Glucose Transport and Transporters in the Endomembranes


 , , , ,
, , , ,
Abstract
:1. Introduction
2. Glucose Transporters
2.1. SGLTs
2.2. GLUTs
2.3. Others
3. Processes Linked to Glucose Transport in the Organelles
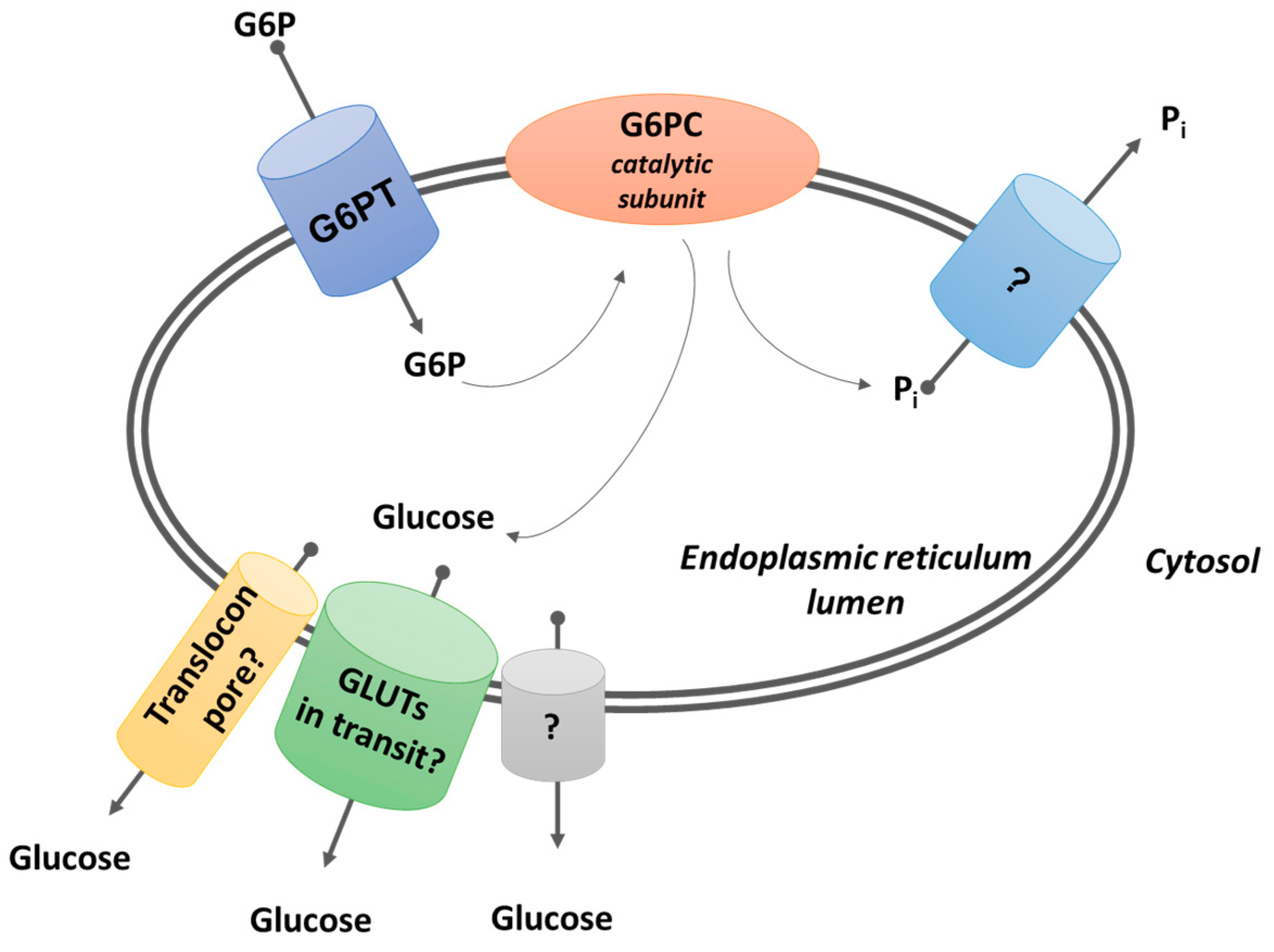
3.1. Glucose Production by Glucose-6-Phosphatases
3.1.1. Glucose-6-Phosphatases
3.1.2. Possible Routes of Glucose Exit from the ER
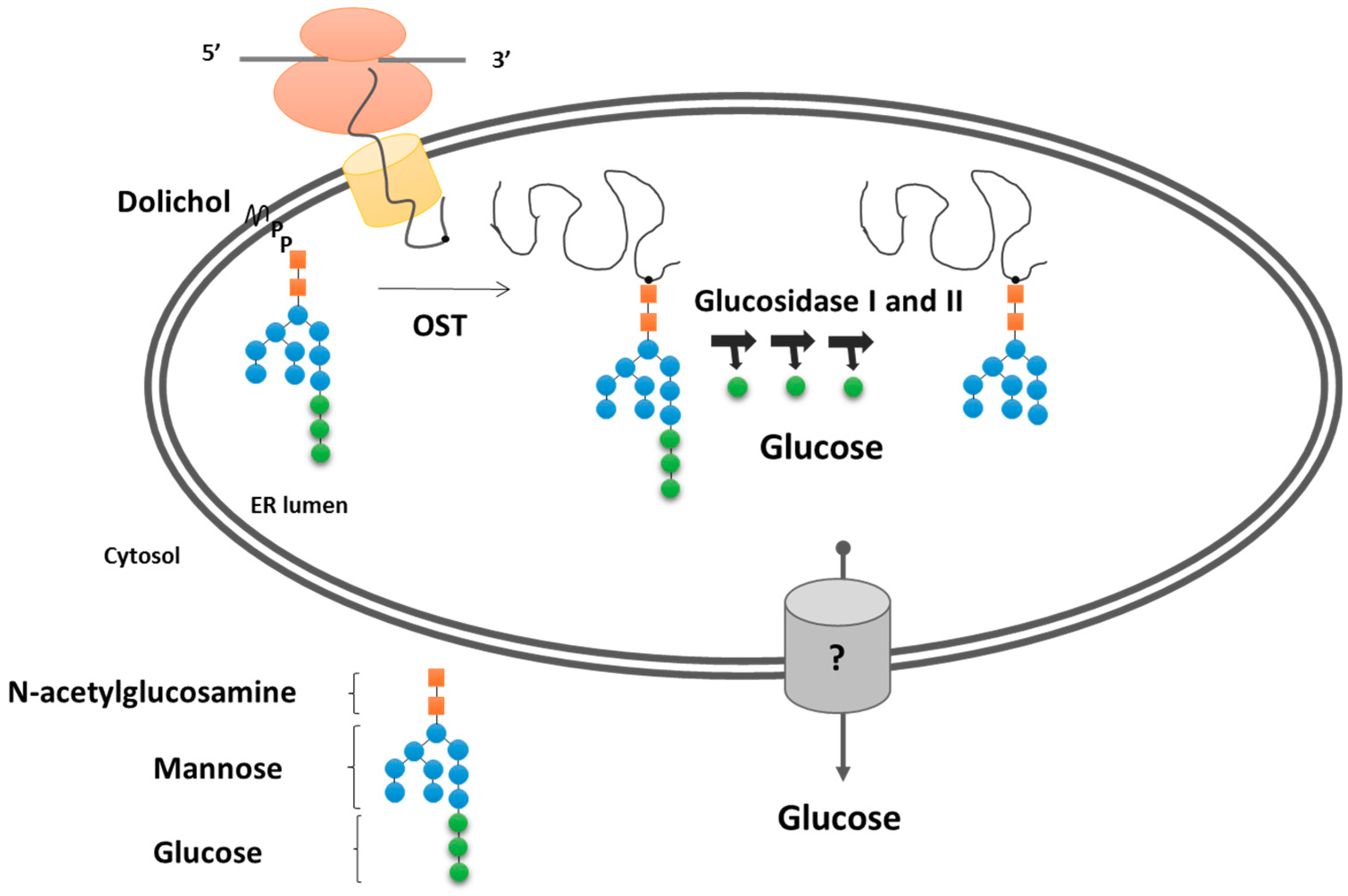
3.2. Protein Glycosylation/Deglycosylation
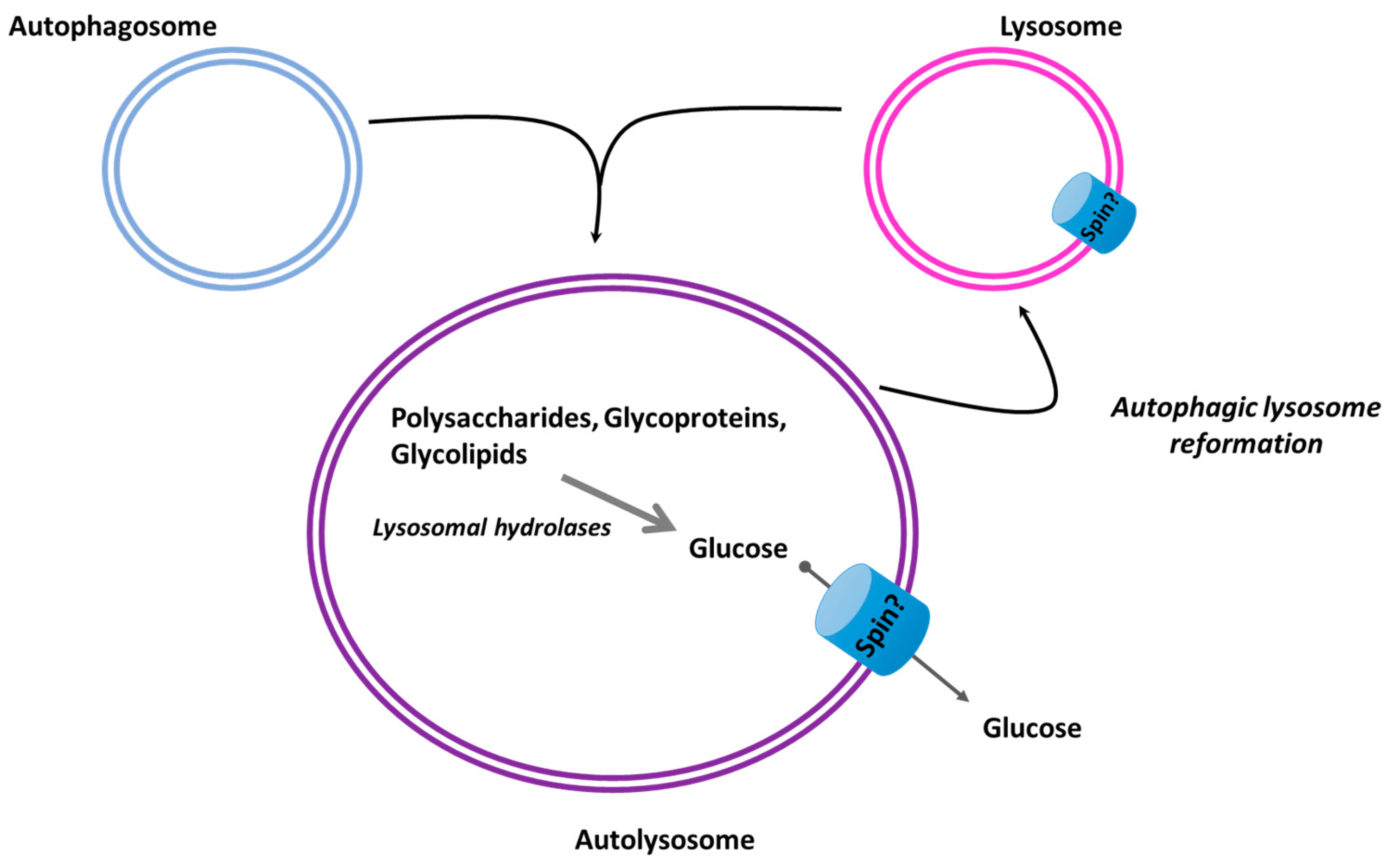
3.3. Autophagy
4. In Silico Predictions of the Localization of Different Glucose Transporters
5. Glucose Transport in the Endomembranes
5.1. GLUT6
5.2. GLUT8
5.3. GLUT10
5.4. Spns1
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA | Ascorbic acid |
| ALR | Autophagic lysosome reformation |
| ATS | Arterial tortuosity syndrome |
| DHA | Dehydroascorbic acid |
| EST | Expressed sequence tag |
| ER | Endoplasmic reticulum |
| HIF | Hypoxia Inducible factor |
| HMIT | Human myoinositol transporter |
| G6P | Glucose-6-phosphate |
| G6PT | Glucose-6-phosphate transporter |
| G6Pase | Glucose-6-phosphatase |
| G6PC | Glucose-6-phosphatase |
| GLUT | Glucose transporter |
| GO | Gene ontology |
| GSD | Glycogen storage disease |
| GSD1rs | Glycogen storage disease 1 related syndrome |
| LPS | Lipopolysaccharide |
| mTOR | Mammalian target of rapamycin |
| Nf-kB | Nuclear factor kappa B |
| OST | Oligosaccharyl-transferase |
| Pi | Inorganic phosphate |
| ROS | Reactive oxygen species |
| SCN4 | Severe congenital neutropenia type 4 |
| SGLT | Sodium-glucose cotransporter |
| SLC | Solute carrier |
| SMIT2 | Sodium-myoinositol cotransporter 2 |
| SPNS | Spinster homologue |
| SVCT | Sodium couplet ascorbic acid transporter |
| SVM | Support vector machine |
| TET | Ten-eleven translocation |
References
- Deng, D.; Yan, N. Glut, sglt, and sweet: Structural and mechanistic investigations of the glucose transporters. Protein Sci. 2016, 25, 546–558. [Google Scholar] [CrossRef]
- Augustin, R. The protein family of glucose transport facilitators: It’s not only about glucose after all. IUBMB Life 2010, 62, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Marcolongo, P.; Fulceri, R.; Gamberucci, A.; Czegle, I.; Banhegyi, G.; Benedetti, A. Multiple roles of glucose-6-phosphatases in pathophysiology: State of the art and future trends. Biochim. Biophys. Acta 2013, 1830, 2608–2618. [Google Scholar] [CrossRef] [PubMed]
- Molinari, M.; Hebert, D.N. Glycoprotein maturation and quality control. Semin. Cell Dev. Biol. 2015, 41, 70. [Google Scholar] [CrossRef] [PubMed]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M. Glucose transport families slc5 and slc50. Mol. Asp. Med. 2013, 34, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Diez-Sampedro, A.; Hirayama, B.A.; Osswald, C.; Gorboulev, V.; Baumgarten, K.; Volk, C.; Wright, E.M.; Koepsell, H. A glucose sensor hiding in a family of transporters. Proc. Natl. Acad. Sci. USA 2003, 100, 11753–11758. [Google Scholar] [CrossRef] [PubMed]
- Tazawa, S.; Yamato, T.; Fujikura, H.; Hiratochi, M.; Itoh, F.; Tomae, M.; Takemura, Y.; Maruyama, H.; Sugiyama, T.; Wakamatsu, A.; et al. Slc5a9/sglt4, a new na+-dependent glucose transporter, is an essential transporter for mannose, 1,5-anhydro-d-glucitol, and fructose. Life Sci. 2005, 76, 1039–1050. [Google Scholar] [CrossRef]
- Grempler, R.; Augustin, R.; Froehner, S.; Hildebrandt, T.; Simon, E.; Mark, M.; Eickelmann, P. Functional characterisation of human sglt-5 as a novel kidney-specific sodium-dependent sugar transporter. FEBS Lett. 2012, 586, 248–253. [Google Scholar] [CrossRef]
- Coady, M.J.; Wallendorff, B.; Gagnon, D.G.; Lapointe, J.Y. Identification of a novel na+/myo-inositol cotransporter. J. Biol. Chem. 2002, 277, 35219–35224. [Google Scholar] [CrossRef]
- Mueckler, M.; Thorens, B. The slc2 (glut) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Q.; Hou, B.H.; Lalonde, S.; Takanaga, H.; Hartung, M.L.; Qu, X.Q.; Guo, W.J.; Kim, J.G.; Underwood, W.; Chaudhuri, B.; et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 2010, 468, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y. Stories of spinster with various faces: From courtship rejection to tumor metastasis rejection. J. Neurogenet. 2019, 33, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Senesi, S.; Marcolongo, P.; Kardon, T.; Bucci, G.; Sukhodub, A.; Burchell, A.; Benedetti, A.; Fulceri, R. Immunodetection of the expression of microsomal proteins encoded by the glucose 6-phosphate transporter gene. Biochem. J. 2005, 389, 57–62. [Google Scholar] [CrossRef]
