1 and dementia. Ann. N. Y. Acad. Sci. 2016, 1367, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.; Hauser, R.; Chen, M. Plasma thiamine deficiency associated with Alzheimer’s disease but not Parkinson’s Disease. Met. Brain Dis. 1998, 13, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Fei, G.; Lu, J.; Jin, L.; Pan, S.; Chen, Z.; Wang, C.; Sang, S.; Liu, H.; Hu, W.; et al. Measurement of blood thiamine metabolites for Alzheimer’s Disease Diagnosis. Ebiomedicine 2016, 3, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Chari, D.; Ali, R.; Gupta, R. Reversible dementia in elderly: Really uncommon? J. Geriatr. Ment. Health 2015, 2, 30–37. [Google Scholar]
- Morris, M.S. The Role of B Vitamins in Preventing and Treating Cognitive Impairment and Decline. Adv. Nutr. 2012, 3, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Bunik, V.I. Thiamin-dependent enzymes: New perspectives from the interface between chemistry and biology. FEBS J. 2013, 280, 6373. [Google Scholar] [CrossRef] [Green Version]
- Szutowicz, A.; Bielarczyk, H.; Jankowska-Kulawy, A.; Pawelczyk, T.; Ronowska, A. Acetyl CoA, the key factor for survival or death of cholinergic neurons in course of neurodegenerative diseases. Neurochem. Res. 2013, 38, 1523–1542. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Gong, N.; Zhao, J.; Yu, Z.; Gu, F.; Chen, J.; Sun, X.; Lei, Z.; Yu, M.; Xu, Z.; et al. Powerful beneficial effects of benfotiamine on cognitive impairment and b-amyloid deposition in amyloid precursor protein/presenilin-1transgenic mice. Brain 2010, 133, 1342–1352. [Google Scholar] [CrossRef]
- Calingasan, N.Y.; Gandy, S.E.; Baker, H.; Sheu, K.F.; Kim, K.S.; Wisniewski, H.M.; Gibson, G.E. Accumulation of amyloid precursor protein-like immunoreactivity in rat brain in response to thiamine deficiency. Brain Res. 1995, 677, 50–60. [Google Scholar] [CrossRef]
- Bettendorff, L.; Wins, P. Biological functions of thiamine derivatives: Focus on non-coenzyme roles. Biochemistry 2013, 1, 10. [Google Scholar]
- Parkhomenko, Y.M.; Pavlova, A.S.; Mezhenskaya, O.A. Mechanisms responsible for the high sensitivity of neural cells to vitamin B1 deficiency. Neurophysiology 2016, 48, 429–448. [Google Scholar] [CrossRef]
- Mkrtchyan, G.; Aleshin, V.; Parkhomenko, Y.M.; Kaehne, T.; Disalvo, M.L.; Parroni, A.; Contestabile, R.; Vovk, A.; Bettendorf, L.; Bunik, V. Molecular mechanisms of the non-coenzyme action of thiamin in brain: Biochemical, structural and pathway analysis. Sci. Rep. 2015, 27, 12583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frederich, M.; Delvaux, D.; Gigliobianc, T.; Gangolf, M.; Dive, G.; Mazzucchelli, G. Thiaminylated adenine nucleotides. Chemical synthesis, structural characterization and natural occurrence. FEBS J. 2009, 276, 3256–3268. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, J.A.; Parrott, J. New considerations on the neuromodulatory role of thiamine. Pharmcology 2012, 89, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Geng, M.Y.; Saito, H.; Katsuki, H. The effects of thiamine and oxythiamine on the survival of cultured brain neurons. Jpn. J. Pharmacol. 1995, 68, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Langlais, P.J.; Zhang, S.X. Extracellular Glutamate Is Increased in Thalamus During Thiamine Deficiency-Induced Lesions and Is Blocked by MK-801. J. Neurochem. 1993, 61, 2175–2182. [Google Scholar] [CrossRef]
- Hazell, A.S.; Butterworth, R.F.; Hakim, A.M. Cerebral vulnerability is associated with selective increase in extracellular glutamate concentration in experimental thiamine deficiency. J. Neurochem. 1993, 61, 1155–1158. [Google Scholar] [CrossRef]
- Chou, W.P.; Chang, Y.H.; Lin, H.C.; Chang, Y.H.; Chen, Y.Y.; Ko, C.H. Thiamine for preventing dementia development among patients with alcohol use disorder: A nationwide population-based cohort study. Clin. Nutr. 2019, 38, 1269–1273. [Google Scholar] [CrossRef]
- Marashly, E.T.; Bohlega, S.A. Riboflavin has neuroprotective potential: Focus on Parkinson’s disease and migraine. Front. Neurol. 2017, 8, 333. [Google Scholar] [CrossRef]
- Thakur, K.; Tomar, S.K.; Singh, A.K.; Mandal, S.; Arora, S. Riboflavin and health: A review of recent human Research. Crit. Rev. Food Sci. Nutr. 2017, 57, 3650–3660. [Google Scholar] [CrossRef] [PubMed]
- Ashoori, M.; Saedisomeolia, A. Riboflavin (Vitamin B2) and Oxidative Stress: A Review. Br. J. Nutr. 2014, 111, 1985–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saedisomeolia, A.; Ashoori, M. Riboflavin in Human Health: A Review of Current Evidences. Adv. Food Nutr. Res. 2018, 83, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Nadh, T.; To, H.; Christensen, H.N. Riboflavin Can Protect Tissue from Oxidative Injury. Nutr. Rev. 1993, 51, 149–150. [Google Scholar] [CrossRef]
- Sanches, S.C.; Ramalho, L.N.Z.; Mendes-Braz, M.; Terra, V.A.V.A.; Cecchini, R.; Augusto, M.J.; Ramalho, F.S. Riboflavin (Vitamin B-2) Reduces Hepatocellular Injury Following Liver Ischaemia and Reperfusion in Mice. Food Chem. Toxicol. 2014, 67, 65–71. [Google Scholar] [CrossRef]
- Betz, A.L.; Ren, X.D.; Ennis, S.R.; Hultquist, D.E. Riboflavin Reduces Edema in Focal Cerebral Ischemia. Brain Edema IX 1994, 60, 314–317. [Google Scholar]
- Peraza, A.V.; Guzmán, D.C.; Brizuela, N.O.; Herrera, M.O.; Olguín, H.J.; Silva, M.L.; Tapia, B.J.; Mejía, G.B. Riboflavin and pyridoxine restore dopamine levels and reduce oxidative stress in brain of rats. BMC Neurosci. 2018, 19, 71. [Google Scholar] [CrossRef]
- Moat, S.J.; Ashfield-watt, P.A.L.; Powers, H.J.; Newcombe, R.G.; Mcdowell, I.F.W. Effect of Riboflavin Status on the Homocysteine-Lowering Effect of Folate in Relation to the MTHFR (C677T) Genotype. Clin. Chem. 2003, 302, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.J.; Wu, W.M.; Yang, F.L.; Hsu, G.S.W.; Huang, C.Y. Vitamin B2 Inhibits Glutamate Release from Rat Cerebrocortical Nerve Terminals. Neuroreport 2008, 19, 1335–1338. [Google Scholar] [CrossRef]
- Lin, Y.; Desbois, A.; Jiang, S.; Hou, S.T. Group B Vitamins Protect Murine Cerebellar Granule Cells from Glutamate/NMDA Toxicity. Neuroreport 2004, 15, 2241–2244. [Google Scholar] [CrossRef]
- Mazur-Bialy, A.I.; Pocheć, E. HMGB1 Inhibition During Zymosan-Induced Inflammation: The Potential Therapeutic Action of Riboflavin. Arch. Immunol. Ther. Exp. 2016, 64, 171–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, K.; Hoey, L.; Hughes, C.F.; Ward, M.; McNulty, H. Causes, Consequences and Public Health Implications of Low B-Vitamin Status in Ageing. Nutrients 2016, 8, 725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boisvert, W.A.; Castaneda, C.; Mendoza, I.; Langeloh, G.; Solomons, N.W.; Gershoff, S.N.; Russell, R.M.; Soloinons, W. Prevalence of Riboflavin Deficiency among Guatemalan Elderly People and Its Relationship to Milk Intake. Am. J. Clin. Nutr. 1993, 58, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Essama-Tjani, J.C.; Guilland, J.C.; Fuchs, F.; Lombard, M.; Richard, D. Changes in Thiamin, Riboflavin, Niacin, Beta-Carotene, Vitamins, C, A, D and E Status of French Elderly Subjects during the First Year of Institutionalization. Int. J. Vitam. Nutr. Res. 2000, 70, 54–64. [Google Scholar] [CrossRef]
- Ter Borg, S.; Verlaan, S.; Hemsworth, J.; Mijnarends, D.M.; Schols, J.M.; Luiking, Y.C.; De Groot, L.C. Micronutrient Intakes and Potential Inadequacies of Community-Dwelling Older Adults: A Systematic Review. Br. J. Nutr. 2015, 113, 1195–1206. [Google Scholar] [CrossRef]
- Powers, H.J. Riboflavin (Vitamin B-2) and Health. Am. J. Clin. Nutr. 2003, 77, 1352–1360. [Google Scholar] [CrossRef]
- Skalka, H.W.; Prchal, J.T. Cataracts and Riboflavin Deficiency. Am. J. Clin. Nutr. 1981, 34, 861–863. [Google Scholar] [CrossRef]
- Leshner, R.T. Riboflavin deficiency—A reversible neurodegenerative disease. Ann. Neurol. 1981, 10, 294–295. [Google Scholar]
- Norton, W.N.; Daskal, I.; Savage, H.E.; Seibert, R.A.; Lane, M. Effects of Riboflavin Deficiency on the Ultrastructure of Rat Sciatic Nerve Fibers. Am. J. Pathol. 1976, 85, 651–660. [Google Scholar] [CrossRef] [Green Version]
- Manole, A.; Jaunmuktane, Z.; Hargreaves, I.; Ludtmann, M.H.R.; Salpietro, V.; Bello, O.D.; Pope, S.; Pandraud, A.; Horga, A.; Scalco, R.S.; et al. Clinical, Pathological and Functional Characterization of Riboflavin-Responsive Neuropathy. Brain 2017, 140, 2820–2837. [Google Scholar] [CrossRef] [Green Version]
- Coimbra, C.G.; Junqueira, V.B.C. High Doses of Riboflavin and the Elimination of Dietary Red Meat Promote the Recovery of Some Motor Functions in Parkinson’s Disease Patients. Braz. J. Med. Biol. Res. 2003, 36, 1409–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, D.F.; Saluja, H.S. Prophylaxis of Migraine Headaches with Riboflavin: A Systematic Review. J. Clin. Pharm. Ther. 2017, 42, 394–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehnke, C.; Reuter, U.; Flach, U.; Schuh-Hofer, S.; Einhaupl, K.M.; Arnold, G. High-Dose Riboflavin Treatment Is Efficacious in Migraine Prophylaxis: An Open Study in a Tertiary Care Centre. Eur. J. Neurol. 2004, 11, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Refsum, H. Homocysteine, B Vitamins, and Cognitive Impairment. Annu. Rev. Nutr. 2016, 36, 211–239. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, A.H.; Yeo, N.E.; Weekman, E.M.; Wilcock, D.M. Homocysteine, Hyperhomocysteinemia and Vascular Contributions to Cognitive Impairment and Dementia (VCID). Biochim. Biophys. Acta 2015, 1862, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Hustad, S.; Ueland, P.M.; Vollset, S.E.; Zhang, Y.; Bjorke-Monsen, A.L.; Schneede, J. Riboflavin as a Determinant of Plasma Total Homocysteine: Effect Modification by the Methylenetetrahydrofolate Reductase C677T Polymorphism. Clin. Chem. 2000, 46, 1065–1071. [Google Scholar]
