An Integrated Analysis of the Rice Transcriptome and Metabolome Reveals Root Growth Regulation Mechanisms in Response to Nitrogen Availability

 ,
,
Abstract
:1. Introduction
2. Results
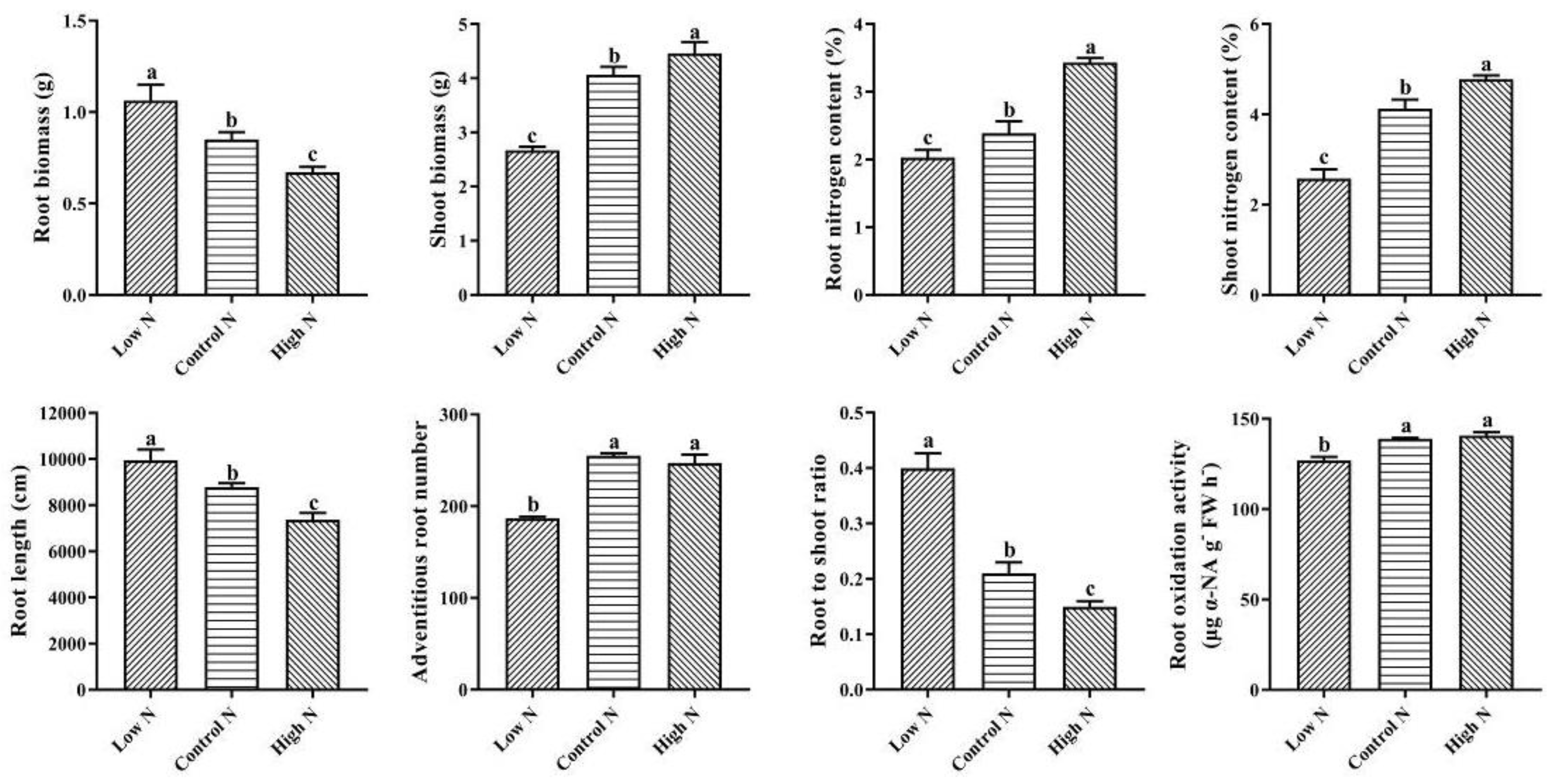
2.1. Nitrogen Availability Affects Rice Root Architectural and Physiological Characteristics
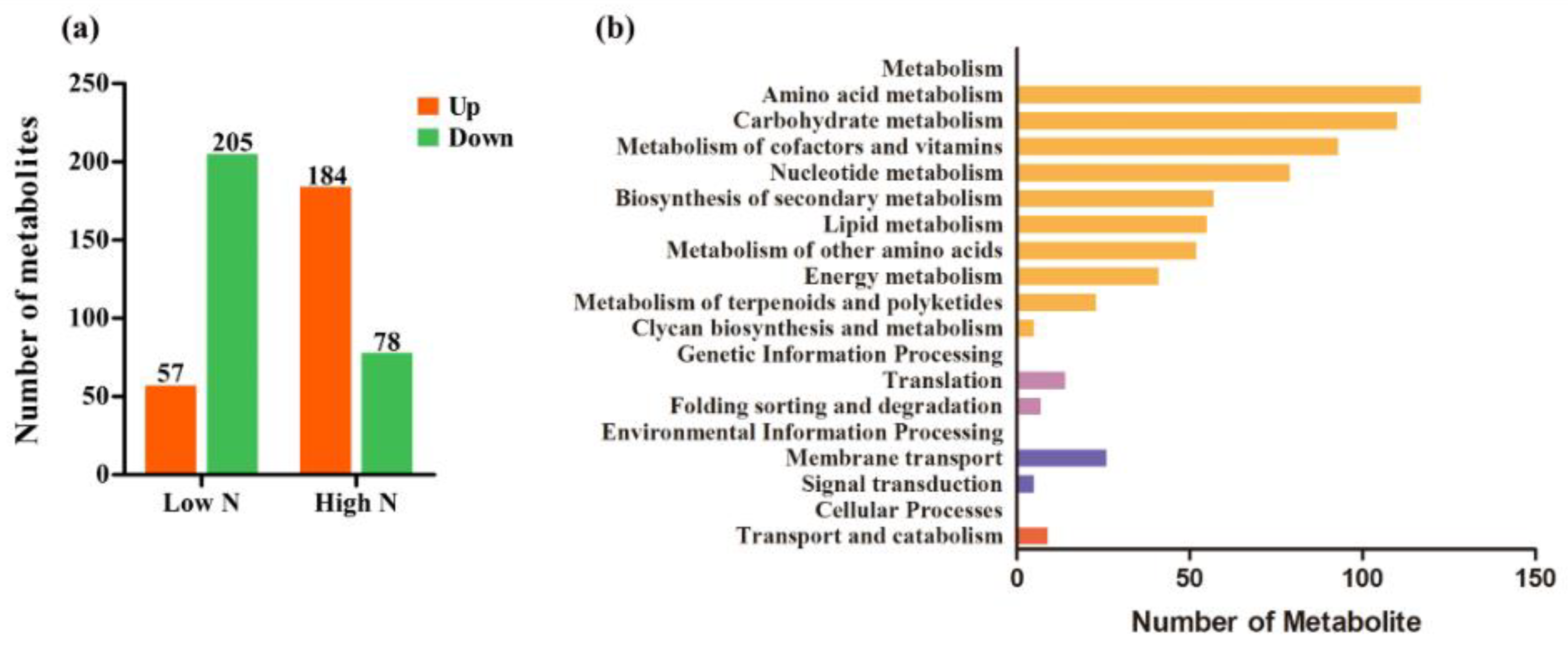
2.2. Metabolite Profiles of Rice Roots in Response to Nitrogen Availability
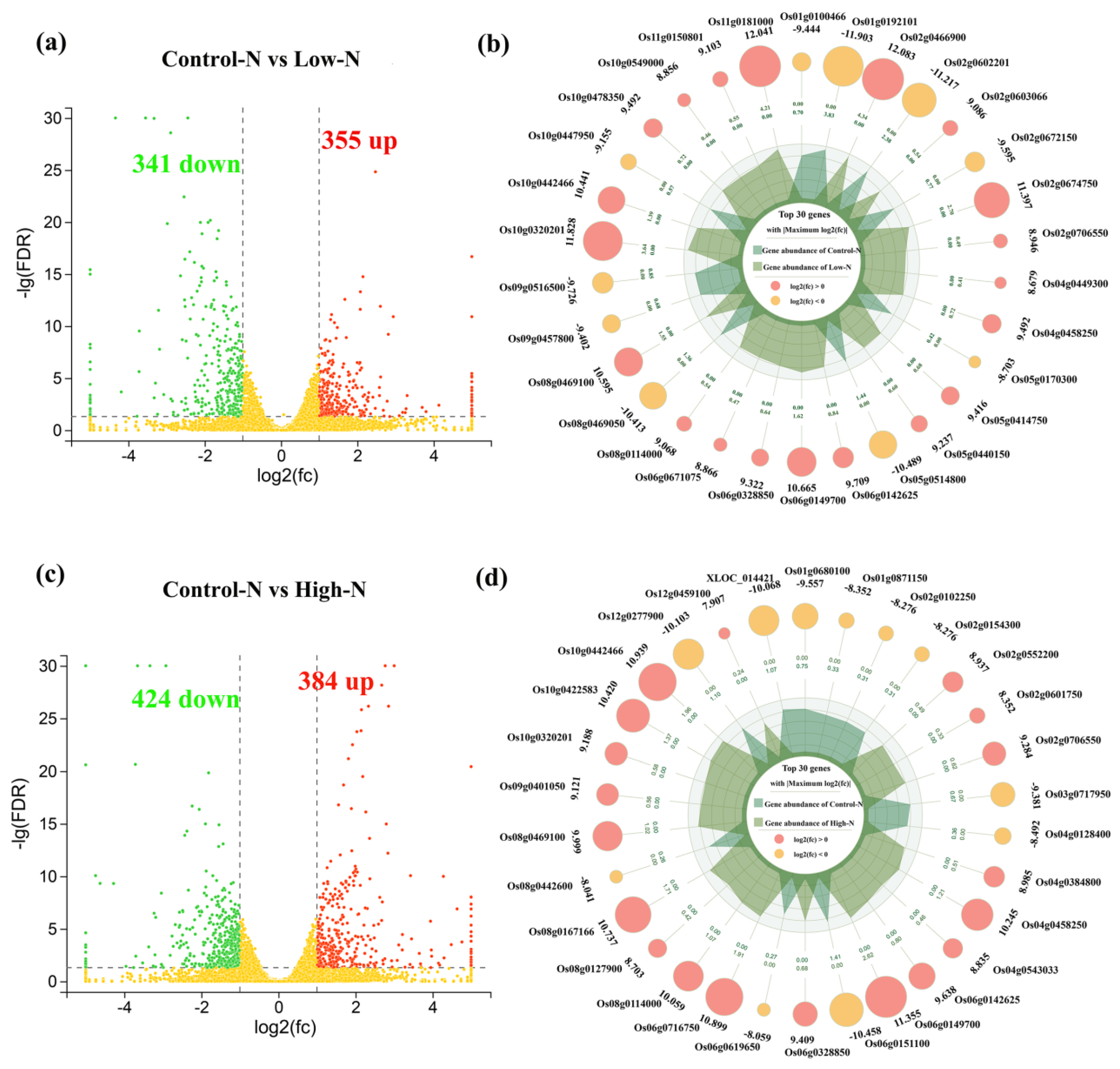
2.3. RNA Sequencing Profiles of Rice Roots in Response to Nitrogen Availability
2.4. Regulation of Differential Gene Expression by Transcription Factors
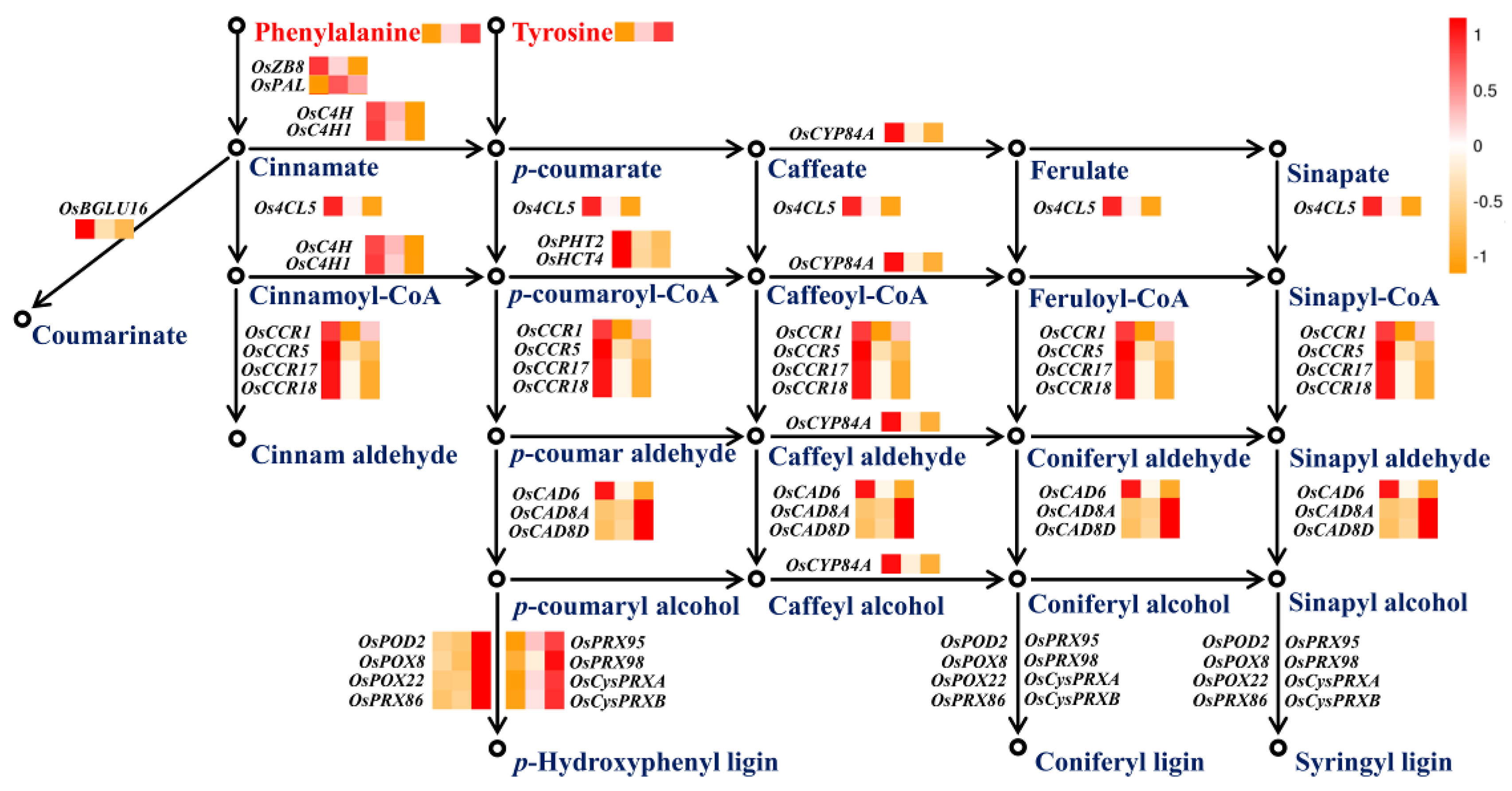
2.5. Phenylpropanoid Metabolism Modulated by Nitrogen Availability
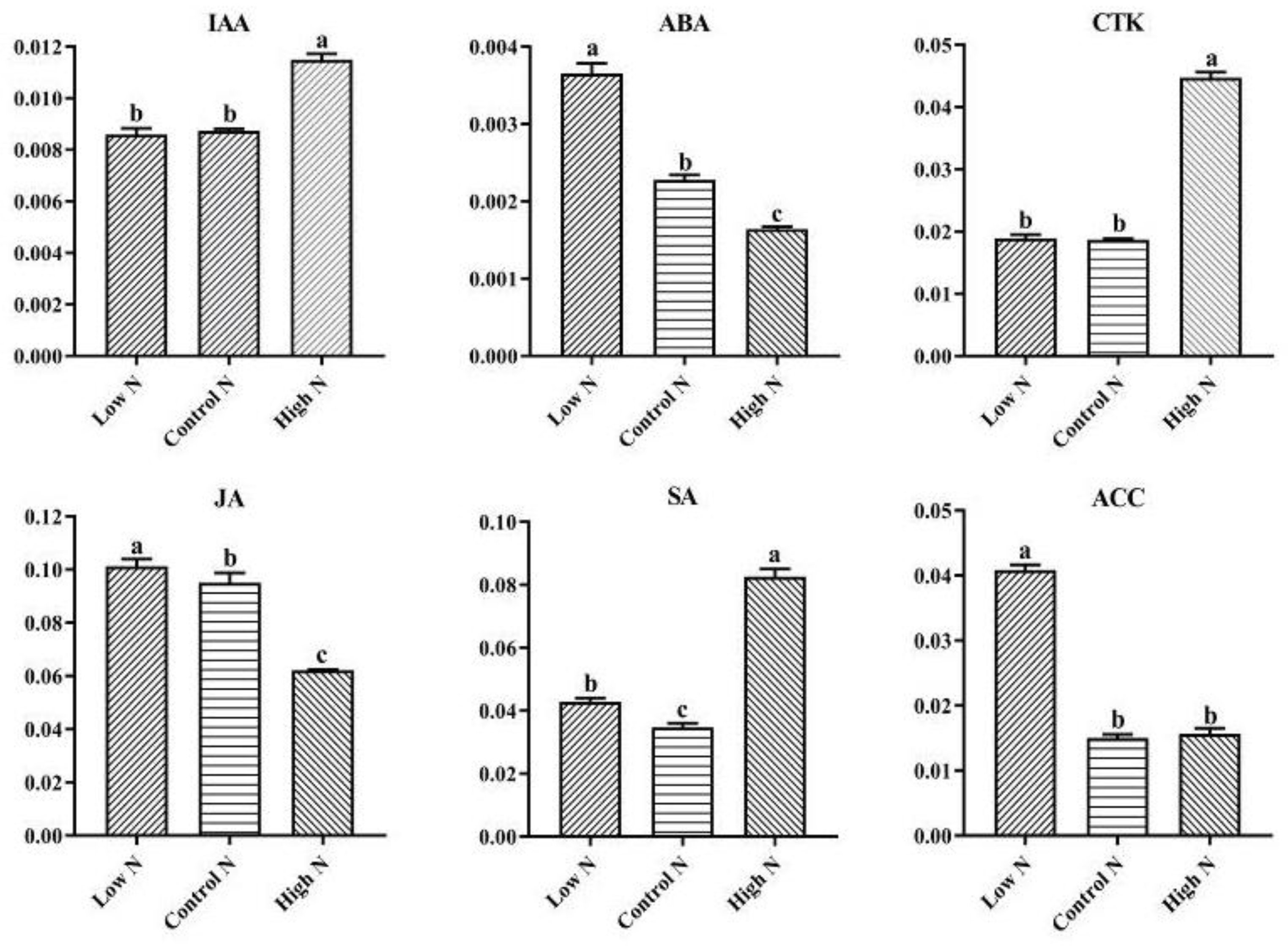
2.6. Nitrogen Availability Affects the Levels of Phytohormones Related to Growth and Stress Responses
3. Discussion
4. Materials and Methods
4.1. Plant Material and Growth Conditions
4.2. Analysis of Root Architecture and Physiological Characteristics
4.3. Metabolite Extraction and Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry (LC-ESI-MS/MS) Analysis
4.4. RNA Extraction and Sequencing
4.5. Confirmation of Transcriptome Data Using qRT-PCR Analysis
4.6. Determination of Phytohormone Content
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, H.; Hu, B.; Chu, C. Nitrogen use efficiency in crops: Lessons from Arabidopsis and rice. J. Exp. Bot. 2017, 68, 2477–2488. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Buresh, R.J.; Huang, J.; Yang, J.; Zou, Y.; Zhong, X.; Wang, G.; Zhang, F. Strategies for overcoming low agronomic nitrogen use efficiency in irrigated rice systems in China. Field Crop. Res. 2006, 96, 37–47. [Google Scholar] [CrossRef]
- Xu, G.; Fan, X.; Miller, A.J. Plant Nitrogen Assimilation and Use Efficiency. Annu. Rev. Plant Biol. 2012, 63, 153–182. [Google Scholar] [CrossRef] [PubMed]