- Annabi, B.; Hiraiwa, H.; Mansfield, B.C.; Lei, K.J.; Ubagai, T.; Polymeropoulos, M.H.; Moses, S.W.; Parvari, R.; Hershkovitz, E.; Mandel, H.; et al. The gene for glycogen-storage disease type 1b maps to chromosome 11q23. Am. J. Hum. Genet. 1998, 62, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Marcolongo, P.; Barone, V.; Priori, G.; Pirola, B.; Giglio, S.; Biasucci, G.; Zammarchi, E.; Parenti, G.; Burchell, A.; Benedetti, A.; et al. Structure and mutation analysis of the glycogen storage disease type 1b gene. FEBS Lett. 1998, 436, 247–250. [Google Scholar] [CrossRef]
- Gerin, I.; Veiga-da-Cunha, M.; Noel, G.; Van Schaftingen, E. Structure of the gene mutated in glycogen storage disease type ib. Gene 1999, 227, 189–195. [Google Scholar] [CrossRef]
- Cappello, A.R.; Curcio, R.; Lappano, R.; Maggiolini, M.; Dolce, V. The physiopathological role of the exchangers belonging to the slc37 family. Front. Chem. 2018, 6, 122. [Google Scholar] [CrossRef]
- Van Schaftingen, E.; Gerin, I. The glucose-6-phosphatase system. Biochem. J. 2002, 362, 513–532. [Google Scholar] [CrossRef]
- Pan, C.J.; Lei, K.J.; Annabi, B.; Hemrika, W.; Chou, J.Y. Transmembrane topology of glucose-6-phosphatase. J. Biol. Chem. 1998, 273, 6144–6148. [Google Scholar] [CrossRef]
- Cori, G.T.; Cori, C.F. Glucose-6-phosphatase of the liver in glycogen storage disease. J. Biol. Chem. 1952, 199, 661–667. [Google Scholar] [PubMed]
- Chou, J.Y.; Matern, D.; Mansfield, B.C.; Chen, Y.T. Type i glycogen storage diseases: Disorders of the glucose-6-phosphatase complex. Curr. Mol. Med. 2002, 2, 121–143. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.Y.; Jun, H.S.; Mansfield, B.C. Glycogen storage disease type i and g6pase-beta deficiency: Etiology and therapy. Nat. Rev. Endocrinol. 2010, 6, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Burchell, A. The molecular basis of the type 1 glycogen storage diseases. Bioessays 1992, 14, 395–400. [Google Scholar] [CrossRef]
- Veiga-da-Cunha, M.; Gerin, I.; Chen, Y.T.; Lee, P.J.; Leonard, J.V.; Maire, I.; Wendel, U.; Vikkula, M.; Van Schaftingen, E. The putative glucose 6-phosphate translocase gene is mutated in essentially all cases of glycogen storage disease type i non-a. Eur. J. Hum. Genet. 1999, 7, 717–723. [Google Scholar] [CrossRef]
- Galli, L.; Orrico, A.; Marcolongo, P.; Fulceri, R.; Burchell, A.; Melis, D.; Parini, R.; Gatti, R.; Lam, C.; Benedetti, A.; et al. Mutations in the glucose-6-phosphate transporter (g6pt) gene in patients with glycogen storage diseases type 1b and 1c. FEBS Lett. 1999, 459, 255–258. [Google Scholar] [CrossRef]
- Wang, Y.; Martin, C.C.; Oeser, J.K.; Sarkar, S.; McGuinness, O.P.; Hutton, J.C.; O’Brien, R.M. Deletion of the gene encoding the islet-specific glucose-6-phosphatase catalytic subunit-related protein autoantigen results in a mild metabolic phenotype. Diabetologia 2007, 50, 774–778. [Google Scholar] [CrossRef]
- Kuijpers, T.W.; Maianski, N.A.; Tool, A.T.; Smit, G.P.; Rake, J.P.; Roos, D.; Visser, G. Apoptotic neutrophils in the circulation of patients with glycogen storage disease type 1b (gsd1b). Blood 2003, 101, 5021–5024. [Google Scholar] [CrossRef]
- Chou, J.Y.; Jun, H.S.; Mansfield, B.C. Type i glycogen storage diseases: Disorders of the glucose-6-phosphatase/glucose-6-phosphate transporter complexes. J. Inherit. Metab. Dis. 2015, 38, 511–519. [Google Scholar] [CrossRef]
- Jun, H.S.; Weinstein, D.A.; Lee, Y.M.; Mansfield, B.C.; Chou, J.Y. Molecular mechanisms of neutrophil dysfunction in glycogen storage disease type ib. Blood 2014, 123, 2843–2853. [Google Scholar] [CrossRef]
- Hayee, B.; Antonopoulos, A.; Murphy, E.J.; Rahman, F.Z.; Sewell, G.; Smith, B.N.; McCartney, S.; Furman, M.; Hall, G.; Bloom, S.L.; et al. G6pc3 mutations are associated with a major defect of glycosylation: A novel mechanism for neutrophil dysfunction. Glycobiology 2011, 21, 914–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veiga-da-Cunha, M.; Chevalier, N.; Stephenne, X.; Defour, J.P.; Paczia, N.; Ferster, A.; Achouri, Y.; Dewulf, J.P.; Linster, C.L.; Bommer, G.T.; et al. Failure to eliminate a phosphorylated glucose analog leads to neutropenia in patients with g6pt and g6pc3 deficiency. Proc. Natl. Acad. Sci. USA 2019, 116, 1241–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.Y.; Pan, C.J.; Nandigama, K.; Mansfield, B.C.; Ambudkar, S.V.; Chou, J.Y. The glucose-6-phosphate transporter is a phosphate-linked antiporter deficient in glycogen storage disease type ib and ic. FASEB J. 2008, 22, 2206–2213. [Google Scholar] [CrossRef] [PubMed]
- Gerin, I.; Noel, G.; Van Schaftingen, E. Novel arguments in favor of the substrate-transport model of glucose-6-phosphatase. Diabetes 2001, 50, 1531–1538. [Google Scholar] [CrossRef] [Green Version]
- Marcolongo, P.; Fulceri, R.; Giunti, R.; Margittai, E.; Banhegyi, G.; Benedetti, A. The glucose-6-phosphate transport is not mediated by a glucose-6-phosphate/phosphate exchange in liver microsomes. FEBS Lett. 2012, 586, 3354–3359. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.Y.; Mansfield, B.C. The slc37 family of sugar-phosphate/phosphate exchangers. Curr. Top. Membr 2014, 73, 357–382. [Google Scholar]