- Udhayabanu, T.; Manole, A.; Rajeshwari, M.; Varalakshmi, P.; Houlden, H.; Ashokkumar, B. Riboflavin Responsive Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Clin. Med. 2017, 6, 52. [Google Scholar] [CrossRef] [Green Version]
- Xiu, L.; Lee, M.; Wahlqvist, M.L.; Chen, R.C.; Huang, Y.; Chen, K.; Li, D. Low and High Homocysteine Are Associated with Mortality Independent of B Group Vitamins but Interactive with Cognitive Status in a Free-Living Elderly Cohort. Nutr. Res. 2012, 32, 928–939. [Google Scholar] [CrossRef]
- McNeill, G.; Jia, X.; Whalley, L.J.; Fox, H.C.; Corley, J.; Gow, A.J.; Brett, C.E.; Starr, J.M.; Deary, I.J. Antioxidant and B Vitamin Intake in Relation to Cognitive Function in Later Life in the Lothian Birth Cohort 1936. Eur. J. Clin. Nutr. 2011, 65, 619–626. [Google Scholar] [CrossRef]
- Kim, H.; Kim, G.; Jang, W.; Kim, S.Y.; Chang, N. Association between Intake of B Vitamins and Cognitive Function in Elderly Koreans with Cognitive Impairment. Nutr. J. 2014, 13, 118. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.; Kang, S.A.; Lee, H.O.; Lee, B.H.; Park, J.S.; Kim, J.H.; Jung, I.K.; Park, Y.J.; Lee, J.E. Relationships between Dietary Intake and Cognitive Function Level in Korean Elderly People. Public Health 2001, 115, 133–138. [Google Scholar] [CrossRef]
- Cunha, N.M.D.; Georgousopoulou, E.N.; Boyd, L.; Sturm, J.; Brien, B.O.; Lucock, M.; Mckune, A.J.; Mellor, D.; Roach, P.D.; Naumovski, N.; et al. Relationship Between B-Vitamin Biomarkers and Dietary Intake with Apolipoprotein E Є4 in Alzheimer’s Disease Dietary Intake with Apolipoprotein E ¾ 4 in. J. Nutr. Gerontol. Geriatr. 2019, 38, 173–195. [Google Scholar] [CrossRef] [PubMed]
- Tucker, D.M.; Penland, J.G.; Sandstead, H.H.; Milne, D.B.; Heck, D.G.; Klevay, L.M.; Tucker, D.M.; Pen, J.G.; Heck, G.; Klevay, L.M. Nutrition Status and Brain Function in Aging. Am. J. Clin. Nutr. 1990, 52, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Ortega, R.M.; Requejo, A.M.; Andrés, P.; Sobaler, A.M.L.; Quintas, M.E.; Redondo, M.R.; Navia, B.; Rivas, T. Dietary Intake and Cognitive Function in A Group of Elderly People. Am. J. Clin. Nutr. 1997, 66, 803–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubitz, I.; Ricny, J.; Atrakchi-Baranes, D.; Shemesh, C.; Kravitz, E.; Liraz-zaltsman, S.; Maksin-matveev, A.; Cooper, I.; Leibowitz, A.; Uribarri, J.; et al. High Dietary Advanced Glycation End Products Are Associated with Poorer Spatial Learning and Accelerated Aβ Deposition in an Alzheimer Mouse Model. Aging Cell 2016, 15, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Jacques, P.F.; Kalmbach, R.; Bagley, P.J.; Russo, G.T.; Rogers, G.; Wilson, P.W.F.; Rosenberg, I.H.; Selhub, J. The Relationship between Riboflavin and Plasma Total Homocysteine in the Framingham Offspring Cohort Is Influenced by Folate Status and the C677T Transition in the Methylenetetrahydrofolate Reductase Gene. J. Nutr. 2002, 132, 283–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, K.; Apostolopoulos, V. B Vitamins and Ageing. Subcell. Biochem. 2018, 90, 451–470. [Google Scholar] [CrossRef]
- Pantoni, L. Cerebral Small Vessel Disease: From Pathogenesis and Clinical Characteristics to Therapeutic Challenges. Lancet Neurol. 2010, 9, 689–701. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy Metabolism and Inflammation in Brain Aging and Alzheimer’s Disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef] [Green Version]
- Makarov, M.V.; Trammell, S.A.J.; Migaud, M.E. The Chemistry of the Vitamin B3 Metabolome. Biochem. Soc. Trans. 2019, 47, 131–147. [Google Scholar] [CrossRef]
- Kirkland, J.B.; Meyer-Ficca, M.L. Niacin. Adv. Food Nutr. Res. 2018, 83, 83–149. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chong, Z.Z.; Maiese, K. Navigating novel mechanisms of cellular plasticity with the nad+ precursor and nutrient nicotinamide. Front. Biosci. 2004, 9, 2500–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, D.O. B Vitamins and the Brain: Mechanisms, Dose and Efficacy—A Review. Nutrients 2016, 8, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, B.; Sannegowda, R.B.; Jain, R.; Dubey, P.; Prakash, S. A rare case of alcoholic pellagra encephalopathy with startle myoclonus and marked response to niacin therapy: Time for a new dictum? BMJ Case Rep. 2013. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Lee, J.H.; Moon, J.H.; Nazim, U.M.; Lee, Y.J.; Seol, J.W.; Hur, J.; Eo, S.K.; Lee, J.H.; Park, S.Y. Niacin alleviates TRAIL-mediated colon cancer cell death via autophagy flux activation. Oncotarget 2016, 7, 4356–4368. [Google Scholar] [CrossRef] [Green Version]
- Kelley, P. Niacin and niacinamide accumulation by rabbit brain slices and choroid plexus. J. Neurochem. 1979, 33, 291–298. [Google Scholar]
- Murthy, M.R.; Rappoport, D.A. Biochemistry of the developing rat brain. Iv. Effect of nicotinamide on brain and liver mitochondria. Biochim. Biophys. Acta 1963, 78, 71–76. [Google Scholar] [CrossRef]
- Deguchi, T.; Ichiyama, A.; Nishizuka, Y.; Hayaishi, O. Studies on the biosynthesis of nicotinamide-adenine dinucleotide in the brain. Biochim. Biophys. Acta 1968, 158, 382–393. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z. Nicotinamide: Necessary Nutrient Emerges as a Novel Cytoprotectant for the Brain. Trends Pharmacol. Sci. 2003, 24, 228–232. [Google Scholar] [CrossRef]
- Park, J.H.; Long, A.; Owens, K.; Kristian, T. Nicotinamide Mononucleotide Inhibits Post-Ischemic NAD+ Degradation and Dramatically Ameliorates Brain Damage Following Global Cerebral Ischemia. Neurobiol. Dis. 2016, 95, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Klaidman, L.; Morales, M.; Kem, S.; Yang, J.; Adams, J.D. Nicotinamide Offers Multiple Protective Mechanisms in Stroke as a Precursor for NAD+, as a PARP Inhibitor and by Partial Restoration of Mitochondrial Function. Pharmacology 2003, 69, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Sadanaga-Akiyoshi, F.; Yao, H.; Tanuma, S.; Nakahara, T.; Hong, J.S.; Ibayashi, S.; Uchimura, H.; Fujishima, M. Nicotinamide Attenuates Focal Ischemic Brain Injury in Rats: With Special Reference to Changes in Nicotinamide and NAD+ Levels in Ischemic Core and Penumbra. Neurochem. Res. 2003, 28, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Wu, T.; Chang, G.; Li, C.; Chen, T.; Chen, H.; Maynard, K.I. Delayed Treatment with Nicotinamide Inhibits Brain Energy Depletion, Improves Cerebral Microperfusion, Reduces Brain Infarct Volume, but Does Not Alter Neurobehavioral Outcome Following Permanent Focal Cerebral Ischemia in Sprague Dawley Rats. Curr. Nurovasc. Res. 2006, 3, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Klaidman, L.K.; Ling, M.; Kem, S.; Sugawara, T.; Chan, P.; Adams, J.D. Nicotinamide Therapy Protects against Both Necrosis and Apoptosis in a Stroke Model. Pharmacol. Biochem. Behav. 2002, 73, 901–910. [Google Scholar] [CrossRef]
- Yang, J.; Klaidman, L.K.; Nalbandian, A.; Oliver, J.; Chang, M.L.; Chan, P.H.; Adams, J.D. The effects of nicotinamide on energy metabolism following transient focal cerebral ischemia in Wistar rats. Neurosci. Lett. 2002, 333, 91–94. [Google Scholar] [CrossRef]
- Hoane, M.R.; Akstulewicz, S.L.; Toppen, J. Treatment with Vitamin B 3 Improves Functional Recovery and Reduces GFAP Expression Following Traumatic Brain Injury in Rats. J. Neurotrauma 2003, 20, 1189–1199. [Google Scholar] [CrossRef]
- Shear, D.A.; Dixon, C.E.; Bramlett, H.M.; Mondello, S.; Dietrich, W.D.; Deng-Bryant, Y.; Schmid, K.E.; Wang, K.K.W.; Hayes, R.L.; Povlishock, J.T.; et al. Nicotinamide Treatment in Traumatic Brain Injury: Operation Brain Trauma Therapy. J. Neurotrauma 2015, 33, 523–537. [Google Scholar] [CrossRef]
- Hoane, M.R.; Gilbert, D.R.; Holland, M.A.; Pierce, J.L. Nicotinamide Reduces Acute Cortical Neuronal Death and Edema in the Traumatically Injured Brain. Neurosci. Lett. 2006, 408, 35–39. [Google Scholar] [CrossRef]
- Goffus, A.M.; Anderson, G.D.; Hoane, M.R. Sustained Delivery of Nicotinamide Limits Cortical Injury and Improves Functional Recovery Following Traumatic Brain Injury. Oxidative Med. Cell. Longev. 2010, 3, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Haar, C.V.; Anderson, G.D.; Hoane, M.R. Continuous Nicotinamide Administration Improves Behavioral Recovery and Reduces Lesion Size Following Bilateral Frontal Controlled Cortical Impact Injury. Behav. Brain Res. 2011, 224, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Klaidman, L.K.; Mukherjee, S.K.; Adams, J.D., Jr. Oxidative Changes in Brain Pyridine Nucleotides and Neuroprotection Using Nicotinamide. Biochim. Biophys. Acta 2001, 1525, 136–148. [Google Scholar] [CrossRef]