- Hakeem, K.R.; Ahmad, A.; Iqbal, M.; Gucel, S.; Ozturk, M. Nitrogen-efficient rice cultivars can reduce nitrate pollution. Environ. Sci. Pollut. Res. 2011, 18, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yoon, J.; Choi, H.; Fan, Y.; Chen, R.; An, G. Transcriptome analysis of nitrogen-starvation-responsive genes in rice. BMC Plant Biol. 2015, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Buresh, R.J.; Wang, Z.; Zhang, H.; Liu, L.; Yang, J.; Zhang, J. Root and shoot traits for rice varieties with higher grain yield and higher nitrogen use efficiency at lower nitrogen rates application. Field Crop. Res. 2015, 175, 47–55. [Google Scholar] [CrossRef]
- Fan, J.; Mcconkey, B.; Wang, H.; Janzen, H. Root distribution by depth for temperate agricultural crops. Field Crop. Res. 2016, 189, 68–74. [Google Scholar] [CrossRef]
- Francisco, R.H.; Shane, M.W.; López-Bucio, J. Nutritional regulation of root development. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 431–443. [Google Scholar]
- Walch-Liu, P. Nitrogen Regulation of Root Branching. Ann. Bot. 2006, 97, 875–881. [Google Scholar] [CrossRef]
- Hirai, M.Y.; Yano, M.; Goodenowe, D.B.; Kanaya, S.; Kimura, T.; Awazuhara, M.; Arita, M.; Fujiwara, T.; Saito, K. From The Cover: Integration of transcriptomics and metabolomics for understanding of global responses to nutritional stresses in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2004, 101, 10205–10210. [Google Scholar] [CrossRef]
- Hirai, M.Y.; Klein, M.; Fujikawa, Y.; Yano, M.; Goodenowe, D.B.; Yamazaki, Y.; Kanaya, S.; Nakamura, Y.; Kitayama, M.; Suzuki, H. Elucidation of gene-to-gene and metabolite-to-gene networks in arabidopsis by integration of metabolomics and transcriptomics. J. Biol. Chem. 2005, 280, 25590–25595. [Google Scholar]
- Dhokane, D.; Shailesh Karre, S.; Kushalappa, A.C.; Cartney, C.M. Integrated Metabolo-Transcriptomics Reveals Fusarium Head Blight Candidate Resistance Genes in Wheat QTL-Fhb2. PLoS ONE 2016, 11, e0155851. [Google Scholar] [CrossRef] [PubMed]