- Meissner, G.; Allen, R. Evidence for two types of rat liver microsomes with differing permeability to glucose and other small molecules. J. Biol. Chem. 1981, 256, 6413–6422. [Google Scholar]
- Marcolongo, P.; Fulceri, R.; Giunti, R.; Burchell, A.; Benedetti, A. Permeability of liver microsomal membranes to glucose. Biochem. Biophys. Res. Commun. 1996, 219, 916–922. [Google Scholar] [CrossRef]
- Banhegyi, G.; Marcolongo, P.; Burchell, A.; Benedetti, A. Heterogeneity of glucose transport in rat liver microsomal vesicles. Arch. Biochem. Biophys. 1998, 359, 133–138. [Google Scholar] [CrossRef]
- Fehr, M.; Takanaga, H.; Ehrhardt, D.W.; Frommer, W.B. Evidence for high-capacity bidirectional glucose transport across the endoplasmic reticulum membrane by genetically encoded fluorescence resonance energy transfer nanosensors. Mol. Cell Biol. 2005, 25, 11102–11112. [Google Scholar] [CrossRef] [Green Version]
- Guillam, M.T.; Burcelin, R.; Thorens, B. Normal hepatic glucose production in the absence of glut2 reveals an alternative pathway for glucose release from hepatocytes. Proc. Natl. Acad. Sci. USA 1998, 95, 12317–12321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Gall, S.; Neuhof, A.; Rapoport, T. The endoplasmic reticulum membrane is permeable to small molecules. Mol. Biol. Cell 2004, 15, 447–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takanaga, H.; Frommer, W.B. Facilitative plasma membrane transporters function during er transit. FASEB J. 2010, 24, 2849–2858. [Google Scholar] [CrossRef] [PubMed]
- Lizak, B.; Czegle, I.; Csala, M.; Benedetti, A.; Mandl, J.; Banhegyi, G. Translocon pores in the endoplasmic reticulum are permeable to small anions. Am. J. Physiol. Cell Physiol. 2006, 291, C511–C517. [Google Scholar] [CrossRef]
- Roy, A.; Wonderlin, W.F. The permeability of the endoplasmic reticulum is dynamically coupled to protein synthesis. J. Biol. Chem. 2003, 278, 4397–4403. [Google Scholar] [CrossRef] [Green Version]
- Takanaga, H.; Chaudhuri, B.; Frommer, W.B. Glut1 and glut9 as major contributors to glucose influx in hepg2 cells identified by a high sensitivity intramolecular fret glucose sensor. Biochim. Biophys. Acta 2008, 1778, 1091–1099. [Google Scholar] [CrossRef] [Green Version]
- Gamberucci, A.; Marcolongo, P.; Nemeth, C.E.; Zoppi, N.; Szarka, A.; Chiarelli, N.; Hegedus, T.; Ritelli, M.; Carini, G.; Willaert, A.; et al. Glut10-lacking in arterial tortuosity syndrome-is localized to the endoplasmic reticulum of human fibroblasts. Int J. Mol. Sci. 2017, 18, 1820. [Google Scholar] [CrossRef] [Green Version]
- Santer, R.; Groth, S.; Kinner, M.; Dombrowski, A.; Berry, G.T.; Brodehl, J.; Leonard, J.V.; Moses, S.; Norgren, S.; Skovby, F.; et al. The mutation spectrum of the facilitative glucose transporter gene slc2a2 (glut2) in patients with fanconi-bickel syndrome. Hum. Genet. 2002, 110, 21–29. [Google Scholar] [CrossRef]
- Cherepanova, N.; Shrimal, S.; Gilmore, R. N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 2016, 41, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Tannous, A.; Pisoni, G.B.; Hebert, D.N.; Molinari, M. N-linked sugar-regulated protein folding and quality control in the er. Semin. Cell Dev. Biol. 2015, 41, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Villagran, M.; Munoz, M.; Inostroza, E.; Venegas, C.; Ruminot, I.; Parra-Valencia, E.; Maldonado, M.; Del Pozo, R.; Rivas, C.I.; Vera, J.C.; et al. Glut1 and glut8 support lactose synthesis in golgi of murine mammary epithelial cells. J. Physiol. Biochem. 2019, 75, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, P.; Juhasz, G. Autophagosome-lysosome fusion. J. Mol. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef]
- Sun, A. Lysosomal storage disease overview. Ann. Transl. Med. 2018, 6, 476. [Google Scholar] [CrossRef]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mtor. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef]
- Rong, Y.; McPhee, C.K.; Deng, S.; Huang, L.; Chen, L.; Liu, M.; Tracy, K.; Baehrecke, E.H.; Yu, L.; Lenardo, M.J. Spinster is required for autophagic lysosome reformation and mtor reactivation following starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 7826–7831. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, H.; Miyashita, T.; Nakano, Y.; Yamamoto, D. Hspin1, a transmembrane protein interacting with bcl-2/bcl-xl, induces a caspase-independent autophagic cell death. Cell Death Differ. 2003, 10, 798–807. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Lian, S.; Qi, J.; Bayliss, P.E.; Carr, C.E.; Johnson, J.L.; Guha, S.; Kobler, P.; Catz, S.D.; Gill, M.; et al. Aberrant autolysosomal regulation is linked to the induction of embryonic senescence: Differential roles of beclin 1 and p53 in vertebrate spns1 deficiency. PLoS Genet. 2014, 10, e1004409. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Lian, S.; Khan, A.; Llop, J.R.; Samuelson, A.V.; Chen, W.; Klionsky, D.J.; Kishi, S. Autolysosome biogenesis and developmental senescence are regulated by both spns1 and v-atpase. Autophagy 2017, 13, 386–403. [Google Scholar] [CrossRef]