- Kamat, J.P.; Devasagayam, T.P.A. Nicotinamide (Vitamin B 3) as an Effective Antioxidant against Oxidative Damage in Rat Brain Mitochondria. Redox Rep. 1999, 4, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shetty, P.K.; Galef, F.; Turner, D.A. Neurobiology of Disease Nicotinamide Pre-Treatment Ameliorates NAD (H) Hyperoxidation and Improves Neuronal Function after Severe Hypoxia. Neurobiol. Dis. 2014, 62, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, E.; Okuda, H.; Nishida, K.; Fujimoto, S.; Nagasawa, K. Protective Effect of Nicotinamide against Poly (ADP-Ribose) Polymerase-1-Mediated Astrocyte Death Depends on Its Transporter-Mediated Uptake. Life Sci. 2010, 86, 676–682. [Google Scholar] [CrossRef]
- Ungerstedt, J.S.; Blombäck, M.; Södeström, T. Nicotinamide Is a Potent Inhibitor of Proinflammatory Cytokines. Clin. Exp. Immunol. 2003, 6, 48–52. [Google Scholar] [CrossRef]
- Vaur, P.; Brugg, B.; Mericskay, M.; Li, Z.; Schmidt, M.S.; Vivien, D.; Orset, C.; Jacotot, E.; Brenner, C.; Duplus, E. Nicotinamide Riboside, a Form of Vitamin B3, Protects against Excitotoxicity-Induced Axonal Degeneration. FASEB J. 2017, 31, 5440–5452. [Google Scholar] [CrossRef] [Green Version]
- Gasperi, V.; Sibilano, M.; Savini, I.; Catani, M.V. Niacin in the Central Nervous System: An Update of Biological Aspects and Clinical Applications. Int. J. Mol. Sci. 2019, 20, 974. [Google Scholar] [CrossRef] [Green Version]
- Hathorn, T.; Synder-Keller, A.; Messer, A. Nicotinamide Improves Motor Deficits and Upregulates PGC-1α and BDNF Gene Expression in a Mouse Model of Huntington’s Disease. Neurobiol. Disord. 2011, 41, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, T.; Kaetsu, A.; Lim, H.; Moriyama, M. Possible Role of 1-Methylnicotinamide in the Pathogenesis of Parkinson’s Disease. Exp. Toxic Pathol. 2001, 43, 469–473. [Google Scholar] [CrossRef]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide Riboside Restores Cognition through an Upregulation of Proliferator-Activated Receptor-g Coactivator 1 a Regulated b -Secretase 1 Degradation and Mitochondrial Gene Expression in Alzheimer’s Mouse Models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef] [Green Version]
- Fricker, R.A.; Green, E.L.; Jenkins, S.I.; Griffin, S.M. The Influence of Nicotinamide on Health and Disease in the Central Nervous System. Int. J. Tryptophan. Res. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakade, C.; Chong, R.; Bradley, E.; Morgan, J.C. Low-Dose Niacin Supplementation Modulates GPR109A, Niacin Index and Ameliorates Parkinson’s Disease Symptoms without Side Effects. Clin. Case Rep. 2015, 3, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Loh, S.H.; Martins, L.M. Enhancing NAD+ salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biol. Open 2017, 6, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aaseth, J.; Dusek, P.; Roos, P.M. Prevention of progression in Parkinson’s disease. BioMetals 2018, 31, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Tan, Z.; Tran, N.D. Chemical Hypoxia-Ischemia Induces Apoptosis in Cerebromicrovascular Endothelial Cells. Brain Res. 2000, 877, 134–140. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Lin, S.H.; Maiese, K. Nicotinamide modulates membrane potential and cysteine protease activity during cerebral vascular endothelial cell injury. J. Vasc. Res. 2002, 39, 131–147. [Google Scholar] [CrossRef]
- Niu, N.; Yu, Y.H.; Wang, Y.; Wang, L.J.; Li, Q.; Guo, L.M. Combined Effects of Niacin and Chromium Treatment on Vascular Endothelial Dysfunction in Hyperlipidemic Rats. Mol. Biol. Rep. 2009, 36, 1275–1281. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Martinez-revollar, G.; Hu, A.; Choi, J. Decline in Sirtuin-1 Expression and Activity Plays a Critical Role in Blood-Brain Barrier Permeability in Aging Neurobiology of Disease. Neurobiol. Dis. 2018, 126, 105–116. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, A.; Wang, Y. SIRT1 in Endothelial Cells as a Novel Target for the Prevention of Early Vascular Aging. J. Cardiovasc. Pharmacol. 2016, 67, 465–473. [Google Scholar] [CrossRef]
- Xu, J.; Jackson, C.W.; Khoury, N.; Escobar, I.; Perez-pinzon, M.A. Brain SIRT1 Mediates Metabolic Homeostasis and Neuroprotection. Front. Endocrinol. 2018, 9, 702. [Google Scholar] [CrossRef]
- De Picciotto, N.E.; Gano, L.B.; Johnson, L.C.; Martens, C.R.; Sindler, A.L.; Mills, K.F.; Imai, S.; Seals, D.R. Nicotinamide Mononucleotide Supplementation Reverses Vascular Dysfunction and Oxidative Stress with Aging in Mice. Aging Cell 2016, 15, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Sawda, C.; Moussa, C.; Turner, R.S. Resveratrol for Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2017, 1403, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Chung, J.W. Associations of Dietary Riboflavin, Niacin, and Retinol with Age-related Hearing Loss: An Analysis of Korean National Health and Nutrition Examination Survey Data. Nutrients 2019, 11, 896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayrakdar, E.T.; Uyanikgil, Y.; Kanit, L.; Koylu, E.; Yalcin, A. Nicotinamide Treatment Reduces the Levels of Oxidative Stress, Apoptosis, and PARP-1 Activity in Aβ (1–42)-Induced Rat Model of Alzheimer’s Disease. Free Rad. Res. 2014, 48, 146–158. [Google Scholar] [CrossRef]
- Benavente, C.A.; Jacobson, E.L. Niacin Restriction Upregulates NADPH Oxidase and ROS in Human Keratinocytes. Free Radic. Biol. Med. 2009, 44, 527–537. [Google Scholar] [CrossRef] [Green Version]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef] [Green Version]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in Vascular Pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Gao, Y.; Zeng, M.; Wang, Y.; Wei, T.F.; Lu, Y.B.; Zhang, W.P. Nicotinamide Ribose Ameliorates Cognitive Impairment of Aged and Alzheimer’s Disease Model Mice. Metab. Brain Dis. 2019, 34, 353–366. [Google Scholar] [CrossRef]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD+ Supplementation Normalizes Key Alzheimer’s Features and DNA Damage Responses in a New AD Mouse Model with Introduced DNA Repair Deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Scherr, P.A.; Tangney, C.C.; Hebert, L.E.; Bennett, D.A.; Wilson, R.S.; Aggarwal, N. Dietary Niacin and the Risk of Incident Alzheimer’s Disease and of Cognitive Decline. J. Neurol. Neurosur. Psychiatry 2004, 75, 1093–1100. [Google Scholar] [CrossRef] [Green Version]
- Csiszar, A.; Tarantini, S.S.; Yabluchanskiy, A.; Balasubramanian, P.; Kiss, T.; Farkas, E.; Baur, J.A.; Ungvari, Z. Role of endothelial NAD+ deficiency in age-related vascular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1253–H1266. [Google Scholar] [CrossRef] [PubMed]
- Venco, P.; Dusi, S.; Valletta, L.; Tiranti, V. Alteration of the Coenzyme a Biosynthetic Pathway in Neurodegeneration with Brain Iron Accumulation Syndromes. Biochem. Soc. Trans. 2014, 42, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Kunugi, H.; Ali, A.M. Royal Jelly and Its Components Promote Healthy Aging and Longevity: From Animal Models to Humans. Int. J. Mol. Sci. 2019, 20, 4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patassini, S.; Begley, P.; Xu, J.; Church, S.J.; Kureishy, N.; Reid, S.J.; Waldvogel, H.J.; Faull, R.L.M.; Snell, R.G.; Unwin, R.D.; et al. Cerebral Vitamin B5 (D-Pantothenic Acid) Deficiency as a Potential Cause of Metabolic Perturbation and Neurodegeneration in Huntington’s Disease. Metabolites 2019, 9, 113. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, R.; Zhang, Y.; Rock, C.O.; Jackowski, S. Progress in Lipid Research Coenzyme A: Back in Action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef]
- Slyshenkov, V.S.; Dymkowska, D.; Wojtczak, L. Pantothenic Acid and Pantothenol Increase Biosynthesis of Glutathione by Boosting Cell Energetics. FEBS Lett. 2004, 569, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Shedid, S.M.; Saada, H.N.; Eltahawy, N.A.; Hammad, A.S. Curative Role of Pantothenic Acid in Brain Damage of Gamma Irradiated Rats. Indian J. Clin. Biochem. 2018, 33, 314–321. [Google Scholar] [CrossRef]
- Wojtczak, L.; Slyshenkov, V.S. Protection by Pantothenic Acid against Apoptosis and Cell Damage by Oxygen Free Radicals—The Role of Glutathione. Biofactors 2003, 17, 61–73. [Google Scholar] [CrossRef]
- Jung, S.; Kim, M.K.; Choi, B.Y. The Long-Term Relationship between Dietary Pantothenic Acid (Vitamin B 5) Intake and C-Reactive Protein Concentration in Adults Aged 40 Years and Older. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 806–816. [Google Scholar] [CrossRef]
- Naruta, E.; Buko, V. Hypolipidemic Effect of Pantothenic Acid Derivatives in Mice with Hypothalamic Obesity Induced by Aurothioglucose. Exp. Toxic Pathol. 2001, 53, 393–398. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.; Rumberger, J.A.; Azumano, I.; Napolitano, J.J.; Citrolo, D.; Kamiya, T. Pantethine, a Derivative of Vitamin B5, Favorably Alters Total, LDL and Non-HDL Cholesterol in Low to Moderate Cardiovascular Risk Subjects Eligible for Statin Therapy: A Triple-Blinded Placebo and Diet-Controlled Investigation. Vasc. Health Risk Manag. 2014, 10, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcrae, M.P. Treatment of Hyperlipoproteinemia with Pantethine: A Review and Analysis of Efficacy and Tolerability. Nutr. Res. 2005, 25, 319–333. [Google Scholar] [CrossRef]
- Lanska, D.J. The Discovery of Niacin, Biotin, and Pantothenic Acid. Ann. Nutr. Metab. 2012, 61, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Ito, K.; Ohtsuki, S.; Kubo, Y.; Suzuki, T.; Terasaki, T. Major Involvement of Na+ -Dependent Multivitamin Transporter (SLC5A6/SMVT) in Uptake of Biotin and Pantothenic Acid by Human Brain Capillary Endothelial Cells. J. Neurochem. 2015, 134, 97–112. [Google Scholar] [CrossRef]