- Agarrwal, R.; Padmakumari, A.P.; Bentur, J.S.; Nair, S. Metabolic and transcriptomic changes induced in host during hypersensitive response mediated resistance in rice against the Asian rice gall midge. Rice 2016, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Zhao, X.; Qi, G.; Bai, Z.; Wang, Y.; Wang, S.; Ma, Y.; Liu, Q.; Hu, R.; Zhou, G. Integrated analysis of transcriptome and metabolites reveals an essential role of metabolic flux in starch accumulation under nitrogen starvation in duckweed. Biotechnol. Biofuels 2017, 10, 167. [Google Scholar] [CrossRef]
- Yanagisawa, S.; Akiyama, A.; Kisaka, H.; Uchimiya, H.; Miwa, T. Metabolic engineering with Dof1 transcription factor in plants: Improved nitrogen assimilation and growth under low-nitrogen conditions. Proc. Natl. Acad. Sci. USA 2004, 101, 7833–7838. [Google Scholar] [CrossRef]
- Zhang, H.; Xue, Y.; Wang, Z.; Yang, J.; Zhang, J. An alternate wetting and moderate soil drying regime improves root and shoot growth in rice. Crop Sci. 2009, 49, 2246–2260. [Google Scholar] [CrossRef]
- Lynch, J.P. Steep, cheap and deep: An ideotype to optimize water and N acquisition by maize root systems. Ann. Bot. 2013, 112, 347–357. [Google Scholar] [CrossRef]
- Yang, C.; Yang, L.; Yang, Y.; Ouyang, Z. Rice root growth and nutrient uptake as influenced by organic manure in continuously and alternately flooded paddy soils. Agric. Water Manag. 2004, 70, 67–81. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Y.; Kobayashi, K.; Zhu, J.; Huang, J.; Yang, H.; Wang, Y.; Dong, G.; Liu, G.; Han, Y.; et al. Seasonal changes in the effects of free-air CO2 enrichment (FACE) on growth, morphology and physiology of rice root at three levels of nitrogen fertilization. Glob. Chang. Biol. 2008, 14, 1–10. [Google Scholar] [CrossRef]