- Yanagisawa, H.; Ishii, T.; Endo, K.; Kawakami, E.; Nagao, K.; Miyashita, T.; Akiyama, K.; Watabe, K.; Komatsu, M.; Yamamoto, D.; et al. L-leucine and spns1 coordinately ameliorate dysfunction of autophagy in mouse and human niemann-pick type c disease. Sci. Rep. 2017, 7, 15944. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Joost, H.G.; Schurmann, A. Glut8, the enigmatic intracellular hexose transporter. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E614–E618. [Google Scholar] [CrossRef] [PubMed]
- Maedera, S.; Mizuno, T.; Ishiguro, H.; Ito, T.; Soga, T.; Kusuhara, H. Glut6 is a lysosomal transporter that is regulated by inflammatory stimuli and modulates glycolysis in macrophages. FEBS Lett. 2019, 593, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shevchenko, A.; Jensen, O.N.; Podtelejnikov, A.V.; Sagliocco, F.; Wilm, M.; Vorm, O.; Mortensen, P.; Shevchenko, A.; Boucherie, H.; Mann, M. Linking genome and proteome by mass spectrometry: Large-scale identification of yeast proteins from two dimensional gels. Proc. Natl. Acad. Sci. USA 1996, 93, 14440–14445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using targetp, signalp and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef] [PubMed]
- Szarka, A.; Balogh, T. In silico aided thoughts on mitochondrial vitamin c transport. J. Theor. Biol. 2015, 365, 181–189. [Google Scholar] [CrossRef]
- Nakai, K.; Horton, P. Psort: A program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci. 1999, 24, 34–36. [Google Scholar] [CrossRef]
- Briesemeister, S.; Rahnenfuhrer, J.; Kohlbacher, O. Yloc—An interpretable web server for predicting subcellular localization. Nucleic Acids Res. 2010, 38, W497–W502. [Google Scholar] [CrossRef]
- Yu, C.S.; Cheng, C.W.; Su, W.C.; Chang, K.C.; Huang, S.W.; Hwang, J.K.; Lu, C.H. Cello2go: A web server for protein subcellular localization prediction with functional gene ontology annotation. PLoS ONE 2014, 9, e99368. [Google Scholar] [CrossRef] [Green Version]
- Kc, S.; Carcamo, J.M.; Golde, D.W. Vitamin c enters mitochondria via facilitative glucose transporter 1 (glut1) and confers mitochondrial protection against oxidative injury. FASEB J. 2005, 19, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Q.; Cheung, L.S.; Feng, L.; Tanner, W.; Frommer, W.B. Transport of sugars. Annu. Rev. Biochem. 2015, 84, 865–894. [Google Scholar] [CrossRef] [PubMed]
- Stuart, C.A.; Howell, M.E.; Zhang, Y.; Yin, D. Insulin-stimulated translocation of glucose transporter (glut) 12 parallels that of glut4 in normal muscle. J. Clin. Endocrinol. Metab. 2009, 94, 3535–3542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uldry, M.; Ibberson, M.; Horisberger, J.D.; Chatton, J.Y.; Riederer, B.M.; Thorens, B. Identification of a mammalian h(+)-myo-inositol symporter expressed predominantly in the brain. EMBO J. 2001, 20, 4467–4477. [Google Scholar] [CrossRef]
- Doege, H.; Bocianski, A.; Joost, H.G.; Schurmann, A. Activity and genomic organization of human glucose transporter 9 (glut9), a novel member of the family of sugar-transport facilitators predominantly expressed in brain and leucocytes. Biochem. J. 2000, 350 Pt 3, 771–776. [Google Scholar] [CrossRef]
- Lisinski, I.; Schurmann, A.; Joost, H.G.; Cushman, S.W.; Al-Hasani, H. Targeting of glut6 (formerly glut9) and glut8 in rat adipose cells. Biochem. J. 2001, 358, 517–522. [Google Scholar] [CrossRef]
- Porpaczy, E.; Bilban, M.; Heinze, G.; Gruber, M.; Vanura, K.; Schwarzinger, I.; Stilgenbauer, S.; Streubel, B.; Fonatsch, C.; Jaeger, U. Gene expression signature of chronic lymphocytic leukaemia with trisomy 12. Eur. J. Clin. Investig. 2009, 39, 568–575. [Google Scholar] [CrossRef]
- Byrne, F.L.; Poon, I.K.; Modesitt, S.C.; Tomsig, J.L.; Chow, J.D.; Healy, M.E.; Baker, W.D.; Atkins, K.A.; Lancaster, J.M.; Marchion, D.C.; et al. Metabolic vulnerabilities in endometrial cancer. Cancer Res. 2014, 74, 5832–5845. [Google Scholar] [CrossRef]
- Caruana, B.T.; Byrne, F.L.; Knights, A.J.; Quinlan, K.G.R.; Hoehn, K.L. Characterization of glucose transporter 6 in lipopolysaccharide-induced bone marrow-derived macrophage function. J. Immunol. 2019, 202, 1826–1832. [Google Scholar] [CrossRef]
- Byrne, F.L.; Olzomer, E.M.; Brink, R.; Hoehn, K.L. Knockout of glucose transporter glut6 has minimal effects on whole body metabolic physiology in mice. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E286–E293. [Google Scholar] [CrossRef]