- Spector, R. Development and Characterization of Pantothenic Acid Transport in Brain. J. Neurochem. 1986, 47, 563–568. [Google Scholar] [CrossRef]
- Spector, R.; Boose, B. Accumulation of Pantothenic Acid by the Isolated Choroid Plexus and Brain Slices In Vitro. J. Neurochem. 1984, 43, 472–478. [Google Scholar] [CrossRef]
- Spector, R. Pantothenic Acid Transport and Metabolism in the Central Nervous System. Am. J. Physiol. 1986, 250, 292–297. [Google Scholar] [CrossRef]
- Spector, R.; Sivesind, C.; Kinzenbaw, D. Pantothenic Acid Transport Through the Blood-Barrier. J. Neurochem. 1986, 47, 966–971. [Google Scholar] [CrossRef]
- Rajalakshmi, R.; Nakhasi, H.L. Effects of neonatal pantothenic acid deficiency on brain lipid composition in rats. J. Neurochem. 1975, 24, 979–981. [Google Scholar] [CrossRef]
- Elbaum, D.; Beconi, M.G.; Monteagudo, E.; Di Marco, A.; Quinton, S.; Lyons, K.A.; Vaino, A.; Harper, S. Fosmetpantotenate (RE-024), a Phosphopantothenate Replacement Therapy for Pantothenate Kinase-Associated Neurodegeneration: Mechanism of Action and Efficacy in Nonclinical Models. PLoS ONE 2018, 13, e0192028. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, S.J. Defective Pantothenate Metabolism and Neurodegenration. Biochem. Soc. Trans. 2018, 42, 1063–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Meo, I.; Carecchio, M.; Tiranti, V. Inborn Errors of Coenzyme a Metabolism and Neurodegeneration. J. Inherit. Metab. Dis. 2019, 42, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zizioli, D.; Tiso, N.; Guglielmi, A.; Saraceno, C.; Busolin, G.; Giuliani, R.; Khatri, D.; Monti, E.; Borsani, G.; Argenton, F.; et al. Neurobiology of Disease Knock-down of Pantothenate Kinase 2 Severely Affects the Development of the Nervous and Vascular System in Zebra Fish, Providing New Insights into PKAN Disease. Neurobiol. Dis. 2016, 85, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ahn, S.; Lee, H.A.; Won, K.S.; Chang, H.W.; Oh, J.S.; Kim, H.W. Dietary Intake of Pantothenic Acid Is Associated with Cerebral Amyloid Burden in Patients with Cognitive Impairment. Food Nutr. Res. 2018, 62. [Google Scholar] [CrossRef]
- Leonardi, R.; Jackowski, S. Biosynthesis of Pantothenic Acid and Coenzyme A. EcoSal Plus 2007, 2. [Google Scholar] [CrossRef] [Green Version]
- Nakahiro, M.; Mochizuki, D.; Uchida, S.; Yoshida, H. Effect of the ‘antidementia drug’ pantoyl-GABA on high affinity transport of choline and on the contents of choline and acetylcholine in rat brain. Br. J. Pharmacol. 1988, 95, 1303–1307. [Google Scholar] [CrossRef] [Green Version]
- Stover, P.J.; Fields, M.S. Vitamin B-6. Adv. Nutr. 2015, 6, 132–133. [Google Scholar] [CrossRef]
- Ueland, P.M.; Mccann, A.; Midttun, Ø.; Ulvik, A. Inflammation, Vitamin B6 and Related Pathway. Mol. Asp. Med. 2016, 53, 20–27. [Google Scholar] [CrossRef]
- Lotto, V.; Choi, S.; Friso, S. Vitamin B6: A Challenging Link between Nutrition and Inflammation in CVD. Br. J. Nutr. 2011, 106, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Meydani, N.S.; Ribaya-mercado, J.; Russel, M.R.; Sahyoun, N.; Morrow, D.F.; Gershoff, N.S. Vitamin B-6 Deficiency Impairs Interleukin 2 Production and Lymphocyte Proliferation in Elderly Adults. Am. J. Clin. Nutr. 1991, 53, 1275–1280. [Google Scholar] [CrossRef]
- Brown, M.; Beier, K. Vitamin B6 Deficiency (Pyridoxine); StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Sudduth, T.L.; Powell, D.K.; Smith, C.D.; Greenstein, A.; Wilcock, D.M. Induction of Hyperhomocysteinemia Models Vascular Dementia by Induction of Cerebral Microhemorrhages and Neuroinflammation. J. Cereb. Blood Flow Metab. 2013, 33, 708–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungert, A.; Neuhauser-Berthold, M. Longitudinal Analysis on Determinants of Vitamin B6 Status in Community-Dwelling Older Adults over a Period of 18 Years. J. Gerontol. A Biol. Sci. Med. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.S.; Picciano, M.F.; Jacques, P.F.; Selhub, J. Plasma Pyridoxal 5′-Phosphate in the US Population: The National Health and Nutrition Examination Survey, 2003–2004. Am. J. Clin. Nutr. 2008, 87, 1446–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullegaddi, R.; Powers, H.J.; Gariballa, S.E. B-Group Vitamin Supplementation Mitigates Oxidative Damage after Acute Ischaemic Stroke. Clin. Sci. 2004, 484, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Shen, L. Associations between B Vitamins and Parkinson’s Disease. Nutrients 2015, 7, 7197–7208. [Google Scholar] [CrossRef]
- Friso, S.; Jacques, P.F.; Wilson, P.W.F.; Rosenberg, I.H.; Selhub, J. Clinical Investigation and Reports Low Circulating Vitamin B 6 Is Associated with Elevation of the Inflammation Marker C-Reactive Protein Independently of Plasma Homocysteine Levels. Circulation 2015, 103, 2788–2791. [Google Scholar] [CrossRef] [Green Version]
- Höhn, A.; Weber, D.; Jung, T.; Ott, C.; Hugo, M.; Kochlik, B.; Kehm, R.; König, J.; Grune, T.; Castro, J.P. Happily (n)ever after: Aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. 2017, 11, 482–501. [Google Scholar] [CrossRef]
- Potter, M.C.; Wozniak, K.M.; Callizot, N.; Slusher, B.S. Glutamate carboxypeptidase II inhibition behaviorally and physiologically improves pyridoxine-induced neuropathy in rats. PLoS One. 2014, 9, e102936. [Google Scholar] [CrossRef]
- Wilson, M.P.; Plecko, B.; Mills, P.B.; Clayton, P.T.; Medicine, G. Disorders Affecting Vitamin B 6 Metabolism. J. Inherit. Metab. Dis. 2019, 42, 629–646. [Google Scholar] [CrossRef] [Green Version]
- Barichello, T.; Generoso, J.S.; Simões, L.R.; Ceretta, R.A.; Dominguini, D.; Ferrari, P.; Gubert, C.; Jornada, L.K.; Budni, J.; Kapczinski, F.; et al. Vitamin B6 Prevents Cognitive Impairment in Experimental Pneumococcal Meningitis. Exp. Biol. Med. 2014, 239, 1360–1365. [Google Scholar] [CrossRef]
- Zysset-Burri, D.C.; Bellac, C.L.; Leib, S.L.; Wittwer, M. Vitamin B6 Reduces Hippocampal Apoptosis in Experimental Pneumococcal Meningitis. BMC Infect. Dis. 2013, 13, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, K.; Miyake, Y.; Sasaki, S.; Tanaka, K.; Fukushima, W.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; et al. Dietary Intake of Folate, Vitamin B6, Vitamin B12 and Riboflavin and Risk of Parkinson’s Disease: A Case-Control Study in Japan. Br. J. Nutr. 2010, 104, 757–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lau, L.M.L.; Koudstaal, P.J.; Witteman, J.C.M.; Hofman, A. Dietary Folate, Vitamin B 12, and Vitamin B 6 and the Risk of Parkinson Disease. Neurology 2006, 67, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Hunt, B.J.; O’Sullivan, M.; Bell, R.; D’Souza, R.; Jeffery, S.; Bamford, J.M.; Markus, H.S. Homocysteine Is a Risk Factor for Cerebral Small Vessel Disease, Acting via Endothelial Dysfunction. Brain 2004, 127, 212–219. [Google Scholar] [CrossRef] [Green Version]
- Bertsch, T.; Mielke, O.; Höly, S.; Casarin, W.; Aufenanger, J.; Walter, S.; Muehlhauser, F.; Kuehl, S.; Ragoschke, A.; Fassbender, K. Homocysteine in Cerebrovascular Disease: An Independent Risk Factor for Subcortical Vascular Encephalopathy. Clin. Chem. 2001, 39, 721–724. [Google Scholar] [CrossRef] [Green Version]
- Nuru, M.; Muradashvili, N.; Kalani, A.; Lominadze, D.; Tyagi, N. High Methionine, Low Folate and Low Vitamin B6/B12 (HM-LF-LV) Diet Causes Neurodegeneration and Subsequent Short-Term Memory Loss. Metab. Brain Dis. 2018, 33, 1923–1924. [Google Scholar] [CrossRef]
- Moretti, R.; Caruso, P. The Controversial Role of Homocysteine in Neurology: From Labs to Clinical Practice. Int. J. Mol. Sci. 2019, 20, 231. [Google Scholar] [CrossRef] [Green Version]
- Ansari, R.; Mahta, A.; Mallack, E.; Luo, J. Hyperhomocysteinemia and Neurologic Disorders: A Review. J. Clin. Neurol. 2014, 10, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Toda, N. Hyperhomocysteinemia Impairs Regional Blood Flow: Involvements of Endothelial and Neuronal Nitric Oxide. Pflügers Arch. 2016, 468, 1517–1525. [Google Scholar] [CrossRef]
- Endo, N.; Nishiyama, K.; Okabe, M.; Matsumoto, M.; Kanouchi, H.; Oka, T. Vitamin B 6 Suppresses Apoptosis of NM-1 Bovine Endothelial Cells Induced by Homocysteine and Copper. Biochim. Biophys. Acta 2007, 1770, 571–577. [Google Scholar] [CrossRef]
- Alam, M.M.; Mohammad, A.A.; Shuaib, U.; Wang, C.; Ghani, U.; Schwindt, B.; Todd, K.G.; Shuaib, A. Homocysteine Reduces Endothelial Progenitor Cells in Stroke Patients through Apoptosis. J. Cereb. Blood Flow Metab. 2009, 29, 157–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahfouz, M.M.; Zhou, S.Q.; Kummerow, F.A. Vitamin B 6 Compounds Are Capable of Reducing the Superoxide Radical and Lipid Peroxide Levels Induced by H2O2 in Vascular Endothelial Cells in Culture. Int. J. Vitam. Nutr. Res. 2009, 79, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Liu, Z.; Lu, H.; Zhang, W.; Mi, Q. Pyridoxine Inhibits Endothelial NOS Uncoupling Induced by Oxidized Low-Density Lipoprotein via the PKC Signaling Pathway in Human Umbilical Vein Endothelial Cells. Br. J. Pharmacol. 2012, 165, 754–764. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Diao, J.; Han, Y.; Huang, Y.; Bai, H.; Chen, Q.; Fan, L.; Ferro, A. Pyridoxine Prevents Dysfunction of Endothelial Cell Nitric Oxide Production in Response to Low-Density Lipoprotein. Atherosclerosis 2006, 188, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Ford, T.C.; Downey, L.A.; Simpson, T.; Mcphee, G.; Oliver, C.; Stough, C. The Effect of a High-Dose Vitamin B Multivitamin Supplement on the Relationship between Brain Metabolism and Blood Biomarkers of Oxidative Stress: A Randomized Control Trial. Nutrients 2018, 10, 1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onorato, J.M.; Jenkins, A.J.; Thorpe, S.R.; Baynes, J.W. Pyridoxamine, an Inhibitor of Advanced Glycation Reactions, Also Inhibits Advanced Lipoxidation Reactions. J. Biol. Chem. 2000, 275, 21177–21184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelso, B.G.; Brower, J.B.; Targovnik, J.H.; Caplan, M.R. Pyridoxine Restores Endothelial Cell Function in High Glucose. Metab. Syndr. Relat. Disord. 2011, 9, 63–68. [Google Scholar] [CrossRef]