- Xin, W.; Zhang, L.; Zhang, W.; Gao, J.; Yi, J.; Zhen, X.; Li, Z.; Zhao, Y.; Peng, C.; Zhao, C. An Integrated Analysis of the Rice Transcriptome and Metabolome Reveals Differential Regulation of Carbon and Nitrogen Metabolism in Response to Nitrogen Availability. Int. J. Mol. Sci. 2019, 20, 2349. [Google Scholar] [CrossRef]
- Hua, L.; Wang, D.R.; Tan, L.B.; Fu, Y.C.; Liu, F.X.; Xiao, L.T.; Zhu, Z.F.; Fu, Q.; Sun, X.Y.; Gu, P.; et al. LABA1, a domestication gene associated with long, barbed awns in wild rice. Plant Cell. 2015, 27, 1875–1888. [Google Scholar] [CrossRef] [PubMed]
- Cong, F.; Diehl, B.G.; Hill, J.L.; Brown, N.R.; Tien, M. Covalent bond formation between amino acids and lignin: Cross-coupling between proteins and lignin. Phytochemistry 2013, 96, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Chen, F.; Inoue, K.; Blount, I.K.; Dixon, R.A. Downregulation of caffeic acid 3-O-methyltransferase and caffeoyl CoA 3-O-methyltransferase in transgenic alfalfa impacts on lignin structure and implications for the biosynthesis of G and S lignin. Plant Cell. 2001, 13, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, E.; Hawkins, S.; Doorsselaere, J.V.; Piquemal, J.; Goffner, D.; Poeydomenge, O.; Boudet, A.M.; Grima-Pettenati, J. Cinnamoyl CoA reductase, the first committed enzyme of the lignin branch biosynthetic pathway: Cloning, expression and phylogenetic relationships. Plant J. 1997, 11, 429–441. [Google Scholar] [CrossRef]
- Fan, F.; Li, J.; Zhan, Q.; Wang, L.; Liu, X. Advances in cloning of plant cinnamoyl-CoA reductase gene. China Biotechnol. 2015, 35, 96–102. [Google Scholar]
- Trevisan, S.; Manoli, A.; Ravazzolo, L.; Botton, A.; Pivato, M.; Masi, A.; Quaggiotti, S. Nitrate sensing by the maize root apex transition zone: A merged transcriptomic and proteomic survey. J. Exp. Bot. 2015, 66, 3699–3715. [Google Scholar] [CrossRef]
- Qin, L.; Walk, T.C.; Han, P.; Chen, L.; Zhang, S.; Li, Y.; Hu, X.; Xie, L.; Yang, Y.; Liu, J.; et al. Adaption of Roots to Nitrogen Deficiency Revealed by 3D Quantification and Proteomic Analysis. Plant Physiol. 2019, 179, 329–347. [Google Scholar] [CrossRef]