- Ibberson, M.; Uldry, M.; Thorens, B. Glutx1, a novel mammalian glucose transporter expressed in the central nervous system and insulin-sensitive tissues. J. Biol. Chem. 2000, 275, 4607–4612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doege, H.; Schurmann, A.; Bahrenberg, G.; Brauers, A.; Joost, H.G. Glut8, a novel member of the sugar transport facilitator family with glucose transport activity. J. Biol. Chem. 2000, 275, 16275–16280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joost, H.G.; Thorens, B. The extended glut-family of sugar/polyol transport facilitators: Nomenclature, sequence characteristics, and potential function of its novel members (review). Mol. Membr. Biol. 2001, 18, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Corpe, C.P.; Eck, P.; Wang, J.; Al-Hasani, H.; Levine, M. Intestinal dehydroascorbic acid (dha) transport mediated by the facilitative sugar transporters, glut2 and glut8. J. Biol. Chem. 2013, 288, 9092–9101. [Google Scholar] [CrossRef] [Green Version]
- Augustin, R.; Riley, J.; Moley, K.H. Glut8 contains a [de]xxxl[li] sorting motif and localizes to a late endosomal/lysosomal compartment. Traffic 2005, 6, 1196–1212. [Google Scholar] [CrossRef] [PubMed]
- Aerni-Flessner, L.B.; Otu, M.C.; Moley, K.H. The amino acids upstream of nh(2)-terminal dileucine motif play a role in regulating the intracellular sorting of the class iii transporters glut8 and glut12. Mol. Membr. Biol. 2011, 28, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, U.; Briese, S.; Leicht, K.; Schurmann, A.; Joost, H.G.; Al-Hasani, H. Endocytosis of the glucose transporter glut8 is mediated by interaction of a dileucine motif with the beta2-adaptin subunit of the ap-2 adaptor complex. J. Cell Sci. 2006, 119, 2321–2331. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, I.V.; Martinez-Arca, S.; Valdueza, J.; Palacios, S.; Holman, G.D. Distinct reading of different structural determinants modulates the dileucine-mediated transport steps of the lysosomal membrane protein limpii and the insulin-sensitive glucose transporter glut4. J. Biol. Chem. 2000, 275, 39874–39885. [Google Scholar] [CrossRef] [Green Version]
- Uldry, M.; Thorens, B. The slc2 family of facilitated hexose and polyol transporters. Pflug. Arch. Eur. J. Physiol. 2004, 447, 480–489. [Google Scholar] [CrossRef] [Green Version]
- Wilson-O’Brien, A.L.; Dehaan, C.L.; Rogers, S. Mitogen-stimulated and rapamycin-sensitive glucose transporter 12 targeting and functional glucose transport in renal epithelial cells. Endocrinology 2008, 149, 917–924. [Google Scholar] [CrossRef] [Green Version]
- Shin, B.C.; McKnight, R.A.; Devaskar, S.U. Glucose transporter glut8 translocation in neurons is not insulin responsive. J. Neurosci. Res. 2004, 75, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Widmer, M.; Uldry, M.; Thorens, B. Glut8 subcellular localization and absence of translocation to the plasma membrane in pc12 cells and hippocampal neurons. Endocrinology 2005, 146, 4727–4736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashima, M.; Chiba, Y.; Murakami, R.; Uemura, N.; Matsumoto, K.; Kawauchi, M.; Kanenishi, K.; Hata, T.; Ueno, M. Glucose transporter 8 immunoreactivity in astrocytic and microglial cells in subependymal areas of human brains. Neurosci. Lett. 2017, 636, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Gomez, O.; Terrado, J.; Mesonero, J.E. Expression of glut8 in mouse intestine: Identification of alternative spliced variants. J. Cell Biochem. 2009, 106, 1068–1078. [Google Scholar] [CrossRef]
- DeBosch, B.J.; Chi, M.; Moley, K.H. Glucose transporter 8 (glut8) regulates enterocyte fructose transport and global mammalian fructose utilization. Endocrinology 2012, 153, 4181–4191. [Google Scholar] [CrossRef]
- Carayannopoulos, M.O.; Chi, M.M.; Cui, Y.; Pingsterhaus, J.M.; McKnight, R.A.; Mueckler, M.; Devaskar, S.U.; Moley, K.H. Glut8 is a glucose transporter responsible for insulin-stimulated glucose uptake in the blastocyst. Proc. Natl. Acad. Sci. USA 2000, 97, 7313–7318. [Google Scholar] [CrossRef] [Green Version]
- Maria, Z.; Campolo, A.R.; Lacombe, V.A. Diabetes alters the expression and translocation of the insulin-sensitive glucose transporters 4 and 8 in the atria. PLoS ONE 2015, 10, e0146033. [Google Scholar] [CrossRef]
- Maria, Z.; Campolo, A.R.; Scherlag, B.J.; Ritchey, J.W.; Lacombe, V.A. Dysregulation of insulin-sensitive glucose transporters during insulin resistance-induced atrial fibrillation. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2018, 1864, 987–996. [Google Scholar] [CrossRef]
- Debosch, B.J.; Chen, Z.; Saben, J.L.; Finck, B.N.; Moley, K.H. Glucose transporter 8 (glut8) mediates fructose-induced de novo lipogenesis and macrosteatosis. J. Biol. Chem. 2014, 289, 10989–10998. [Google Scholar] [CrossRef] [Green Version]
- Mayer, A.L.; Higgins, C.B.; Heitmeier, M.R.; Kraft, T.E.; Qian, X.; Crowley, J.R.; Hyrc, K.L.; Beatty, W.L.; Yarasheski, K.E.; Hruz, P.W.; et al. Slc2a8 (glut8) is a mammalian trehalose transporter required for trehalose-induced autophagy. Sci. Rep. 2016, 6, 38586. [Google Scholar] [CrossRef]