- Morris, M.S.; Sakakeeny, L.; Jacques, P.F.; Picciano, M.F.; Selhub, J. Vitamin B-6 Intake Is Inversely Related to, and the Requirement Is Affected by Inflammation Status. J. Nutr. 2009, 140, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Ifrea, R.A.T.; Ozlea, L.C.; Arașca, E.C. The Impact of C Reactive Protein on Global Cardiovascular Risk on Patients with Coronary Artery Disease. Curr. Health Sci. J. 2013, 39, 225–231. [Google Scholar]
- Rall, L.C.; Meydani, S.N. Vitamin B, and Immune Competence. Nutr. Rev. 1993, 51, 217–225. [Google Scholar] [CrossRef]
- Kelly, P.J.; Shih, V.E.; Kistler, J.P.; Barron, M.; Lee, H.; Mandell, R.; Furie, K.L. Low Vitamin B6 but Not Homocyst(e)ine Is Associated with Increased Risk of Stroke and Transient Ischemic Attack in the Era of Folic Acid Grain Fortification. Stroke 2003, 34, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Polyak, Z.; Berner, Y.N.; Sela, B.; Gomori, J.M.; Doolman, R. Hyperhomocysteinemia and Vitamin Score: Correlations with Silent Brain Ischemic Lesions and Brain Atrophy. Dement. Geriatr. Cogn. Disord. 2003, 16, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Mulder, C.; Scheltens, P.; Barkhof, F.; Gundy, C.; Verstraeten, R.A.; de Leeuw, F.E. Low B Vitamin B6 Levels Are Associated with White Matter Lesions in Alzheimer’s Disease. J. Am. Geriatr. Soc. 2005, 53, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Mulder, C.; van der Flier, W.M.; Veerhuis, R.; Bouwman, F.; Jakobs, C.; Verhoeven, N.M.; Barkhof, F.; Scheltens, P.; Blankenstein, M.A. Association Between Vitamin B6 and White Matter Hyperintensities in Patients with Alzheimer’s Disease Not Mediated by Homocysteine Metabolism. J. Am. Geriatr. Soc. 2007, 55, 956–958. [Google Scholar] [CrossRef]
- Miller, J.W.; Green, R.; Mungas, D.M.; Reed, B.R.; Jagust, W.J. Homocysteine, Vitamin B 6, and Vascular Disease in AD Patients. Neurology 2002, 58, 1471–1476. [Google Scholar] [CrossRef]
- Malaguarnera, M.; Ferri, R.; Alagona, G.; Carnemolla, A.; Pennisi, G. Homocysteine, Vitamin B 12 and Folate in Vascular Dementia and in Alzheimer Disease. Clin. Chem. Lab. Med. 2004, 42, 1032–1035. [Google Scholar] [CrossRef]
- Nelson, C.; Wengreen, H.J.; Munger, R.G.; Corcoran, C.D. Dietary folate, vitamin B-12, vitamin B-6 and incident Alzheimer’s disease: The cache county memory, health, and aging study. J. Nutr. Health Ageing 2009, 13, 899–905. [Google Scholar] [CrossRef]
- Toole, J.F.; Malinow, M.R.; Chambless, L.E.; Spence, J.D.; Pettigrew, L.C.; Howard, V.J.; Sides, E.G.; Wang, C.H.; Stampfer, M. Lowering Homocysteine in Patients with Ischemic Stroke to Prevent Recurrent Stroke, Myocardial Infarction, and Death. The Vitamin Intervention for Stroke Prevention (VISP) Randomized Controlled Trial. JAMA 2004, 291, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Schwammenthal, Y.; Tanne, D. Homocysteine, B-Vitamin Supplementation, and Stroke Prevention: From Observational to Interventional Trials. Lancet Neurol. 2004, 3, 493–495. [Google Scholar] [CrossRef]
- The VITATOPS Trial Study Group. B Vitamins in Patients with Recent Transient Ischemic Attack or Stroke in the VITAmins TO Prevent Stroke (VITATOPS) Trial: A Randomized, Double-Blind, Parallel, Placebo-Controlled Trial. Lancet Neurol. 2010, 9, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Cavalieri, M.; Schmidt, R.; Chen, C.; Mok, V.; De Freitas, G.R.; Song, S.; Yi, Q.; Ropele, S.; Grazer, A.; Homayoon, N.; et al. B Vitamins and Magnetic Resonance Imaging—Detected Ischemic Brain Lesions in Patients with Recent Transient Ischemic Attack or Stroke. Stroke 2012, 43, 3266–3270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, I.K.; Suever, L.B.; Prakash, S.R.; Colcombe, S.J.; McAuley, E.; Kramer, A.F. Greater Intake of Vitamins B6 and B12 Spares Gray Matter in Healthy Elderly: A Voxel-Based Morphometry Study. Brain Res. 2009, 1553, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jannusch, K.; Jockwitz, C.; Bidmon, H.; Moebus, S.; Amunts, K.; Caspers, S. A Complex Interplay of Vitamin B1 and B6 Metabolism with Cognition, Brain Structure, and Functional Connectivity in Older Adults. Front. Neurosci. 2017, 11, 596. [Google Scholar] [CrossRef] [PubMed]
- Twisk, J.W.R.; Prevoo, W.; Rauwerda, J.A. Effect of Homocysteine-Lowering Treatment with Folic Acid plus Vitamin B 6 on Cerebrovascular Atherosclerosis and White Matter Abnormalities as Determined by MRA and MRI: A Placebo-Controlled, Randomized Trial. Eur. J. Clin. Investig. 2004, 34, 256–261. [Google Scholar]
- Palacios, N.; Scott, T.; Sahasrabudhe, N.; Gao, X.; Tucker, K.L. Lower Plasma Vitamin B-6 Is Associated with 2-Year Cognitive Decline in the Boston Puerto Rican Health Study. J. Nutr. 2019, 149, 635–641. [Google Scholar] [CrossRef]
- Riggs, M. Relations of Vitamin B-12, Homocysteine to Cognitive Aging Study1 Performance in the Normative. Am. J. Clin. Nutr. 1996, 63, 306–314. [Google Scholar] [CrossRef]
- Qin, B.; Xun, P.; Jacobs, D.R., Jr.; Zhu, N.; Daviglus, M.L.; Reis, J.P.; Steffen, L.M.; van Horn, L.; Sidney, S.; He, K. Intake of Niacin, Folate, Vitamin B-6, and Vitamin B-12 through Young Adulthood and Cognitive Function in Midlife: The Coronary Artery Risk Development in Young Adults (CARDIA) Study. Am. J. Clin. Nutr. 2017, 106, 1032–1040. [Google Scholar] [CrossRef] [Green Version]
- Tucker, K.L.; Qiao, N.; Scott, T.; Rosenberg, I.; Iii, A.S. High Homocysteine and Low B Vitamins Predict Cognitive Decline in Aging Men: The Veterans Affairs Normative Aging Study. Am. J. Clin. Nutr. 2005, 82, 627–635. [Google Scholar] [CrossRef]
- Aisen, P.S.; Schneider, L.S.; Sano, M.; Diaz-Arrastia, R.; van Dyck, C.H.; Weiner, M.F.; Bottiglieri, T.; Jin, S.; Stokes, K.T.; Thomas, R.G.; et al. High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: A randomized controlled trial. JAMA 2012, 300, 1774–1783. [Google Scholar] [CrossRef]
- Sun, Y.; Lu, C.J.; Chien, K.L.; Chen, S.T.; Chen, R.C. Efficacy of Multivitamin Supplementation Containing Vitamins B6 and B12 and Folic Acid as Adjunctive Treatment with a Cholinesterase Inhibitor in Alzheimer’s Disease: A 26-Week, Randomized, Double-Blind, Placebo-Controlled Study in Taiwanese Patients. Clin. Ther. 2007, 29. [Google Scholar] [CrossRef]
- Stott, D.J.; MacIntosh, G.; Lowe, G.D.; Rumley, A.; McMahon, A.D.; Langhorne, P.; Tait, R.C.; O’Reilly, D.S.J.; Spilg, E.G.; MacDonald, J.B.; et al. Randomized Controlled Trial of Homocysteine-Lowering Vitamin Treatment in Elderly Patients with Vascular Disease. Am. J. Clin. Nutr. 2005, 82, 1320–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Ye, J.; Mu, J. Efficacy of Vitamin B Supplementation on Cognition in Elderly Patients with Cognitive-Related Diseases: A Systematic Review and Meta-Analysis. J. Geriatr. Psychiatry 2017, 30, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Bryan, J.; Calvaresi, E.; Hughes, D. Short-Term Folate, Vitamin B-12 or Vitamin B-6 Supplementation Slightly Affects Memory Performance but Not Mood in Women of Various Ages. J. Nutr. 2002, 132, 1345–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deijen, J.B.; van der Beck, E.J.; Orlebeke, J.F.; van der Berg, H. Vitamin B-6 Supplementation in Elderly Men: Effects on Mood, Memory, Performance and Mental Effort. Psychopharmacology 1992, 109, 489–496. [Google Scholar] [CrossRef]
- Malouf, R.; Evans, J.G. The Effect of Vitamin B6 on Cognition. Cochrane Database Syst. Rev. 2003, 4, CD004393. [Google Scholar] [CrossRef]
- Hassel, B.; Rogne, A.G.; Hope, S. Intellectual Disability Associated with Pyridoxine-Responsive Epilepsies: The Need to Protect Cognitive Development. Front. Psychiatry 2019, 10, 116. [Google Scholar] [CrossRef]
- Tourbah, A.; Lebrun-Frenay, C.; Edan, G.; Clanet, M.; Papeix, C.; Vukusic, S.; De Sèze, J.; Debouverie, M.; Gout, O.; Clavelou, P.; et al. MD1003 (High-Dose Biotin) for the Treatment of Progressive Multiple Sclerosis: A Randomised, Double-Blind, Placebo-Controlled Study. Mult. Scler. J. 2016, 22, 1719–1731. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, K.; Stojanovska, L.; Apostolopoulos, V. The Effects of Vitamin B in Depression. Curr. Med. Chem. 2016, 23, 4317–4337. [Google Scholar] [CrossRef] [Green Version]
- Zempleni, J.; Wijeratne, S.S.K.; Hassan, Y.I. Biotin. Biofactors 2016, 35, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Dakshinamurti, K. Biotin—A Regulator of Gene Expression B. J. Nutr. Biochem. 2005, 16, 419–423. [Google Scholar] [CrossRef]
- Zempleni, J. Uptake, Localization and Noncarboxylase Roles of Biotin. Annu. Rev. Nutr. 2005, 25, 175–196. [Google Scholar] [CrossRef] [PubMed]