- Dunand, C.; Crèvecoeur, M.; Penel, C. Distribution of superoxide and hydrogen peroxide in Arabidopsis root and their influence on root development: Possible interaction with peroxidases. N. Phytol. 2007, 174, 332–341. [Google Scholar] [CrossRef]
- Raggi, S.; Ferrarini, A.; Delledonne, M.; Dunand, C.; Ranocha, P.; De Lorenzo, G.; Cervone, F.; Ferrari, S. The Arabidopsis class III peroxidase AtPRX71 negatively regulates growth under physiological conditions and in response to cell wall damage. Plant Phytol. 2015, 169, 2513–2525. [Google Scholar] [CrossRef]
- Shani, Z.; Dekel, M.; Tsabary, G.; Shoseyov, O. Cloning and characterization of elongation specific endo-1, 4-beta-glucanase (cel1) from Arabidopsis thaliana. Plant Mol. Biol. 1997, 34, 837–842. [Google Scholar] [CrossRef]
- Wu, S.C.; Blumer, J.M.; Darvill, A.G.; Albersheim, P. Characterization of an endo-beta-1, 4-glucanase gene induced by auxin in elongating pea epicotyls. Plant Physiol. 1996, 110, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Shani, Z.; Dekel, M.; Roiz, L.; Horowitz, M.; Kolosovski, N.; Lapidot, S.; Alkan, S.; Koltai, H.; Tsabary, G.; Goren, R.; et al. Expression of endo-1,4-beta-glucanase (cel1) in Arabidopsis thaliana is associated with plant growth, xylem development and cell wall thickening. Plant Cell Rep. 2006, 25, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Matthysse, A.G.; Thomas, D.L.; White, A.R. Mechanism of cellulose synthesis in Agrobacterium tumefaciens. J. Bacteriol. 1995, 177, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Castaings, L.; Camargo, A.; Pocholle, D.; Gaudon, V.; Texier, Y.; Boutet-Mercey, S.; Taconnat, L.; Renou, J.P.; Daniel-Vedele, F.; Fernandez, E.; et al. The nodule inception-like protein 7 modulates nitrate sensing and metabolism in Arabidopsis. Plant J. 2009, 57, 426–435. [Google Scholar]