- McVie-Wylie, A.J.; Lamson, D.R.; Chen, Y.T. Molecular cloning of a novel member of the glut family of transporters, slc2a10 (glut10), localized on chromosome 20q13.1: A candidate gene for niddm susceptibility. Genomics 2001, 72, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Mychaleckyj, J.C.; Fossey, S.C.; Mihic, S.J.; Craddock, A.L.; Bowden, D.W. Sequence and functional analysis of glut10: A glucose transporter in the type 2 diabetes-linked region of chromosome 20q12-13.1. Mol. Genet. Metab. 2001, 74, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Coucke, P.J.; Willaert, A.; Wessels, M.W.; Callewaert, B.; Zoppi, N.; De Backer, J.; Fox, J.E.; Mancini, G.M.; Kambouris, M.; Gardella, R.; et al. Mutations in the facilitative glucose transporter glut10 alter angiogenesis and cause arterial tortuosity syndrome. Nat. Genet. 2006, 38, 452–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drera, B.; Guala, A.; Zoppi, N.; Gardella, R.; Franceschini, P.; Barlati, S.; Colombi, M. Two novel slc2a10/glut10 mutations in a patient with arterial tortuosity syndrome. Am. J. Med. Genet. A 2007, 143A, 216–218. [Google Scholar] [CrossRef]
- Callewaert, B.L.; Willaert, A.; Kerstjens-Frederikse, W.S.; De Backer, J.; Devriendt, K.; Albrecht, B.; Ramos-Arroyo, M.A.; Doco-Fenzy, M.; Hennekam, R.C.; Pyeritz, R.E.; et al. Arterial tortuosity syndrome: Clinical and molecular findings in 12 newly identified families. Hum. Mutat. 2008, 29, 150–158. [Google Scholar] [CrossRef]
- Faiyaz-Ul-Haque, M.; Zaidi, S.H.; Al-Sanna, N.; Alswaid, A.; Momenah, T.; Kaya, N.; Al-Dayel, F.; Bouhoaigah, I.; Saliem, M.; Tsui, L.C.; et al. A novel missense and a recurrent mutation in slc2a10 gene of patients affected with arterial tortuosity syndrome. Atherosclerosis 2009, 203, 466–471. [Google Scholar] [CrossRef]
- Zaidi, S.H.; Meyer, S.; Peltekova, V.D.; Lindinger, A.; Teebi, A.S.; Faiyaz-Ul-Haque, M. A novel non-sense mutation in the slc2a10 gene of an arterial tortuosity syndrome patient of kurdish origin. Eur. J. Pediatr. 2009, 168, 867–870. [Google Scholar] [CrossRef]
- Faiyaz-Ul-Haque, M.; Zaidi, S.H.; Wahab, A.A.; Eltohami, A.; Al-Mureikhi, M.S.; Al-Thani, G.; Peltekova, V.D.; Tsui, L.C.; Teebi, A.S. Identification of a p.Ser81arg encoding mutation in slc2a10 gene of arterial tortuosity syndrome patients from 10 qatari families. Clin. Genet. 2008, 74, 189–193. [Google Scholar] [CrossRef]
- Castori, M.; Ritelli, M.; Zoppi, N.; Molisso, L.; Chiarelli, N.; Zaccagna, F.; Grammatico, P.; Colombi, M. Adult presentation of arterial tortuosity syndrome in a 51-year-old woman with a novel homozygous c.1411+1g>a mutation in the slc2a10 gene. Am. J. Med. Genet. A 2012, 158A, 1164–1169. [Google Scholar] [CrossRef]
- Karakurt, C.; Kocak, G.; Elkiran, O.; Coucke, P.J.; Van Maldergem, L. Arterial tortuosity syndrome: Case report. Genet. Couns. 2012, 23, 477–482. [Google Scholar]
- Takahashi, Y.; Fujii, K.; Yoshida, A.; Morisaki, H.; Kohno, Y.; Morisaki, T. Artery tortuosity syndrome exhibiting early-onset emphysema with novel compound heterozygous slc2a10 mutations. Am. J. Med. Genet. A 2013, 161A, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Ritelli, M.; Chiarelli, N.; Dordoni, C.; Reffo, E.; Venturini, M.; Quinzani, S.; Monica, M.D.; Scarano, G.; Santoro, G.; Russo, M.G.; et al. Arterial tortuosity syndrome: Homozygosity for two novel and one recurrent slc2a10 missense mutations in three families with severe cardiopulmonary complications in infancy and a literature review. BMC Med. Genet. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyens, A.; Albuisson, J.; Boel, A.; Al-Essa, M.; Al-Manea, W.; Bonnet, D.; Bostan, O.; Boute, O.; Busa, T.; Canham, N.; et al. Arterial tortuosity syndrome: 40 new families and literature review. Genet. Med. 2018, 20, 1236–1245. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, M.T.; Urrutia, R.; Cousin, M.A.; Oliver, G.R.; Klee, E.W. Assessing human genetic variations in glucose transporter slc2a10 and their role in altering structural and functional properties. Front. Genet. 2018, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Huang, H.Y.; Chang, C.J.; Cheng, C.H.; Chen, Y.T. Mitochondrial glut10 facilitates dehydroascorbic acid import and protects cells against oxidative stress: Mechanistic insight into arterial tortuosity syndrome. Hum. Mol. Genet. 2010, 19, 3721–3733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Montesino, C.; Roa, F.J.; Pena, E.; Gonzalez, M.; Sotomayor, K.; Inostroza, E.; Munoz, C.A.; Gonzalez, I.; Maldonado, M.; Soliz, C.; et al. Mitochondrial ascorbic acid transport is mediated by a low-affinity form of the sodium-coupled ascorbic acid transporter-2. Free Radic Biol. Med. 2014, 70, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Segade, F. Glucose transporter 10 and arterial tortuosity syndrome: The vitamin c connection. FEBS Lett. 2010, 584, 2990–2994. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, C.E.; Marcolongo, P.; Gamberucci, A.; Fulceri, R.; Benedetti, A.; Zoppi, N.; Ritelli, M.; Chiarelli, N.; Colombi, M.; Willaert, A.; et al. Glucose transporter type 10-lacking in arterial tortuosity syndrome-facilitates dehydroascorbic acid transport. FEBS Lett. 2016, 590, 1630–1640. [Google Scholar] [CrossRef] [Green Version]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martinez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin c induces tet-dependent DNA demethylation and a blastocyst-like state in es cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef]
- Butler, J.S.; Koutelou, E.; Schibler, A.C.; Dent, S.Y. Histone-modifying enzymes: Regulators of developmental decisions and drivers of human disease. Epigenomics 2012, 4, 163–177. [Google Scholar] [CrossRef] [Green Version]
- Pastor, W.A.; Aravind, L.; Rao, A. Tetonic shift: Biological roles of tet proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banhegyi, G.; Benedetti, A.; Margittai, E.; Marcolongo, P.; Fulceri, R.; Nemeth, C.E.; Szarka, A. Subcellular compartmentation of ascorbate and its variation in disease states. Biochim. Biophys. Acta 2014, 1843, 1909–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, C.E.; Nemoda, Z.; Low, P.; Szabo, P.; Horvath, E.Z.; Willaert, A.; Boel, A.; Callewaert, B.L.; Coucke, P.J.; Colombi, M.; et al. Decreased nuclear ascorbate accumulation accompanied with altered genomic methylation pattern in fibroblasts from arterial tortuosity syndrome patients. Oxid. Med. Cell Longev. 2019, 2019, 8156592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoppi, N.; Chiarelli, N.; Cinquina, V.; Ritelli, M.; Colombi, M. Glut10 deficiency leads to oxidative stress and non-canonical alphavbeta3 integrin-mediated tgfbeta signalling associated with extracellular matrix disarray in arterial tortuosity syndrome skin fibroblasts. Hum. Mol. Genet. 2015, 24, 6769–6787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novakovic, B.; Gordon, L.; Robinson, W.P.; Desoye, G.; Saffery, R. Glucose as a fetal nutrient: Dynamic regulation of several glucose transporter genes by DNA methylation in the human placenta across gestation. J. Nutr. Biochem. 2013, 24, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Dermaut, B.; Norga, K.K.; Kania, A.; Verstreken, P.; Pan, H.; Zhou, Y.; Callaerts, P.; Bellen, H.J. Aberrant lysosomal carbohydrate storage accompanies endocytic defects and neurodegeneration in drosophila benchwarmer. J. Cell Biol. 2005, 170, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, Y.; Fujitani, K.; Kurihara, J.; Ragan, J.; Usui-Aoki, K.; Shimoda, L.; Lukacsovich, T.; Suzuki, K.; Sezaki, M.; Sano, Y.; et al. Mutations in the novel membrane protein spinster interfere with programmed cell death and cause neural degeneration in drosophila melanogaster. Mol. Cell Biol. 2001, 21, 3775–3788. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, S.T.; Davis, G.W. Unrestricted synaptic growth in spinster-a late endosomal protein implicated in tgf-beta-mediated synaptic growth regulation. Neuron 2002, 36, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Young, R.M.; Marty, S.; Nakano, Y.; Wang, H.; Yamamoto, D.; Lin, S.; Allende, M.L. Zebrafish yolk-specific not really started (nrs) gene is a vertebrate homolog of the drosophila spinster gene and is essential for embryogenesis. Dev. Dyn. 2002, 223, 298–305. [Google Scholar] [CrossRef]
- Johnson, A.E.; Shu, H.; Hauswirth, A.G.; Tong, A.; Davis, G.W. Vcp-dependent muscle degeneration is linked to defects in a dynamic tubular lysosomal network in vivo. eLife 2015, 4. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Transporter | Location | PsortII | yLoc | Cello2GO |
|---|---|---|---|---|
| Glut1 | Plasma membrane | 65.2% | 93.9% | 4.937 (98.74%) |
| ER | 17.4% | 0.2% | ||
| Vacuolar pathway | 8.7% | 0.2% | ||
| Peroxisome | 5.9% | |||
| Cytoplasm | 0.014 (0.28%) | |||
| Nucleus | 0.009 (0.18%) | |||
| Glut2 | Plasma membrane | 69.6% | 86.3% | 4.963 (99.26%) |
| ER | 26.1% | 0.8% | ||
| Extracellular space | 12.7% | |||
| Peroxisome | 0.007 (0.14%) | |||
| Mitochondrion | 4.3% | |||
| Nucleus | 0.007 (0.14%) | |||
| Glut3 | Plasma membrane | 78.3% | 90.0% | 4.930 (98.6%) |
| ER | 17.4% | 0.5% | ||
| Extracellular space | 9.4% | |||
| Lysosome | 0.011 (0.22%) | |||
| Peroxisome | 0.026 (0.52%) | |||
| Mitochondrion | 4.3% | |||
| Glut4 | Plasma membrane | 60.9% | 88.3% | 4.957 (99.14%) |
| ER | 17.4% | |||
| Extracellular space | 0.011 (0.22%) | |||
| Vacuolar | 8.7% | |||
| Peroxisome | 10.9% | 0.005 (0.1%) | ||
| Cytoplasm | 0.4% | |||
| Glut5 | Plasma membrane | 52.2% | 53.6% | 4.970 (99.4%) |