- Mccarty, M.F.; Dinicolantonio, J.J. Neuroprotective Potential of High-Dose Biotin. Med. Hypotheses 2017, 109, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Magied, N.; Shedid, S.M.; Ahmed, A.G. Mitigating Effect of Biotin against Irradiation-Induced Cerebral Cortical and Hippocampal Damage in the Rat Brain Tissue. Environ. Sci. Pollut. Res. 2019, 26, 13441–13452. [Google Scholar] [CrossRef] [PubMed]
- Madsen, C.T.; Sylvestersen, K.B.; Young, C.; Larsen, S.C.; Poulsen, J.W.; Andersen, M.A.; Palmqvist, E.A.; Hey-Mogensen, M.; Jensen, P.B.; Treebak, J.T.; et al. Biotin starvation causes mitochondrial protein hyperacetylation and partial rescue by the SIRT3-like deacetylase Hst4p. Nat. Commun. 2015, 6, 7726. [Google Scholar] [CrossRef] [Green Version]
- Watanabe-Kamiyama, M.; Kamiyama, S.; Horiuchi, K.; Ohinata, K.; Shirakawa, H.; Furukawa, Y.; Komai, M. Antihypertensive Effect of Biotin in Stroke-Prone Spontaneously Hypertensive Rats. Br. J. Nutr. 2008, 99, 756–763. [Google Scholar] [CrossRef] [Green Version]
- Suormala, T.; Wiesmann, U.N.; Cruz, F.; Wolf, A.; Daschner, M.; Limat, A.; Fowler, B.; Baumgartner, E.R. Biotin-Dependent Carboxylase Activities in Different CNS and Skin-Derived Cells, and Their Sensitivity to Biotin-Depletion. Int. J. Vitam. Nutr. Res. 2002, 72, 278–286. [Google Scholar] [CrossRef]
- Fukuwatari, T.; Wada, H.; Shibata, K.; Ukuwatari, T.F.; Ada, H.W.; Hibata, K.S. Age-Related Alterations of B-Group Vitamin Contents in Urine, Blood and Liver from Rats. J. Nutr. Sci. Vitaminol. 2008, 54, 357–362. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Yasumura, S.; Shibata, H. Biotin Status and Its Correlation with Other Biochemical Parameters in the Elderly People of Japan. J. Am. Coll. Nutr. 1998, 17, 37–41. [Google Scholar] [CrossRef]
- McKay, B.E.; Molineux, M.L.; Turner, R.W. Biotin Is Endogenously Expressed in Select Regions of the Rat Central Nervous System. J. Comp. Neurol. 2004, 473, 86–96. [Google Scholar] [CrossRef]
- Spector, R.; Mock, D. Biotin Transport Through the Blood-Brain Barrier. J. Neurochem. 1987, 48, 400–404. [Google Scholar] [CrossRef]
- Lo, W.; Kaldleck, T.; Packman, S. Biotin Transport in the Rat Central Nervous System. J. Nutr. Sci. Vitaminol. 1991, 37, 567–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassem, H.; Wafaie, A.; Alsuhibani, S.; Farid, T. Biotin-Responsive Basal Ganglia Disease: Neuroimaging Features before and after Treatment. AJNR Am. J. Neuroradiol. 2014, 35, 1990–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yatzidis, H.; Kutsicos, D.; Agroyannis, B.; Papastephanidis, C.; Francos-Plemenos, M.; Delatola, Z. Biotin in the Managment of Uremic Neurologic Disorders. Nephron 1984, 36, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Koutsikos, D.; Agroyannis, B.; Tzanatos-Exarchou, H. Biotin for Diabetic Peripheral Neuropathy. Biomed. Pharmacother. 1990, 44, 511–514. [Google Scholar] [CrossRef]
- Birnbaum, G.; Stulc, J. High Dose Biotin as Treatment for Progressive Multiple Sclerosis. Mult. Scler. Relat. Disord. 2017, 18, 141–143. [Google Scholar] [CrossRef]
- Sedel, F.; Bernard, D.; Mock, D.M.; Tourbah, A. Targeting Demyelination and Virtual Hypoxia with High-Dose Biotin as a Treatment for Progressive Multiple Sclerosis. Neuropharmacology 2015, 110, 644–653. [Google Scholar] [CrossRef] [Green Version]
- Melichar, V.O.; Behr-Roussel, D.; Zabel, U.; Uttenthal, L.O.; Rodrigo, J.; Rupin, A.; Verbeuren, T.J.; Kumar, H.S.A.; Schmidt, H.H.H.W. Reduced cGMP Signaling Associated with Neointimal Proliferation and Vascular Dysfunction in Late-Stage Atherosclerosis. Proc. Natl. Acad. Sci. USA 2004, 101, 16671–16676. [Google Scholar] [CrossRef]
- Ahluwalia, A.; Foster, P.; Scotland, R.S.; Mclean, P.G.; Mathur, A.; Perretti, M.; Moncada, S.; Hobbs, A.J. Antiinflammatory Activity of Soluble Guanylate Cyclase: cGMP-Dependent Down-Regulation of P-Selectin Expression and Leukocyte Recruitment. Proc. Natl. Acad. Sci. USA 2004, 101, 1386–1391. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, Y.; Huang, Q.; Su, Y.; Zhang, Y.; Wang, G.; Li, F. NF-kB Activation and Cell Death after Intracerebral Hemorrhage in Patients. Neurol. Sci. 2014, 35, 1097–1102. [Google Scholar] [CrossRef]
- Gonos, E.S.; Kapetanou, M.; Sereikaite, J.; Bartosz, G.; Naparło, K.; Grzesik, M.; Sadowska-Bartosz, I. Origin and pathophysiology of protein carbonylation, nitration and chlorination in age-related brain diseases and aging. Aging 2018, 10, 868–901. [Google Scholar] [CrossRef]
- Moretti, R.; Dal Ben, M.; Gazzin, S.; Tiribelli, C. Homocysteine in Neurology: From Endothelium to Neurodegeneration. Curr. Nutr. Food Sci. 2017, 13, 163–175. [Google Scholar] [CrossRef]
- Barber, R.C.; Lammer, E.J.; Shaw, G.M.; Greer, K.A.; Finnell, R.H. The role of folate transport and metabolism in neural tube defect risk. Mol. Genet. Metab. 1999, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kamen, B.A.; Smith, A.K. A review of folate receptor alpha cycling and 5-methyltetrahydrofolate accumulation with an emphasis on cell models in vitro. Adv. Drug Deliv. Rev. 2004, 56, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, L. Folic acid and vitamin B12 deficiency. J. Health Res. Rev. Dev. Ctries. 2014, 1, 5–9. [Google Scholar] [CrossRef]
- Wang, X.; Qin, X.; Demirtas, H.; Li, J.; Mao, G.; Huo, Y.; Sun, N.; Liu, L.; Xu, X. Efficacy of folic acid supplementation in stroke prevention: A meta-analysis. Lancet 2007, 369, 1876–1882. [Google Scholar] [CrossRef]
- McNulty, H. Folate requirements for health in different population groups. Br. J. Biomed. Sci. 1995, 52, 110–119. [Google Scholar]
- Stolzenberg, R. Possible folate deficiency with postsurgical infection. Nutr. Clin. Pract. Off. Publ. Am. Soc. Parenter. Nutr. 1994, 9, 247–250. [Google Scholar] [CrossRef]
- Pietrzik, K.F.; Thorand, B. Folate economy in pregnancy. Nutrition 1997, 13, 975–977. [Google Scholar] [CrossRef]
- Hoffbrand, A.V.; Weir, D.G. The history of folic acid. Br. J. Haematol. 2001, 113, 579–589. [Google Scholar] [CrossRef]
- Folate Evidence—Mayo Clinic, N.D. Available online. Available online: http://www.mayoclinic.org/drugs-supplements/folate/evidence/hrb-20059475 (accessed on 15 August 2019).
- Bailey, L.B.; Stover, P.J.; McNulty, H.; Fenech, M.F.; Gregory, J.F.; Mills, J.L.; Pfeiffer, C.M.; Fazili, Z.; Zhang, M.; Ueland, P.M.; et al. Biomarkers of Nutrition for Development-Folate Review. J. Nutr. 2015, 145, 1636S–1680S. [Google Scholar] [CrossRef] [Green Version]
- Froese, D.S.; Fowler, B.; Baumgartner, M.R. Vitamin B12, folate and the methionine remethylation cycle-biochemistry, pathways and regulation. J. Inher. Metab. Disord. 2019, 42, 673–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Overbeek, E.C.; Staals, J.; van Oostenbrugge, R.J. Vitamin B12 and Progression of White Matter Lesions. A 2-Year Follow-Up Study in First-Ever Lacunar Stroke Patients. PLoS ONE 2013, 8, e78100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obeid, R.; Herrmann, W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006, 580, 2994–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson-Ehle, H. Age-related changes in cobalamin (vitamin B12) handling. Implications for therapy. Drugs Aging 1998, 12, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.M.; Allen, E.; Mills, K.; Clarke, R.; Uauy, R.; Dangour, A.D. Vitamin B-12 status and neurologic function in older people: A cross-sectional analysis of baseline trial data from the Older People and Enhanced Neurological Function (OPEN) study. Am. J. Clin. Nutr. 2016, 104, 790–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamparo, C.D.; Marcia, L.A. Diseases of the Human Body, 5th ed.; F. A. Davis Company: Philadelphia, PA, USA, 2011. [Google Scholar]
- Dowd, P.; Shapiro, M.; Kang, K. Letter: The mechanisms of action of vitamin B12. J. Am. Chem. Soc. 1975, 97, 4754–4757. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, E.H. Benefits and risks of folic acid to the nervous system. J. Neurol. Neurosurg. Psychiatry 2002, 72, 567–571. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Ford, E.S.; Li, C.; Greenlund, K.J.; Croft, J.B.; Balluz, L.S. Use of folic Acid and vitamin supplementation among adults with depression and anxiety: A cross-sectional, population-based survey. Nutr. J. 2011, 10, 102. [Google Scholar] [CrossRef] [Green Version]
- Moretti, R.; Torre, P.; Antonello, R.M.; Cazzato, G. Is isolated vitamin B12 deficiency a sufficient causative factor of dementia? Eur. J. Neurol. 2001, 8, 87–88. [Google Scholar] [CrossRef]
- Ubbink, J.B. Should all elderly people receive folate supplements? Drugs Aging 1998, 13, 415–420. [Google Scholar] [CrossRef]
- Mollin, D.L.; Ross, G.I. Serum vitamin B12 concentrations of patients with megaloblastic anemia after treatment with vitamin B12, folic acid, or folinic acid. Br. Med. J. 1953, 2, 640–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucock, M. Is folic acid the ultimate functional food component for disease prevention? BMJ 2004, 328, 211–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malnick, S.; Goland, S. Folic acid as ultimate in disease prevention: Beware of vitamin B12 deficiency. BMJ 2004, 328, 769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietary Supplement Fact Sheet: Vitamin B12—Health Professional Fact Sheet, N.D. Available online: https://ods.od.nih.gov/factsheets/VitaminB12-HealthProfessional/ (accessed on 15 August 2019).