- Kurai, T.; Wakayama, M.; Abiko, T.; Yanagisawa, S.; Aoki, N.; Ohsugi, R. Introduction of the ZmDof1 gene into rice enhances carbon and nitrogen assimilation under low-nitrogen conditions. Plant Biotechnol. J. 2011, 9, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Heerah, S.; Katari, M.S.; Penjor, R.; Coruzzi, G.M.; Marshall-Colon, A. WRKY1 mediates transcriptional regulation of light and nitrogen signaling pathways. Plant Physiol. 2019, 181, 1371–1388. [Google Scholar] [CrossRef] [Green Version]
- Imamura, S.; Kanesaki, Y.; Ohnuma, M.; Inouye, T.; Sekine, Y.; Fujiwara, T.; Kuroiwa, T.; Tanaka, K. R2R3-type MYB transcription factor, CmMYB1, is a central nitrogen assimilation regulator in Cyanidioschyzon merolae. Proc. Natl. Acad. Sci. USA 2009, 106, 12548–12553. [Google Scholar]
- Luo, J.; Zhou, J.; Li, H.; Shi, W.; Polle, A.; Lu, M.; Sun, X.; Luo, Z.B. Global poplar root and leaf transcriptomes reveal links between growth and stress responses under nitrogen starvation and excess. Tree Physiol. 2015, 35, 1283–1302. [Google Scholar] [CrossRef] [Green Version]
- Krouk, G.; Ruffel, S.; Gutiérrez, R.A.; Gojon, A.; Crawford, N.M.; Coruzzil, G.M.; Lacombe, B. A framework integrating plant growth with hormones and nutrients. Trends Plant Sci. 2011, 16, 178–182. [Google Scholar] [CrossRef]
- Krouk, G.; Lacombe, B.; Bielach, A.; Perrine-Walker, F.; Malinska, K.; Mounier, E.; Hoyerova, K.; Tillard, P.; Leon, S.; Ljung, K.; et al. Nitrate-Regulated Auxin Transport by NRT1.1 Defines a Mechanism for Nutrient Sensing in Plants. Dev. Cell. 2010, 18, 927–937. [Google Scholar] [CrossRef]
- Tuteja, N. Abscisic Acid and Abiotic Stress Signaling. Plant Signal. Behav. 