| ER | 34.8% | |||
| Extracellular space | 1.9% | 0.006 (0.12%) | ||
| Peroxisome | 43.7% | 0.006 (0.12%) | ||
| Mitochondrion | 4.3% | |||
| Glut6 | Plasma membrane | 60.9% | 54.0% | 4.970 (99.4%) |
| ER | 17.4% | |||
| Extracellular space | 0.1% | |||
| Vacuolar | 8.7% | |||
| Peroxisome | 45.8% | |||
| Mitochondrion | 0.009 (0.18%) | |||
| Lysosome | 0.009 (0.18%) | |||
| Glut7 | Plasma membrane | 60.9% | 72.5% | 4.943 (98.86%) |
| ER | 21.7% | 0.1% | ||
| Vacuolar | 8.7% | |||
| Peroxisome | 27.4% | 0.019 (0.38%) | ||
| Mitochondrion | 0.012 (0.24%) | |||
| Glut8 | Plasma membrane | 69.6% | 98.8% | 4.949 (98.98%) |
| ER | 26.1% | |||
| Peroxisome | 1.0% | |||
| Mitochondrion | 4.3% | 0.008 (0.16%) | ||
| Lysosome | 0.1% | 0.018 (0.36%) | ||
| Glut9 | Plasma membrane | 73.9% | 2.0% | 4.961 (99.22%) |
| ER | 21.7% | |||
| Peroxisome | 94.1% | 0.010 (0.2%) | ||
| Mitochondrion | 4.3% | |||
| Cytoplasm | 3.2% | |||
| Lysosome | 0.006 (0.12%) | |||
| Glut10 | Plasma membrane | 43.5% | 99.8% | 4.853 (97.06%) |
| ER | 39.1% | 0.1% | ||
| Extracellular space | 0.1% | 0.061 (1.22%) | ||
| Lysosome | 0.028 (0.56%) | |||
| Mitochondrion | 4.3% | |||
| Glut11 | Plasma membrane | 44.4% | 75.1% | 4.924 (98.48%) |
| ER | 55.6% | 19.6% | ||
| Extracellular space | 5.3% | |||
| Peroxisome | 0.014 (0.28%) | |||
| Mitochondrion | 0.017 (0.34%) | |||
| Glut12 | Plasma membrane | 82.6% | 45.3% | 4.958 (99.16%) |
| ER | 17.4% | 0.4% | ||
| Peroxisome | 54.1% | |||
| Mitochondrion | 0.005 (0.1%) | |||
| Lysosome | 0.005 (0.1%) | |||
| Glut13 | Plasma membrane | 65.2% | 14.6% | 4.879 (97.58%) |
| ER | 17.4% | |||
| Vacuolar | 8.7% | 0.023 (0.46%) | ||
| Peroxisome | 77.9% | |||
| Mitochondrion | 0.016 (0.32%) | |||
| Cytoplasm | 5.2% | |||
| Glut14 | Plasma membrane | 65.2% | 99.5% | 4.971 (99.42%) |
| ER | 26.1% | |||
| Extracellular space | 0.4% | |||
| Peroxisome | 0.1% | 0.011 (0.22%) | ||
| Nucleus | 4.3% | |||
| Lysosome | 0.004 (0.08%) |
| Transporter | Location | PSORT II | yLoc | Cello |
|---|---|---|---|---|
| SGLT1 | Plasma membrane | 73.9% | 99.2% | 4.969 (99.38%) |
| ER | 13.0% | 0.3% | 0.004 (0.08%) | |
| Vacuolar | 8.7% | |||
| Peroxisome | 0.3% | |||
| Nucleus | 0.005 (0.1%) | |||
| SGLT2 | Plasma membrane | 69.6% | 99.8% | 4.966 (99.32%) |
| ER | 13.0% | |||
| Vacuolar | 8.7% | |||
| Peroxisome | 0.1% | |||
| Mitochondrion | 0.003 (0.06%) | |||
| Lysosome | 0.006 (0.12%) | |||
| SGLT3 | Plasma membrane | 69.6% | 99.5% | 4.981 (99.62%) |
| ER | 13.0% | 0.3% | 0.003 (0.06%) | |
| Vacuolar | 8.7% | |||
| Lysosome | 0.2% | 0.002 (0.04%) | ||
| SGLT4 | Plasma membrane | 82.6% | 99.9% | 4.967 (99.34%) |
| ER | 13.0% | |||
| Peroxisome | 0.004 (0.08%) | |||
| Mitochondrion | 4.3% | 0.004 (0.08%) | ||
| SGLT5 | Plasma membrane | 69.6% | 99.9% | 4.968 (99.36%) |
| ER | 13.0% | |||
| Vacuolar | 8.7% | |||
| Nucleus | 0.007 (0.14%) | |||
| Lysosome | 0.008 (0.16%) |
| Transporter | Location | PSORT II | yLoc | Cello |
|---|---|---|---|---|
| SPNS1 | Plasma membrane | 73.9% | 74.2% | 4.970 (99.4%) |
| ER | 21.7% | |||
| Extracellular space | 0.007 (0.14%) | |||
| Peroxisome | 25.3% | 0.004 (0.08%) | ||
| Mitochondrion | 4.3% | |||
| Cytoplasm | 0.3% | |||
| SPNS2 | Plasma membrane | 60.9% | 78.8% | 4.897 (97.94%) |
| ER | 21.7% | |||
| Vacuolar | 8.7% | |||
| Peroxisome | 20.9% | 0.016 (0.32%) | ||
| Mitochondrion | 0.023 (0.46%) | |||
| Nucleus | 0.3% |
| Transporter | Location | PSORT II | yLoc | Cello |
|---|---|---|---|---|
| hSWEET1 | Plasma membrane | 22.2% | 80.6% | 4.881 (97.62%) |
| ER | 33.3% | |||
| Extracellular space | 18.4% | 0.020 (0.4%) | ||
| Vacuolar | 22.2% | |||
| Nucleus | 0.030 (0.6%) | |||
| Lysosome | 0.6% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lizák, B.; Szarka, A.; Kim, Y.; Choi, K.-s.; Németh, C.E.; Marcolongo, P.; Benedetti, A.; Bánhegyi, G.; Margittai, É. Glucose Transport and Transporters in the Endomembranes. Int. J. Mol. Sci. 2019, 20, 5898. https://doi.org/10.3390/ijms20235898
Lizák B, Szarka A, Kim Y, Choi K-s, Németh CE, Marcolongo P, Benedetti A, Bánhegyi G, Margittai É. Glucose Transport and Transporters in the Endomembranes. International Journal of Molecular Sciences. 2019; 20(23):5898. https://doi.org/10.3390/ijms20235898
Chicago/Turabian StyleLizák, Beáta, András Szarka, Yejin Kim, Kyu-sung Choi, Csilla E. Németh, Paola Marcolongo, Angelo Benedetti, Gábor Bánhegyi, and Éva Margittai. 2019. "Glucose Transport and Transporters in the Endomembranes" International Journal of Molecular Sciences 20, no. 23: 5898. https://doi.org/10.3390/ijms20235898
APA StyleLizák, B., Szarka, A., Kim, Y., Choi, K. -s., Németh, C. E., Marcolongo, P., Benedetti, A., Bánhegyi, G., & Margittai, É. (2019). Glucose Transport and Transporters in the Endomembranes. International Journal of Molecular Sciences, 20(23), 5898. https://doi.org/10.3390/ijms20235898