- Elmadfa, I.; Singer, I. Vitamin B12 and homocysteine status among vegetarians: A global perspective. Am. J. Clin. Nut. 2009, 89, S1693–S1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Institute of Medicine. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Panthotenic Acid, Biotin, and Choline; National Academy Press: Washington, DC, USA, 1999. [Google Scholar]
- Meadows, M.E.; Kaplan, R.F.; Bromfield, E.B. Cognitive recovery with vitamin B12 therapy: A longitudinal neuropsychological assessment. Neurology 1994, 44, 1764–1765. [Google Scholar] [CrossRef] [PubMed]
- Eastley, R.; Wilcock, G.K.; Bucks, R.S. Vitamin B12 deficiency in dementia and cognitive impairment: The effects of treatment on neuropsychological function. Int. J. Geriatr. Psychiatry 2000, 15, 226–233. [Google Scholar] [CrossRef]
- Teunisse, S.; Bollen, A.E.; van Gool, W.A.; Walstra, G.J. Dementia and subnormal levels of vitamin B12: Effects of replacement therapy on dementia. J. Neurol. 1996, 243, 522–529. [Google Scholar] [CrossRef]
- Wahlin, T.B.R.; Wahlin, A.; Winblad, B.; Bäckman, L. The influence of serum vitamin B12 and folate status on cognitive functioning in very old age. Biol. Psychol. 2001, 56, 247–265. [Google Scholar] [CrossRef]
- Fioravanti, M.; Ferrario, E.; Massaia, M.; Cappa, G.; Rivolta, G.; Grossi, E.; Buckley, A.E. Low folate levels in the cognitive decline of elderly patients and the efficacy of folate as a treatment for improving memory deficits. Arch. Gerontol. Geriatr. 1998, 26, 1–13. [Google Scholar] [CrossRef]
- Hassing, L.; Wahlin, A.; Winblad, B.; Bäckman, L. Further evidence on the effects of vitamin B12 and folate levels on episodic memory functioning: A population-based study of healthy very old adults. Biol. Psychiatry 1999, 45, 1472–1480. [Google Scholar] [CrossRef]
- Eussen, S.J.P.M.; Ferry, M.; Hininger, I.; Haller, J.; Matthys, C.; Dirren, H. Five year changes in mental health and associations with vitamin B12/folate status of elderly Europeans. J. Nutr. Health Aging 2002, 6, 43–50. [Google Scholar] [PubMed]
- Nilsson, K.; Gustafson, L.; Hultberg, B. Improvement of cognitive functions after cobalamin/folate supplementation in elderly patients with dementia and elevated plasma homocysteine. Int. J. Geriatr. Psychiatry 2001, 16, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, E.H. Folic acid, ageing, depression, and dementia. BMJ 2002, 324, 1512–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blom, H.J. Folic acid, methylation and neural tube closure in humans. Birth Defects Res. A Clin. Mol. Teratol. 2009, 85, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Pitkin, R.M. Folate and neural tube defects. Am. J. Clin. Nutr. 2007, 85, 285S–288S. [Google Scholar] [CrossRef] [Green Version]
- Frosst, P.; Blom, H.J.; Milos, R.; Goyette, P.; Sheppard, C.A.; Matthews, R.G.; Boers, G.J.; den Heijer, M.; Kluijtmans, L.A.; van den Heuvel, L.P.; et al. A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 1995, 10, 111–113. [Google Scholar] [CrossRef]
- Bottiglieri, T.; Laundy, M.; Crellin, R.; Toone, B.K. Homocysteine, folate, methylation, and monoamine metabolism in depression. J. Neurol. Neurosurg. Psychiatry 2000, 69, 228–232. [Google Scholar] [CrossRef]
- Bottiglieri, T.; Crellin, R.; Reynolds, E.H. Folate and neuropsychiatry. In Folate Health Dis.; Marcel Dekker: New York, NY, USA, 1995; pp. 435–462. [Google Scholar]
- Botez, M.I.; Reynolds, E.H. Folic Acid in Neurology, Psychiatry and Internal Medicine; Raven Press: New York, NY, USA, 1979. [Google Scholar]
- Maxwell, C.J.; Hogan, D.B.; Ebly, E.M. Serum folate levels and subsequent adverse cerebrovascular outcomes in elderly persons. Dement. Geriatr. Cogn. Disord. 2002, 13, 225–234. [Google Scholar] [CrossRef]
- Snowdon, D.A.; Tully, C.L.; Smith, C.D.; Riley, K.P.; Markesbery, W.R. Serum folate and the severity of atrophy of the neocortex in Alzheimer disease: Findings from the Nun study. Am. J. Clin. Nutr. 2000, 71, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Stanhewicz, A.E.; Kenney, W.L. Role of folic acid in nitric oxide bioavailability and vascular endothelial function. Nutr. Rev. 2017, 75, 61–70. [Google Scholar] [CrossRef]
- Ma, F.; Wu, T.; Zhao, J.; Song, A.; Liu, H.; Xu, W.; Huang, G. Folic acid supplementation improves cognitive function by reducing the levels of peripheral inflammatory cytokines in elderly Chinese subjects with MCI. Sci Rep. 2016, 6, 37486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudd, S.H.; Cantoni, G.L. Activation of methionine for transmethylation. III. The methionine-activating enzyme of Bakers’ yeast. J. Biol. Chem. 1958, 231, 481–492. [Google Scholar] [PubMed]
- Mato, J.M.; Alvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280. [Google Scholar] [CrossRef] [Green Version]
- Blom, H.J.; Smulders, Y. Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J. Inherit. Metab. Dis. 2011, 34, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loscalzo, J.; Handy, D.E. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. 2013 Grover Conference Series. Pulm. Circ. 2014, 482, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Weir, D.G.; Keating, S.; Molloy, A.; McPartlin, J.; Kennedy, S.; Blachflower, J.; Kennedy, D.G.; Rice, D.; Scott, J.M. Methylation deficiency causes vitamin B12-associated neuropathy in the pig. J. Neurochem. 1988, 51, 1949–1952. [Google Scholar] [CrossRef]
- Harvey, R.A.; Ferrier, D.R. Biochemistry. In Lippincott’s Illustrated Reviews, 5th ed.; Rhyner, S., Ed.; Wolters Kluwer Health: Philadelphia, PA, USA, 2011; pp. 264–265. [Google Scholar]
- Khan, U.; Crossley, C.; Kalra, L.; Rudd, A.; Wolfe, C.D.; Collinson, P.; Markus, H.S. Homocysteine and its relationship to stroke subtypes in a UK black population: The South London Ethnicity and Stroke Study. Stroke 2008, 39, 2943–2949. [Google Scholar] [CrossRef] [Green Version]
- Selhub, J.; Bagley, L.C.; Miller, J.; Rosenberg, I.H. B vitamins, homocysteine, and neurocognitive function in the elderly. Am. J. Clin. Nutr. 2000, 71, 614S–620S. [Google Scholar] [CrossRef]
- Li, Z.; Sun, L.; Zhang, H.; Liao, Y.; Wang, D.; Zhao, B.; Zhu, Z.; Zhao, J.; Ma, A.; Han, Y.; et al. Elevated plasma homocysteine was associated with hemorrhagic and ischemic stroke, but methylenetetrahydrofolate reductase gene c677t polymorphism was a risk factor for thrombotic stroke a multicenter case-control study in China. Stroke 2003, 34, 2085–2090. [Google Scholar] [CrossRef] [Green Version]
- Ali, Z.; Troncoso, J.C.; Fowler, D.R. Recurrent cerebral venous thrombosis associated with heterozygote methylenetetrahydrofolate reductase C677T mutation and sickle cell trait without homocysteinemia: An autopsy case report and review of literature. Forensic Sci. Int. 2014, 242, e52–e55. [Google Scholar] [CrossRef]
- Que, X.; Hung, M.Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Määttä, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, K.A.; Sanders, L.M.; Fischer, L.M.; Zeisel, S.H. Docosahexaenoic acid in plasma phosphatidylcholine may be a potential marker for in vivo phosphatidylethanolamine N-methyltransferase activity in humans. Am. J. Clin. Nutr. 2011, 93, 968–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLong, C.J.; Shen, Y.J.; Thomas, M.J.; Cui, Z. Molecular distinction of phosphatidylcholine synthesis between the CDP-choline pathway and phosphatidylethanolamine methylation pathway. J. Biol. Chem. 1999, 274, 29683–29688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pynn, J.; Henderson, N.G.; Clark, H.; Koster, G.; Bernhard, W.; Postle, A.D. The specificity and rate of human and mouse liver and plasma phosphatidylcholine synthesis analysed in vivo. J. Lipid Res. 2011, 52, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blusztajn, J.K.; Zeisel, S.H.; Wurtman, R.J. Synthesis of lecithin (phosphatidylcholine) from phosphatidylethanolamine in bovine brain. Brain Res. 1979, 179, 319–327. [Google Scholar] [CrossRef]
- Blusztajn, J.K.; Holbrook, P.G.; Lakher, M.; Liscovitch, M.; Maire, J.-C.; Mauron, C.; Richardson, U.I.; Tacconi, M.T.; Wurtman, R.J. Relationships between acetylcholine release and membrane phosphatidylcholine turnover in brain and in cultured cholinergic neurons. In Phospholipids in the Nervous System: Biochemical and Molecular Pharmacology; Springer: Berlin, Germany, 1986; pp. 283–290. [Google Scholar]
- Vance, D.E. Phospholipid methylation in mammals: From biochemistry to physiological function. Biochim. Biophys. Acta 2014, 1838, 1477–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudd, S.H.; Brosnan, J.T.; Brosnan, M.E.; Jacobs, R.L.; Stabler, S.P.; Allen, R.H.; Vance, D.E.; Wagner, C. Methyl balance and transmethylation fluxes in humans. Am. J. Clin. Nutr. 2007, 85, 19–25. [Google Scholar] [CrossRef]
- Selley, M.L. A metabolic link between S-adenosylhomocysteine and polyunsaturated fatty acid metabolism in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1834–1839. [Google Scholar] [CrossRef]
- Clark, W.M.; Williams, B.J.; Selzer, K.A.; Zweifler, R.M.; Sabounjian, L.A.; Gammans, R.E. A randomized efficacy trial of citicoline in patients with acute ischemic stroke. Stroke 1999, 30, 2592–2597. [Google Scholar] [CrossRef] [Green Version]