2007, 2, 135–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.A.; Benková, E. Cytokinin Cross-talking During Biotic and Abiotic Stress Responses. Front. Plant Sci. 2013, 4, 451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, H.; Chinnusamy, V.; Rodrigues, A.; Rubio, S.; Antoni, R.; Park, S.Y.; Cutler, S.R.; Sheen, J.; Rodriguez, P.L.; Zhu, J.K. In vitro reconstitution of an abscisic acid signalling pathway. Nature 2009, 462, 660–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Szostkiewicz, I.; Korte, A.; Moes, D.; Yang, Y.; Christmann, A.; Grill, E. Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science 2009, 324, 1064–1068. [Google Scholar] [CrossRef]
- Kiba, T.; Kudo, T.; Kojima, M.; Sakakibara, H. Hormonal control of nitrogen acquisition: Roles of auxin, abscisic acid, and cytokinin. J. Exp. Bot. 2011, 62, 1399–1409. [Google Scholar] [CrossRef]
- Li, Y.; Yang, X.; Ren, B.; Shen, Q.; Guo, S. Why nitrogen use efficiency decreases under high nitrogen supply in rice (Oryza sativa L.) seedlings. J. Plant Growth Regul. 2012, 31, 47–52. [Google Scholar] [CrossRef]
- Ramasamy, S.; Berge, H.F.M.; Purushothaman, S. Yield formation in rice in response to drainage and nitrogen application. Field Crops Res. 1997, 51, 65–82. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Qi, D.; Huo, H.; Dong, X.; Tian, L.; Zhang, X.; Liu, C.; Cao, Y. Postharvest metabolomic changes in Pyrus ussuriensis Maxim. Wild accession ‘Zaoshu Shanli’. J. Sep. Sci. 2018, 41, 4001–4013. [Google Scholar] [CrossRef]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef]
- Zhu, Z.J.; Schultz, A.W.; Wang, J.; Johnson, C.H.; Yannone, S.M.; Patti, G.J.; Siuzdak, G. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat. Protoc. 2013, 8, 451–460. [Google Scholar] [CrossRef]
- Cosgrove, D.J. Plant cell wall extensibility: Connecting plant cell growth with cell wall structure, mechanics, and the action of wall-modifying enzymes. J. Exp. Bot. 2015, 67, 463–476. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TF Family | Low-N | High-N | ||
|---|---|---|---|---|
| Up | Down | Up | Down | |
| bHLH | 6 | 1 | 1 | 1 |
| bZIP | 1 | 1 | 1 | 1 |
| C2H2 | 4 | 0 | 0 | 0 |
| CO-like | 1 | 0 | 1 | 0 |
| DBB | 0 | 2 | 0 | 0 |
| E2F/DP | 0 | 1 | 1 | 0 |
| EIL | 0 | 0 | 0 | 1 |
| ERF | 4 | 1 | 1 | 0 |
| G2-like | 0 | 1 | 1 | 0 |
| GRAS | 1 | 0 | 1 | 0 |
| HD-ZIP | 1 | 1 | 0 | 0 |
| HSF | 1 | 0 | 0 | 0 |
| LSD | 0 | 1 | 0 | 0 |
| M-type_MADS | 0 | 0 | 1 | 0 |
| MYB | 1 | 0 | 0 | 0 |
| MYB_related | 3 | 1 | 0 | 6 |
| NAC | 5 | 1 | 0 | 3 |
| NF-YA | 1 | 0 | 0 | 0 |
| NF-YC | 0 | 1 | 1 | 0 |
| Nin-like | 0 | 0 | 1 | 1 |
| Whirly | 0 | 1 | 0 | 0 |
| WRKY | 6 | 0 | 0 | 0 |
| Total | 35 | 13 | 10 | 13 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xin, W.; Zhang, L.; Zhang, W.; Gao, J.; Yi, J.; Zhen, X.; Du, M.; Zhao, Y.; Chen, L. An Integrated Analysis of the Rice Transcriptome and Metabolome Reveals Root Growth Regulation Mechanisms in Response to Nitrogen Availability. Int. J. Mol. Sci. 2019, 20, 5893. https://doi.org/10.3390/ijms20235893
Xin W, Zhang L, Zhang W, Gao J, Yi J, Zhen X, Du M, Zhao Y, Chen L. An Integrated Analysis of the Rice Transcriptome and Metabolome Reveals Root Growth Regulation Mechanisms in Response to Nitrogen Availability. International Journal of Molecular Sciences. 2019; 20(23):5893. https://doi.org/10.3390/ijms20235893
Chicago/Turabian StyleXin, Wei, Lina Zhang, Wenzhong Zhang, Jiping Gao, Jun Yi, Xiaoxi Zhen, Ming Du, Yanze Zhao, and Liqiang Chen. 2019. "An Integrated Analysis of the Rice Transcriptome and Metabolome Reveals Root Growth Regulation Mechanisms in Response to Nitrogen Availability" International Journal of Molecular Sciences 20, no. 23: 5893. https://doi.org/10.3390/ijms20235893
APA StyleXin, W., Zhang, L., Zhang, W., Gao, J., Yi, J., Zhen, X., Du, M., Zhao, Y., & Chen, L. (2019). An Integrated Analysis of the Rice Transcriptome and Metabolome Reveals Root Growth Regulation Mechanisms in Response to Nitrogen Availability. International Journal of Molecular Sciences, 20(23), 5893. https://doi.org/10.3390/ijms20235893