- Clark, W.M.; Wechsler, L.R.; Sabounjian, L.A.; Schwiderski, U.E. Citicoline Stroke Study Group. A phase III randomized efficacy trial of 2000 mg citicolie in acute ischemic stroke patients. Neurology 2001, 57, 1595–1602. [Google Scholar] [CrossRef]
- Warach, S.; Pettigrew, L.C.; Dashe, J.F.; Pullicino, P.; Lefkowitz, D.M.; Sabounjian, L.; Harnett, K.; Schwiderski, U.; Gammans, R. Effect of citicoline on ischemic lesions as measured by diffusion-weighted magnetic resonance imaging. Citicoline 010 Investigators. Ann. Neurol. 2000, 48, 713–722. [Google Scholar] [CrossRef]
- Ivarez-Sabin, J.A.; Roman, G.C. Citicoline in Vascular Cognitive Impairment and Vascular Dementia After Stroke. Stroke 2011, 42, S40–S43. [Google Scholar] [CrossRef] [Green Version]
- Ylilauri, M.P.T.; Voutilainen, S.; Lönnroos, E.; Virtanen, H.E.K.; Tuomainen, T.P.; Salonen, J.T.; Virtanen, J.K. Associations of dietary choline intake with risk of incident dementia and with cognitive performance: The Kuopio Ischaemic Heart Disease Risk Factor Study. Am. J. Clin. Nutr. 2019. [Google Scholar] [CrossRef] [PubMed]
- Klancnik, J.M.; Cuénod, M.; Gähwiler, B.H.; Jiang, Z.P.; Do, K.Q. Release of endogenous amino acids, including homocysteic acid and cysteine sulphinic acid, from rat hippocampal slices evoked by electrical stimulation of Schaffer collateral-commissural fibres. Neuroscience 1992, 49, 557–570. [Google Scholar] [CrossRef]
- Lipton, S.A.; Kim, W.K.; Choi, Y.B.; Kumar, S.; D’Emilia, D.M.; Rayudu, P.V.; Arnelle, D.R.; Stamler, J.S. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928. [Google Scholar] [CrossRef] [Green Version]
- Zieminska, E.; Stafiej, A.; Lazarewicz, J.W. Role of group I metabotropic glutamate receptors and NMDA receptors in homocysteine-evoked acute neurodegeneration of cultured cerebellar granule neurons. Neurochem. Int. 2003, 43, 481–492. [Google Scholar] [CrossRef]
- Scarpa, S.; Fuso, A.; D’Anselmi, F.; Cavallaro, R.A. Presenilin 1 gene silencing by S-adenosylmethionine: A treatment for Alzheimer disease? FEBS Lett. 2003, 541, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.; Lu, Q.; Orecchio, L.; Kosik, K.S. Selective phosphorylation of adult tau isoforms in mature hippocampal neurons exposed to fibrillar A beta. Mol. Cell. Neurosci. 1997, 9, 220–234. [Google Scholar] [CrossRef]
- Hasegawa, T.; Ukai, W.; Jo, D.G.; Xu, X.; Mattson, M.P.; Nakagawa, M.; Araki, W.; Saito, T.; Yamada, T. Homocysteic acid induces intraneuronal accumulation of neurotoxic Abeta42: Implications for the pathogenesis of Alzheimer’s disease. J. Neurosci. Res. 2005, 80, 869–876. [Google Scholar] [CrossRef]
- Sai, X.; Kawamura, Y.; Kokame, K.; Yamaguchi, H.; Hirohisa Shiraishi, H.; Suzuki, R.; Suzuki, T.; Kawaichi, M.; Miyata, T.; Kitamura, T.; et al. Endoplasmic reticulum stress-inducible protein, Herp, enhances presenilin-mediated generation of amyloid beta-protein. J. Biol. Chem. 2002, 277, 12915–12920. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Liu, J.; Zhao, J.; Mao, J.; Zhang, X.; Feng, L.; Han, C.; Li, M.; Wang, S.; Wu, D. Homocysteine induces the expression of C-reactive protein via NMDAr-ROS-MAPK-NF-KB signal pathway in rat vascular smooth muscle cells. Atherosclerosis 2014, 236, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Curro, M.; Gugliandolo, A.; Gangemi, C.; Risitano, R.; Ientile, R.; Caccamo, D. Toxic effects of mildy elevated homocysteine conncetrations in neuronal-like cells. Neurochem. Res. 2014, 39, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Pushpakumar, S.; Kundu, S.; Sen, U. Endothelial Dysfunction: The Link Between Homocysteine and Hydrogen Sulfide. Curr. Med. Chem. 2014, 21, 3662–3672. [Google Scholar] [CrossRef]
- Vallance, P.; Chan, N. Endothelial function and nitric oxide: Clinical relevance. Heart 2001, 85, 342–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [CrossRef] [Green Version]
- Perna, A.F.; Ingrosso, D.; De Santo, N.G. Homocysteine and oxidative stress. Amino Acids 2003, 25, 409–417. [Google Scholar] [CrossRef]
- Hoffman, M. Hypothesis: Hyperhomocysteinemia is an indicator of oxidant stress. Med Hypotheses 2011, 77, 1088–1093. [Google Scholar] [CrossRef]
- Sawle, P.; Foresti, R.; Green, C.J.; Motterlini, R. Homocysteine attenuates endothelial heme oxygenase-1 induction by nitric oxide (NO) and hypoxia. FEBS Lett. 2001, 508, 403–406. [Google Scholar] [CrossRef] [Green Version]
- Stuhlinger, M.C.; Tsao, P.S.; Her, J.H.; Kimoto, M.; Balint, R.F.; Cooke, J.P. Homocysteine impairs the nitric oxide synthase pathway: Role of asymmetric dimethylarginine. Circulation 2001, 104, 2569–2575. [Google Scholar] [CrossRef]
- Wakita, H.; Tomimoto, H.; Akiguchi, I.; Kimura, J. Glial activation and white matter changes in the rat brain induced by chronic cerebral hypoperfusion: An immunoistochemical study. Acta Neuropathol. 1994, 87, 484–492. [Google Scholar] [CrossRef]
- Farkas, E.; Donka, G.; de Vous, R.A.I.; Mihaly, A.; Bari, F.; Luiten, P.G.M. Experimental cerebral hypoperfusion induces white matter injury and microglial activation in the rat brain. Acta Neuropathol. 2004, 108, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 2017, 98, 17–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploder, M.; Kurz, K.; Splitter, A.; Neurauter, G.; Roth, E.; Fuch, D. Early increase of plasma Hcy in sepsis patients with poor outcome. Mol. Med. 2010, 16, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Li, Q.; Du, H.P.; Wang, Y.L.; You, S.J.; Wang, F.; Xu, X.S.; Cheng, J.; Cao, Y.J.; Liu, C.F.; et al. Homocysteine Triggers inflammatory responses in macrophages through inhibiting CSE-H2S signaling via DNA hypermethylation of CSE promoter. Int. J. Mol. Sci. 2015, 16, 12560–12577. [Google Scholar] [CrossRef]
- Jiang, Y.-D.; Sun, T.; Zhang, H.-P.; Xiong, J.-T.; Cao, J.; Li, G.-Z.; Wang, S.-R. Folate and ApoE DNA methylation induced by homocysteine in human monocytes. DNA Cell Biol. 2007, 26, 737–744. [Google Scholar]
- Boldyrev, A.; Bryshkova, E.; Mashkina, A.; Vladychenskaya, E. Why is homocysteine toxic for the nervous and immune systems? Curr. Aging Sci. 2013, 6, 29–36. [Google Scholar] [CrossRef]
- Ying, G.; Wang, Y.; Cen, X.M.; Yang, M.; Liang, Y.; Xie, Q.B. Lipid peroxidation-mediated inflammation promotes cell apoptosis through activation of NFK-B pathway in rheumatoid arthritis synovial cells. Med. Infalmm. 2015, 2015, 460310. [Google Scholar]
- Chang, P.Y.; Lu, S.C.; Lee, C.M.; Chen, Y.J.; Dugan, T.A.; Huang, W.H.; Chang, S.F.; Liao, W.S.; Chen, C.H.; Lee, Y.T. Homocysteine inhibits arterial endothelial cell growth through transcriptional downregulation of fibroblast growth factor-2 involving G protein and DNA methylation. Circ. Res. 2008, 102, 933–941. [Google Scholar] [CrossRef]
- Nichols, J. Testing for homocysteine in clinical practice. Nutr. Health 2017, 23, 13–15. [Google Scholar] [CrossRef]
- Vidoni, M.L.; Gabriel, K.P.; Luo, S.T.; Simonsick, E.M.; Day, R.S. Vitamin B12 and Homocysteine Associations with Gait Speed in Older Adults: The Baltimore Longitudinal Study of Aging. J. Nutr. Health Aging 2017, 21, 1321–1328. [Google Scholar] [CrossRef] [Green Version]
- Kramer, C.S.; Szmidt, M.K.; Sicinska, E.; Brzozowska, A.; Santoro, A.; Franceschi, C.; de Groot, L.C.P.G.M.; Berendsen, A.A.M. The Elderly-Nutrient Rich Food Score Is Associated with Biochemical Markers of Nutritional Status in European Older Adults. Front. Nutr. 2019, 6, 150. [Google Scholar] [CrossRef] [PubMed]
- Hankey, G.J.; Ford, A.H.; Yi, Q.; Eikelboom, J.W.; Lees, K.R.; Chen, C.; Xavier, D.; Navarro, J.C.; Ranawaka, U.K.; Uddin, W.; et al. Effect of B-vitamins and lowering homocysteine on cognitive impairment in patients with previous stroke or transient ischemic attack: A prespecified secondary analysis of a randomized, placebo-controlled trial and meta-analysis. Stroke 2013, 44, 2232–2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, S.K.S.; Earnest, A.; Li, H.; Hameed, S.; Chang, H.M.; Chen, C.L.H.; Tan, E.K. B vitamins and cognition in subjects with small vessel disease: A substudy of VITATOPS, a randomized, placebo-controlled trial. J. Neurol. Sci. 2017, 379, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Liu, K.; Chen, S.; Yu, H.; An, Y.; Wang, Y.; Zhang, X.; Wang, Y.; Qin, Z.; Xiao, R. Dietary Intake of Riboflavin and Unsaturated Fatty Acid Can Improve the Multi-Domain Cognitive Function in Middle-Aged and Elderly Populations: A 2-Year Prospective Cohort Study. Front. Aging Neurosci. 2019, 11, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moretti, R.; Peinkhofer, C. B Vitamins and Fatty Acids: What Do They Share with Small Vessel Disease-Related Dementia? Int. J. Mol. Sci. 2019, 20, 5797. https://doi.org/10.3390/ijms20225797
Moretti R, Peinkhofer C. B Vitamins and Fatty Acids: What Do They Share with Small Vessel Disease-Related Dementia? International Journal of Molecular Sciences. 2019; 20(22):5797. https://doi.org/10.3390/ijms20225797
Chicago/Turabian StyleMoretti, Rita, and Costanza Peinkhofer. 2019. "B Vitamins and Fatty Acids: What Do They Share with Small Vessel Disease-Related Dementia?" International Journal of Molecular Sciences 20, no. 22: 5797. https://doi.org/10.3390/ijms20225797
APA StyleMoretti, R., & Peinkhofer, C. (2019). B Vitamins and Fatty Acids: What Do They Share with Small Vessel Disease-Related Dementia? International Journal of Molecular Sciences, 20(22), 5797. https://doi.org/10.3390/ijms20225797